Abstract
Dendritic cell (DC)-based immunotherapy has been shown to be a promising strategy for anti-cancer therapy. Nevertheless, only a low overall clinical response rate has been observed in vaccinated patients with advanced cancer and therefore methods to improve DC immuno-stimulatory functions are currently under intense investigation. In this respect, we exposed human monocyte-derived DCs to a physiological temperature stress of 40°C for up to 24 h followed by analysis for (i) expression of different heat shock proteins, (ii) survival, (iii) cell surface maturation markers, (iv) cytokine secretion, and (v) migratory capacity. Furthermore, we examined the ability of heat-shocked DCs to prime naïve CD8+ T cells after loading with MelanA peptide, by transfection with MelanA RNA, or by transduction with MelanA by an adenovirus vector. The results clearly indicate that in comparison to control DCs, which remained at 37°C, heat-treated cells revealed no differences concerning the survival rate or their migratory capacity. However, DCs exposed to thermal stress showed a time-dependent enhanced expression of the immune-chaperone heat shock protein 70A and both an up-regulation of co-stimulatory molecules such as CD80, CD83, and CD86 and of the inflammatory cytokine TNF-α. Moreover, these cells had a markedly improved capacity to prime autologous naïve CD8+ T cells in vitro in an antigen-specific manner, independent of the method of antigen-loading. Thus, our strategy of heat treatment of DCs offers a promising means to improve DC functions during immune activation which, as a physical method, facilitates straight-forward applications in clinical DC vaccination protocols.
Introduction
Dendritic cells (DCs) are unique antigen presenting cells (APCs) involved in the induction of primary immune responses Citation[1–3], which require the participation of different cells of the immune system, such as T and B lymphocytes, orchestrated by activated DCs. In the steady state, DCs reside in peripheral tissues and lymphoid organs, while several subsets of DCs also circulate in the blood. Upon encounter of pathogens, microbes or other ‘danger’ signals (e.g. microbial products), DCs undergo a complex process of morphological changes Citation[4–6], e.g. the loss of adhesive structures and the acquisition of high cellular motility Citation[7]. The latter comprises the enhanced expression of CC and CXC chemokine receptors like CCR7 and CXCR4 on the cell surface of activated mature DCs. As a consequence, these DCs show an enhanced response to chemokines CCL19 (MIP-3β) and CXCL12 (SDF-1α). Citation[8], Citation[9] The necessity of CCR7 and CXCR4 expression for the migration of human DCs has been demonstrated recently by Prechtel and colleagues Citation[10], showing that HSV-1 mediated down-regulation of CCR7 and CXCR4 abrogates the migratory capacity of mature DCs in vitro. Beyond that activated DCs undergo maturation and develop from an antigen-capturing cell into an APC. Thereby, activated DCs up-regulate co-stimulatory molecules such as CD40, CD80, CD86, as well as the maturation markers CD25 and CD83 Citation[11–13], translocate major histocompatibility complex (MHC) class II compartments to the cell surface Citation[14], Citation[15], and start to secrete cytokines such as interleukin-12p70 (IL-12p70) and TNF-α that differentiate and polarise the attracted immune effectors Citation[16]. Hence, DCs can guide CD4+ T cells to become either a T helper cell (TH) producing interferon-γ (IFN-γ) (TH1), or IL-4, -5 and -13 (TH2), an IL-17-secreting TH17 cell, or a FOXP3+ regulatory T cell (Treg) producing immune suppressive cytokines such as IL-10 and TGF-β Citation[16], Citation[17]. Moreover, DCs are able to present exogenous antigens via MHC class I to cytotoxic CD8+ T cells (CTLs), a process called ‘cross-presentation’, which results in a powerful activation of CTLs Citation[18–20]. Besides its essential activity in natural immune responses, CTL activation by DCs is pivotal for the induction of DC-mediated anti-tumour immune responses during tumour vaccination Citation[21].
For DC vaccination, tumour antigens are provided to DCs in different forms such as (i) incubation with naïve or engineered peptides, (ii) whole tumour cell lysates, (iii) purified proteins, (iv) RNA encoding tumour-associated antigens (TAAs) or total tumour RNA, or (v) viral transduction of TAAs Citation[12], Citation[22–25]. However, despite promising results in selected cases, the clinical efficacy of DC immunotherapy was limited Citation[26], Citation[27]. This might be due to the fact that the majority of tumours, like most normal tissue, lack co-stimulatory properties and release no ‘danger’ signals. Thus, tumour antigens are cross-presented in a tolerising way Citation[28–30], leading to weak T cell responses. Consequently, there is a high need to improve the T cell stimulatory capacity of DCs for tolerance-breaking vaccination protocols. One promising strategy is the application of physiologically relevant mild thermal stress which has been shown recently to be associated with enhanced DC function Citation[31], Citation[32]. For murine DCs, strong evidence arose that mild elevation of temperature has the potential to enhance antigen uptake, activation associated migration, maturation, cytokine expression and T cell stimulatory capacity of DCs Citation[32]. However, comparably little is published so far about the effects of heat treatment on the immunological functions of human DCs, especially considering antigen-specific responses. Regarding the immunological mechanisms, the heat-mediated induction of heat shock proteins seems to play a major role as it has been demonstrated for murine and human DCs that heat treatment increases the expression of Hsp70 and/or Hsp90 thereby enhancing the capacity of DCs to stimulate antigen-specific T cells Citation[31], Citation[33–35]. The assumed underlying mechanisms are heat-mediated DC maturation accompanied by increased expression of co-stimulatory molecules, as well as enhanced processing of tumour-derived antigens to MHC class I molecules resulting in an increased CTL response.
In the light of these recent findings we investigated in the present in vitro study, whether exposure of human monocyte-derived DCs to mild thermal stress not only enhances the expression of different heat shock proteins, but also affects DC functions, particularly during antigen presentation and T cell activation. Moreover, in the context of priming naïve T cells, we addressed the crucial issue of the order of antigen-loading and heat treatment of DCs.
Material and methods
DC generation
Human DCs were generated as described before Citation[12]. In short, peripheral blood mononuclear cells (PBMCs) were prepared from leukapheresis products of healthy donors (obtained following informed consent and approved by the institutional review board) by density centrifugation using Lymphoprep (Alexis-Shield, Oslo, Norway), followed by plastic adherence. The non-adherent cell fraction was removed and the adherent cell fraction was cultured in DC-medium consisting of RPMI 1640 (Lonza, Basle, Switzerland) supplemented with each 1% of heat-inactivated autologous plasma, Penicillin/Streptomycin/L-Glutamine (PAA, Pasching, Austria) and HEPES (Lonza). On days 1 and 4, 5 mL of fresh medium additionally supplemented with recombinant human granulocyte macrophage colony-stimulating factor (GM-CSF) and recombinant IL-4 (both Cell Genix, Freiburg, Germany) were added. Final concentrations for days 1 and 4 were 800 U/mL and 400 U/mL of GM-CSF, respectively, and 250 U/mL of IL-4 on days 1 and 4. On day 6, immature DC (iDCs) were used for the heat shock-treatment, either cultured in medium containing 400 U/mL GM-CSF and 250 U/mL IL-4 (for iDCs) or additionally (for mature DCs) consisting of 200 U/mL IL-1ß, 1000 U/mL IL-6 (both Cell Genix), 10 ng/mL TNF-α (Beromun; Boehringer Ingelheim, Ingelheim am Rhein, Germany) and 1 µg/mL PGE2 (Prostin E2, Pfizer, Karlsruhe, Germany).
Heat shock of DCs
One million DCs per well were seeded in a 12-well tissue culture plate (BD Biosciences, Heidelberg, Germany) and exposed to mild thermal heat of 40°C for 3-, 6-, 12-, or 24 h either in the presence or absence of maturation cocktail (MC). Afterwards, heat-treated cells were harvested directly or after further incubation at 37°C for 24 h and used for further procedures.
Flow cytometric analysis
Cells were stained with specific mAb or appropriate isotype controls for 30 min at 4°C in FACS buffer (Dulbecco's PBS (Lonza) containing 2% FBS (PAA)), washed twice and finally resuspended in cold FACS buffer containing 0.1 µg/mL propidiumiodide (PI) (Carl Roth, Karlsruhe, Germany). Stained cells were immediately analysed with a FACScan flow cytometer (BD Biosciences). Cell debris and dead cells were excluded from the analysis by gating on proper forward and sideward light scatter and on PI negative cells. A minimum of 104 living cells derived from a single well per condition were analysed for each sample and results were exploited using CellQuest software (BD Biosciences). The following monoclonal antibodies (all from BD Biosciences) were used to determine the phenotype of DCs and CD8+ T cells: FITC-labelled mouse anti-human CD3 (HIT3a) and CD8 (RPA-T8), PE-labelled mouse anti-human CD4 (RPA-T4), CD25 (M-A251), CD40 (5C3), CD80 (L307.4), CD83 (HB15e), CD86 (IT2.2), HLA-A,B,C (G46-2.6), HLA-DR (G46.6), CCR7 (150503), and CXCR4 (12G5). Isotype mAb controls (all from BD Biosciences) used were: IgG1-FITC, IgG1-PE (both MOPC-21), IgG2a-PE (G155-178) and IgG2b-PE (Citation[27–35]).
Cytometric bead array
Determination of TH1/TH2- and inflammatory cytokines secreted by heat-treated DCs into the supernatant within 24 to 48 h was assessed by a human inflammatory and a human TH1/TH2 cytokine cytrometric bead array assay (BD Biosciences) according to the manufacturer's protocol.
Western blot analyses
Whole cell extracts for SDS-PAGE were generated as follows: 1 × 106 DCs were washed with ice-cold PBS and then resuspended in 50 µL lysis buffer mixed with NaVO3, NaF and phenylmethylsulfonyl fluoride (PMSF). Lysates were shaken for at least 1.5 h at 4°C before centrifugation. Afterwards supernatants were transferred to a fresh reaction tube and protein concentration was determined by the Bradford method (Bradford reagent, Biorad, Hercules, CA). Sample proteins were prepared by adding 2× SDS-loading buffer containing 2-mercaptoethanol and DTT and were denatured at 95°C for 10 min. For western blot analyses, 30 µg of total protein per lane were loaded onto a 10% SDS-polyacrylamide gel. Proteins separated by SDS-PAGE were transferred onto nitrocellulose filters (Protran, Whatman, Dassel, Germany) with a pore size of 0.2 µm. Membranes were incubated with antibodies (Abs) against HSF-1, Hsp40, Hsp70A (C92F3A-5), Hsp70B', Hsp90 (AC-88), Grp94 (9G10) all from Stressgen (distributed by Biomol, Hamburg, Germany) or beta-actin (Sigma-Aldrich, Taufkirchen, Germany), followed by HRP-conjugated horse anti-mouse IgG, goat anti-rabbit IgG or goat anti-rat IgG Abs (all Cell Signaling Technology, Danvers, MA). Detection was performed with the chemiluminescent Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL) on a high performance chemiluminescence film (GE Healthcare, Chalfont St Giles, Buckinghamshire, UK). Western blot results were further analysed using the AIDA Image Analyzer software (Raytest, Berlin, Germany). Bands corresponding to the proteins of interest were quantified via AIDA 2D densitometry. All values were normalised to the corresponding beta-actin loading control. The corresponding untreated controls (37°C) were set to 1 in order to determine a potential increase or decrease in protein expression.
Transwell migration assay
Transwell migration assays of heat-treated and non-treated immature DCs and mature DCs were performed as described before, using the transwell system with 5 µm pore size (Costar Corning, Bodenheim, Germany) Citation[10], Citation[25].
Recombinant adenoviruses
Ad5Luc1 and Ad5TL are first-generation, E1- and E3-deleted, replication-deficient adenoviruses of serotype 5. Ad5Luc1 contains a cytomegalovirus (CMV)-firefly luciferase cassette and Ad5TL contains both a CMV-firefly luciferase and a CMV-GFP (green fluorescent protein) cassette (both kindly provided by D.T. Curiel, Birmingham, AL) Citation[12]. Replication-deficient Ad5MelA contains the human MelanA (MART-1) cDNA expressed from the CMV promoter Citation[12]. Viruses were amplified in 293 cells and purified by two rounds of CsCl equilibrium density gradient ultracentrifugation. Verification of viral genomes and exclusion of wild-type contamination was performed by polymerase chain reaction. Physical particle concentration (viral particles/mL) was determined photometrically (OD260) and infectious particle concentration was determined using the 50% tissue culture infective dose (TCID50) assay on 293 cells.
Adenoviral transduction of DCs
Immature day 6 DCs were seeded in 12-well tissue culture plates (BD Biosciences) at a concentration of 1 × 106 cells/well in 250 µL DC-medium supplemented with 800 U/mL GM-CSF, 500 U/mL IL-4, and 2-fold concentrated MC. Adenovirus at 500 TCID50/cell in a final volume of 250 µL medium without cytokines was added to the cells. After 1.5 h of incubation at room temperature, 2.5 mL of growth medium, replenished with cytokines as described before, were added per well. If indicated, heat shock was performed immediately after transduction as described above. To determine transduction efficacy, cells were transduced with Ad5TL and the percentage of living green fluorescent cells was assessed by flow cytometric analyses with a FACScan cell analyser (BD Biosciences). Only experiments that yielded transduction efficiencies of more than 70% were evaluated.
Transfection of DCs with MelanA RNA
DCs (1 × 106) were either exposed to 40°C for 24 h or remained at 37°C, both in the presence of MC. Afterwards cells were transfected with 50 µg/mL MelanA RNA or were mock-transfected using the TransMessenger Transfection Reagent from Qiagen (Hilden, Germany) as described before Citation[36]. Cells were then immediately used for a cytotoxic T cell induction assay.
Loading of DCs with MelanA peptide
Mature DCs (2 × 105) either heat-treated or not, were pulsed for 1 h at 37°C in a humidified atmosphere of 5% CO2 with 5 µg/mL of a MelanA-derived HLA-A2-binding analogue peptide ELAGIGILTV. Afterwards cells were harvested, washed once with medium, and then immediately used for a cytotoxic T cell induction assay.
Cytotoxic T cell induction assay
DCs were loaded with antigen and pre-treated as described above. Heat treatment was performed for 24 h at 40°C. CTL induction assays were performed with these DCs as described before Citation[25]. On day 7, T cells were harvested and characterised by tetramer staining and FACS analysis.
Tetramer staining and phenotyping of naïve and antigen-specific CD8+ T cells
Tetramer staining and phenotyping of antigen-specific CD8+ T cells was performed as described Citation[25]. To stain naïve, non-primed CD8+ T cells, either 5 µL of PC5-labelled anti-CD27 (1A4-CD27, Beckman Coulter) or anti-CD28 (CD28.2, Beckman Coulter) or anti-CD62L (DREG56, Beckman Coulter) mouse anti-human Ab were added additionally. After incubation for 30 min on ice, cells were washed and analysed using the CYTOMICS FC500 from Beckman Coulter (Krefeld, Germany). CD8+CCR7+CD45RA+ cells were thereby characterised as naïve phenotype, CD8+CCR7+CD45RA− cells as memory phenotype, CD8+CCR7−CD45RA− cells as effector memory phenotype and CD8+CCR7-CD45RA+ cells as lytic effector phenotype. The naïve, non-primed phenotype was further characterised by being positive for CD27, CD28 and CD62L.
Statistical analysis
The significance of differences was determined using one-way ANOVA and Bonferroni's multiple comparison post hoc test; P-values < 0.05 were considered statistically significant.
Results
Exposure of DCs to mild thermal heat increases Hsp70A expression in a time-dependent manner and does not reduce viability
To determine the influence of mild thermal stress on the expression levels of different crucial heat shock proteins (HSPs), day 6 immature DCs were seeded in DC-medium containing IL-4 and GM-CSF with or without the pro-inflammatory cytokine MC. The next day, cells were transferred to 40°C for 24 h, while control cells remained at 37°C. Afterwards cells were harvested, lysed and Western blotting was performed to detect HSF1, Hsp40, Hsp70A, Hsp70B', Hsp90 and the endoplasmatic reticulum paralogue of Hsp90, Grp94, also known as Gp96. As shown in , protein content of HSF1 was somewhat reduced in the whole cell extract of heat-treated DCs in comparison with control cells, whereas Hsp70A was strongly up-regulated, independent of the presence or absence of MC. Expression levels of other HSPs remained nearly unaffected in both cases. These data were supported by further analysis of Western blot results using AIDA Image Analyzer software (). Cells which were recovered after heat shock for 16 h at 37°C before cell lysis expressed heat shock proteins similar to cells harvested immediately after heat shock (data not shown). Moreover, we assessed the amount of Hsp70A expression over a time course of 24 h. Therefore, day 6 iDCs were cultured in DC-medium containing IL-4, GM-CSF as well as MC, and were incubated at 37°C (control cells) or 40°C for 3-, 6-, 12-, or 24 h, respectively. After generation of whole cell lysates for each time point, western blotting was performed to detect Hsp70A. As indicated in here is no increase in Hsp70A expression for non-heat-treated DCs. In contrast, exposure of DCs to 40°C leads to a clear enhancement of Hsp70A expression which peaked at 12 h and remained stable up to 24 h. Again, these data were supported by further Western blot analyses using AIDA Image Analyzer software (). Regarding the clinical application of our protocol, where the general maturation time of ex vivo generated and modified DCs is 24 h, we decided to use a 24-h heat treatment in the following experiments to ensure the full MC-induced maturation of DCs.
Figure 1. Heat treatment enhances the expression of Hsp70A in a time-dependent manner and does not influence the viability of DCs. (A and C) Western blot analyses: DCs were lysed and 30 µg of whole cell extract proteins were separated by SDS-PAGE. After transfer to a nitrocellulose membrane, blots were incubated with antibodies specific for HSF1 (A), Hsp40 (a), Hsp70A (A and C), Hsp70B' (A), Hsp90 (A), Grp94(A) and β-Actin (A and C). (B and D) Quantification of Western blot results using the AIDA Image Analyzer software. All values were normalised to the corresponding β-Actin loading control. The appropriate untreated controls (37°C) were set to 1 in order to depict the relative increase or decrease in protein expression after heat treatment. Data shown in display one representative experiment out of three (E). To determine the viability of heat-treated versus untreated DCs, cells were directly stained after heat treatment with PI and analysed using a FACScan flow cytometer. Results represent the mean ± SD of three independent donors. P values were calculated with one-way ANOVA and Bonferroni's Multiple Comparison post hoc test. n.s., statistically non significant (P > 0.05).
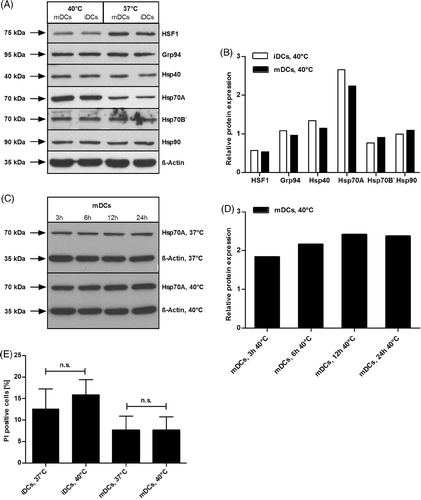
Next, we addressed the viability of heat-treated DCs in comparison to non-treated cells, either in the presence or absence of MC. FACS analyses of cells stained with PI directly after heat treatment revealed that exposure of DCs to mild heat of 40°C for 24 h had no effect on the viability of the cells (), which was the same for DCs which were recovered for 16 h at 37°C (data not shown). In conclusion, these results demonstrate that heat treatment of DCs has no influence on the viability and leads to an enhanced expression of Hsp70A in both immature as well as matured DCs. Furthermore, we show that this heat-induced Hsp70A expression is time-dependent in maturing DCs.
Heat treatment induces a differentiated up-regulation of maturation markers on the surface of DCs
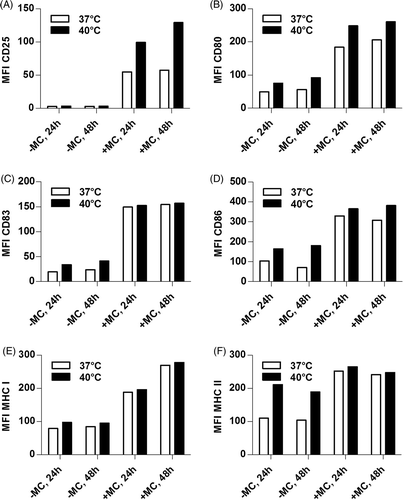
Mild thermal stress in vitro was found to enhance expression levels of MHC I, MHC II, CD80 and CD86 on murine bone marrow-derived DCs Citation[33], Citation[35]. To assess whether this is also the case for human DCs, immature DCs were seeded in medium containing IL-4 and GM-CSF with MC (mature DCs) or without MC (immature DCs), and were immediately exposed to 40°C or, as a control, remained at 37°C for 24 h. DCs were either stained directly after heat shock (24 h values) or were incubated for additional 24 h at 37°C (48 h values) and then analysed by FACS for the DC-maturation markers CD25, CD80, CD83, CD86, HLA-A,B,C (MHC I), and HLA-DR (MHC II) (, S1, and S2). The late maturation marker CD25 remained unaffected by the heat treatment on immature DCs, whereas expression levels on mature DCs enhanced markedly by hyperthermia (A, S1A, and S2A). Expression of CD80 (B, S1B, and S2B) increased on both immature DCs and mature DCs upon mild thermal stress, while CD83, CD86, and MHC class II were slightly up-regulated only on immature DCs (, 2D, 2 F; S1C, S1D, S1F and S2C, S2D, S2F). MHC class I was not affected (E, S1E, and S2E). Taken together, heat treatment resulted in the up-regulation of maturation markers CD80, CD83, CD86, and MHC class II in the absence of MC as well as of CD25 and CD80 in the presence of MC.
Figure 2. Exposure of DCs to 40°C up-regulates maturation markers. Day 6 immature DCs (1 × 106) were incubated in the presence (+MC) or absence (−MC) of maturation cocktail at 40°C or 37°C for 24 h. Subsequently, cells were either stained directly (24 h) or after a recovery time of 24 h at 37°C (48 h) for surface expression of CD25 (A), CD80 (B), CD83 (C), CD86 (D), MHC I (E), MHC II (F) mAbs and were characterised using a FACScan flow cytometer. For each condition 1 × 104 living cells, derived from a single well per condition, were analysed and mean fluorescence intensities (MFI) are shown. The data shown here represent one representative experiment out of three (see supplementary Figures S1 and S2 for the results with DCs from two additional donors).
Mild thermal stress induces the up-regulation of the anti-inflammatory cytokine IL-10 and the pro-inflammatory cytokine TNF-α
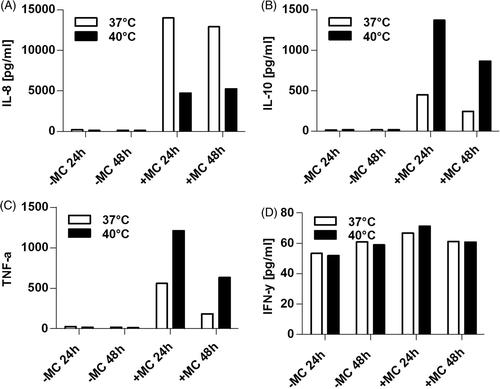
To characterise the cytokine profile of heat-treated immature DCs and mature DCs in comparison to control cells, supernatants derived from DCs described in , were examined for inflammatory and T helper cytokines IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, TNF-α, and IFN-γ by cytometric bead array. Only low quantities of IFN-γ () were secreted, and remained almost unaffected by heat treatment. In contrast, high quantities of IL-8 () were induced by MC but those were markedly decreased by exposure to 40°C. IL-10 and TNF-α were also only observed when MC was added and were strongly increased by hyperthermia (concentrations were tripled for IL-10 and doubled for TNF-α) in MC-treated DCs ( and 3 C, respectively). Note that TNF-α is a component of the MC, but still clearly increased concentrations of TNF-α were detected in supernatants of heat shocked DCs in comparison to control cells which remained at 37°C, thus heat-dependent TNF-α secretion by DCs is underestimated by the results shown in . However, no differences could be observed between heat- and untreated DCs concerning IL-1β, IL-2, IL-4, IL-5, IL-6, and IL-12p70 (data not shown). In summary, hyperthermic treatment of DCs in the presence of MC results in a reduced IL-8, and a strongly enhanced IL-10 and TNF-α secretion.
Figure 3. Exposure to heat influences quantities of cytokines secreted by DCs. Supernatants of DC cultures of the experiment shown in were examined for contents of IL-8 (A), IL-10 (B), TNF-α (C) and IFN-γ (D) by Cytometric Bead Arrays. For each cytokine a standard curve was generated to calculate the quantity of measured cytokines in pg/ml from MFI values. Data of one representative donor out of three is shown.
Stimulation by mild heat does not influence the migratory capacity of DCs in vitro
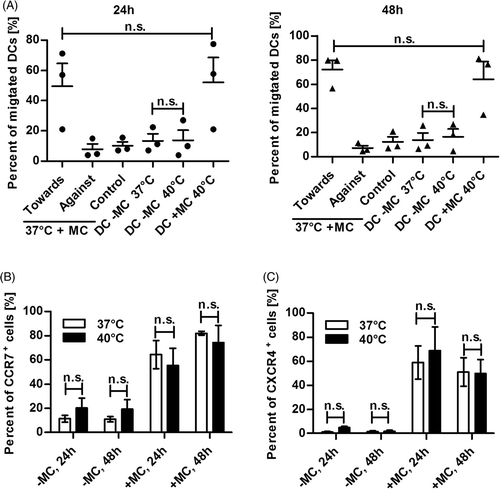
Ostberg and co-workers were the first to suggest that in the murine system mild thermal stress in the form of fever-like whole body hyperthermia (WBH) might have the ability to enhance Langerhans cell activation and migration out of the epidermis Citation[37]. Therefore, we investigated whether human DCs exposed to 40°C are better migrators towards CCL19, the chemokine triggering migration of DCs to the lymph node, and up-regulate its receptor CCR7. Moreover, we analysed the cell surface expression of CXCR4, an additional important chemokine receptor involved in migration of DCs mediated by the chemokine CXCL12 (SDF-1α) Citation[9]. Thus, immature DCs were seeded in tissue culture plates either in the presence or absence of MC. Heat shock was performed at 40°C for 24 h. Afterwards heat-treated and control cells were either incubated for an additional 24 h at 37°C or not. Subsequently, cells were analysed by transwell migration assays for their migratory ability and by FACS staining for the cell surface expression of CCR7 and CXCR4. outlines the migratory capacity of DCs derived from three different donors determined in three independently performed experiments either directly (24 h values) after heat treatment or after an additional recovery time of 24 h at 37°C (48 h values). Thus, heat treatment did not influence the migratory capacity of DCs either with or without MC in comparison to untreated cells (37°C) at both time points. Accordingly, for each time point no significant increase in CCR7 () nor in CXCR4 () cell surface expression between heat-treated (40°C) and control (37°C) cells could be measured either in the presence or absence of MC. The observed enhanced migratory ability and CCR7 expression from 24- to 48 h can be explained by the prolonged maturation time of the DCs. Taken together, these data clearly indicate that exposure of human DCs to mild thermal stress does not influence the cell surface expression of the chemokine receptors CCR7 and CXCR4 nor damps their migratory capacity in vitro.
Figure 4. Heat shocked DCs maintain their migratory capacity as well as CCR7 and CXCR4 expression. Day 6 immature DCs (1 × 106) were incubated at 37°C or 40°C with (+MC) or without MC (−MC) for 24 h. Directly after heat shock (24 h) or 24 h later, after recovery at 37°C (48 h), heat-treated and control DCs were harvested and analysed using a transwell migration assay and by FACS. (A) Harvested DCs were examined for their migratory capacity through a 5 µm mesh towards the chemokine CCL19 in a concentration of 100 ng/mL over a period of 2 h. As controls, DCs were set to migrate away from the chemokine (Against) or in absence of the chemokine (Control). Points (24 h) or triangles (48 h) represent values ± SEM of three different donors from three independently performed experiments. P values were calculated with one-way ANOVA and Bonferroni's multiple comparison post hoc test. n.s., statistically non significant (P > 0.05). (B) The fraction of CCR7 positive cells was determined by surface staining with PE-labelled anti-CCR7 mAb and subsequent FACS analysis. Columns represent the mean ± SD of three independent donors. P values were calculated with one-way ANOVA and Bonferroni's Multiple Comparison post hoc test. n.s., statistically non significant (P > 0.05) (C) The fraction of CXCR4 positive cells was assessed by surface staining with PE-labelled anti-CXCR4 mAb and subsequent FACS analyses. Columns represent the mean ± SD of three independent donors. P values were calculated with one-way ANOVA and Bonferroni's multiple comparison post hoc test. n.s., statistically non significant (P > 0.05).
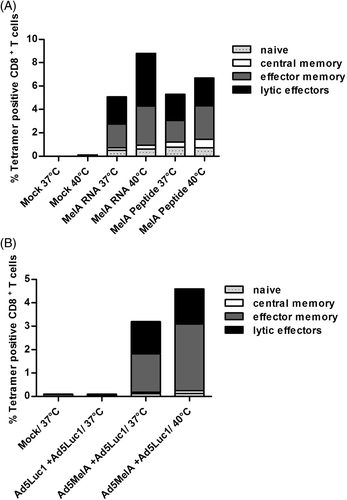
Heat treatment enhances the capacity of human DCs to prime naïve autologous melanoma antigen-specific CD8+ T cells in vitro
Finally, we examined whether heat-treated matured human DCs are better in priming naïve CD8+ T cells in vitro compared to non-heat-treated ones. Thus, HLA-A2+ DCs derived from healthy donors were transduced with an adenovirus vector coding for the tumour antigen MelanA, with a control vector encoding the irrelevant reporter luciferase, or were mock-transduced before heat treatment. In parallel, DCs were first heat shocked and then immediately transfected with MelanA RNA or loaded with MelanA peptide. Non-heat-treated cells were handled the same way, but were cultured at 37°C. Subsequently, DCs were used to prime autologous CD8+ T cells. After one week of stimulation the percentage of MelanA-specific CD8+ T cells was determined by tetramer staining and the T cell phenotype was further characterised by staining of cell surface markers. Notably, heat-treated DCs showed an enhanced capacity to expand melanoma antigen-specific CTL in vitro, independent of the mode of antigen loading and the time point of heat treatment (, S3, and S4). In this respect, as shown in , the percentage of MelanA tetramer positive CD8+ T cells derived from stimulation with DCs transduced with RNA increased from 5.1% (37°C) to 8.8% (40°C) and for peptide-loaded DCs from 5.3% (37°C) to 6.7% (40°C). A similar result was observed when DCs in the presence of MC were transduced with Ad5MelA and then exposed to heat. The percentage of MelanA tetramer positive CD8+ T cells increased from 3.2% to 4.6% in comparison to control cells (). However, the percentage of cells showing a lytic effector phenotype regarding their surface markers was increased by heat treatment for DCs, which were transfected with MelanA RNA only (from 2.3% to 4.5%, ), whereas in all other stimulating conditions the numbers of these cells remained almost unaffected between heated (40°C) and control cells (37°C). The results of the independent analyses of two further donors are shown in Figure S3 and Figure S4. Thus we conclude, that exposure to mild thermal stress of 40°C clearly increases the capacity of antigen-loaded matured human DCs to prime autologous naïve CD8+ T cells in vitro, independent of the method of antigen loading of DCs.
Figure 5. Heat-treated DCs show an enhanced in vitro capacity to prime naïve CD8+ T cells. (A) HLA-A2+ DCs were incubated at 37°C or at 40°C for 24 h both in the presence of MC. Afterwards cells were transfected with MelanA RNA or loaded with 5 µg/mL MelanA peptide or were mock-transfected (Mock) and were then used to prime autologous CD8+ naïve T cells. (B) Immature DCs from a different HLA-A2+ healthy donor were transduced with Ad5MelA or control virus Ad5Luc1 (500 TCID50/cell each) or were mock-transduced (Mock) in the presence of MC. Afterwards, heat shock was performed for 24 h at 40°C and DCs were then used to prime autologous CD8+ naïve T cells. The percentage of MelanA-specific CD8+ T cells was determined by tetramer staining and the cell surface phenotype of the MelanA-tetramer-positive CD8+ T cells was further determined by additional staining for CCR7 and CD45RA (CCR7+/CD45RA+: naïve phenotype; CCR7+/CD45RA−: central memory phenotype; CCR7−/CD45RA−: effector memory phenotype; CCR7-/CD45RA+: lytic effector phenotype). Cells were analysed using the CYTOMICS FC500 from Beckman Coulter. The data shown here represent one representative experiment out of three (see supplementary Figures S3 and S4 for the results with DCs from two additional donors).
Discussion
Elevation of temperatures associated with inflammation or fever has been linked to improved survival from infections, enhanced immunological functions and increased control of tumour growth. However, relatively little is known regarding these effects of thermal stimulation on APCs, especially human DCs. Notably, heat-induced activation of human DCs might be of interest for applications in vaccination against infectious diseases or cancer. Here we report that exposure of human DCs to 40°C for 24 h resulted in an efficiently enhanced biological activity of these cells without any loss of viability (). Consistent with recent findings Citation[31] we demonstrate that this enhanced biological function is accompanied by the time-dependent up-regulation of the heat shock protein 70 A. Furthermore, our data indicate that heat treatment of 40°C for 24 h induces up-regulation of maturation markers CD80, CD83, CD86, and MHC I on immature DCs and of CD25 and CD80 on mature DCs (, S1, and S2), which is different from previous reports Citation[32]. Hatzfeld-Charbonnier and colleagues on the other hand reported in their work that heat-stress did not modify the expression of cell-surface molecules of human immature and matured DCs, nor did it result in the maturation of DCs Citation[38]. Notably, this group followed a different protocol for heat treatment, as DCs were exposed to 41°C on days 0 and 3 and to 41.5°C on day 5 during DC generation, which is in clear contrast to our concept. Ostberg and co-workers described that DCs incubated at 40°C for 7 h with or without LPS showed no difference in MHC class I, MHC class II, CD40L, CD54, CD80, CD83, or CD86 levels when compared to DCs incubated at 37°C Citation[39]. Hence, different protocols of heat treatment influence the biological activity of DCs and therefore cause varying results. Clearly, the protocol we describe in the present study enhances surface expression of co-stimulatory molecules and the functionality of these cells, indicating the heat treatment of DCs for 24 h at 40°C is advantageous for applications in vaccination. Besides changes in phenotypic marker expression, the synthesis and release of important cytokines is another feature of DC maturation. Results herein demonstrated that elevated temperatures of 40°C for 24 h cause a considerably enhanced secretion of the pro-inflammatory cytokine TNF-α compared with DCs incubated at 37°C in the presence of MC (). Noteworthy, also IL-10 expression was clearly increased by heat shock, which is known to down-modulate or skew the immune system towards a TH2 immune response. This phenomenon is in contrast to previous reports Citation[32], Citation[38]. However, this most likely represents a ‘natural control mechanism’ in order to down-modulate strong immune responses once the ‘danger’ is not present any more. Moreover, other groups have shown that especially Hsp70A, which is markedly up-regulated in our system, induces the release of TH1-polarising cytokines such as TNF-α and IL-6 Citation[40–42]. However, in this context the use of the different heat shock protocols has to be considered and in addition different time points regarding cytokine analyses after heat treatment may also play a role.
Maturation of DCs is characterised by a third phenotypic change: after antigen up-take, DCs gain the property to migrate to the draining lymph nodes via a chemokine gradient, where they interact with and activate T cells Citation[43], Citation[44]. However, in ear skin organ cultures, where murine epidermal DCs naturally migrate out of the skin into the media of these cultures, differences between heated and unheated cultures were undetectable, although mRNA levels of CCR7 were up-regulated Citation[39]. Our findings herein show that exposure of human DCs to 40°C does not alter the capability of either immature or mature DCs to migrate along a CCL19/MIP-3β gradient in vitro (). Furthermore, heat-treated DCs either in the presence or absence of the maturation cocktail show no significant differences in CCR7 as well as CXCR4 expression when compared to DCs incubated at 37°C ( and 4 C).
Importantly, these DCs also efficiently enhance the ability to prime naïve CD8+ T cells in an antigen-specific manner, which is the most important test regarding the physiological relevance of thermal activation or maturation of DCs. Recent studies showed that heat-treated bone-marrow-derived DCs from mice had a two-fold enhancement of their ability to stimulate allogeneic T cells Citation[33], Citation[35], Citation[45]. Moreover, heat-treated DCs from patients with metastatic medullary thyroid carcinoma (MTC) loaded with tumour lysate derived from different allogeneic MTC cell lines were reported to be more potent stimulators of cytotoxic T cell responses than control DCs Citation[31]. This is suitable to our results, since we demonstrate that loading of DCs from healthy donors either with MelanA-RNA or -peptides leads to a 1.7- to 1.3-fold increase, respectively, to prime antigen-specific CTL in vitro (, S3, and S4).
Furthermore, our data indicate that these effects are independent of the order of antigen-loading and heat shock, since DCs which were first loaded with MelanA antigen by adenoviral transduction and then heat treated showed also an approximately 1.5-fold increased MelanA-tetramer positive CD8+ T cell population. This effect might be explained in part by the thermal up-regulation of Hsp70A, which is known, amongst other heat shock proteins, to act as a powerful anti-apoptotic protein and as a molecular chaperone inducing adaptive immunity Citation[46–49]. In this respect, Hsp70 might directly interact with MHC class I molecules and MelanA-derived antigenic peptides thereby eliciting CD8+ T cell responses.
In conclusion, our data show that mild thermal stress on DCs of 40°C for 24 h is a promising instrument to enhance the function and immunostimulatory capacity of human DCs. Moreover, it provides the basis for a sophisticated combinatorial approach with immunotherapy strategies, as heat treatment of DCs in vitro is an inexpensive and easily applicable measure. Most important, heat treatment of DCs can be straightforwardly translated into a good manufacturing practice (GMP)-compatible protocol. Generation of DCs and subsequent heat shock treatment on a large scale is uncomplicated and very well feasible. However, to maximise the use of thermal stress in the clinic, various questions must still be answered. For example, what are the mechanisms by which elevations in temperature regulate DC functions? When is the best time to apply heat, and what is the optimal temperature? Will multiple cycles of heat shock be more effective than a single one? Although several questions remain to be answered there is growing evidence that DCs are highly sensitive to their thermal microenvironment. Jun Guo and colleagues demonstrated in their study that intra-tumoural injection of DCs in combination with local hyperthermia induces systemic anti-tumour effects in patients with metastatic melanoma Citation[50]. Taken together, it is evident that further research is needed and understanding these intriguing processes will provide helpful information for the development of new clinical protocols using in vitro heat treatment or in vivo hyperthermia in combination with immunotherapy. The protocol for heat treatment of human DCs described in this study represents a basis for future pre-clinical and clinical studies investigating these issues.
Acknowledgements
The authors thank David T. Curiel (University of Alabama, Birmingham, AL), Rik Scheper (VU Medical Centre, Amsterdam, The Netherlands), and Jeffrey Schlom (National Institutes of Health, Bethesda, MD) for providing valuable research material as well as Kerstin Zander for her helpful comments on the manuscript.
Declaration of interest: This study was supported by the German Research Foundation (Collaborative research Centre SFB643, projects B9 and C1) and the Interdisciplinary Center for Clinical research (IZKF, project J7).The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Supplemental references.
Download PDF (526.8 KB)References
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392: 245–252
- Steinman RM, Gutchinov B, Witmer MD, Nussenzweig MC. Dendritic cells are the principal stimulators of the primary mixed leukocyte reaction in mice. J Exp Med 1983; 157: 613–627
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991; 9: 271–296
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124: 783–801
- Medzhitov R, Janeway CA, Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997; 91: 295–298
- Ueno H, Klechevsky E, Morita R, Aspord C, Cao T, Matsui T, Di PT, Connolly J, Fay JW, Pascual V, Palucka AK, Banchereau J. Dendritic cell subsets in health and disease. Immunol Rev 2007; 219: 118–142
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 2005; 23: 975–1028
- Cavanagh LL, von Andrian UH. Travellers in many guises: The origins and destinations of dendritic cells. Immunol Cell Biol 2002; 80: 448–462
- Gunn MD. Chemokine mediated control of dendritic cell migration and function. Semin Immunol 2003; 15: 271–276
- Prechtel AT, Turza NM, Kobelt DJ, Eisemann JI, Coffin RS, McGrath Y, Hacker C, Ju X, Zenke M, Steinkasserer A. Infection of mature dendritic cells with herpes simplex virus type 1 dramatically reduces lymphoid chemokine-mediated migration. J Gen Virol 2005; 86: 1645–1657
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18: 767–811
- Knippertz I, Hesse A, Schunder T, Kampgen E, Brenner MK, Schuler G, Steinkasserer A, Nettelbeck DM. Generation of human dendritic cells that simultaneously secrete IL-12 and have migratory capacity by adenoviral gene transfer of hCD40L in combination with IFN-gamma. J Immunother 2009; 32: 524–538
- Prechtel AT, Steinkasserer A. CD83: An update on functions and prospects of the maturation marker of dendritic cells. Arch Dermatol Res 2007; 299: 59–69
- Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature 1997; 388: 782–787
- Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, Mellman I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature 1997; 388: 787–792
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature 2007; 449: 419–426
- Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: Balancing immunity and tolerance. Cell Death Differ 2008; 15: 39–50
- Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998; 392: 86–89
- Heath WR, Belz GT, Behrens GM, Smith CM, Forehan SP, Parish IA, Davey GM, Wilson NS, Carbone FR, Villadangos JA. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev 2004; 199: 9–26
- Jung S, Unutmaz D, Wong P, Sano G, De los SK, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity 2002; 17: 211–220
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol 2010; 10: 403–414
- Erdmann M, Dorrie J, Schaft N, Strasser E, Hendelmeier M, Kampgen E, Schuler G, Schuler-Thurner B. Effective clinical-scale production of dendritic cell vaccines by monocyte elutriation directly in medium, subsequent culture in bags and final antigen loading using peptides or RNA transfection. J Immunother 2007; 30: 663–674
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998; 4: 328–332
- Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med 1996; 183: 317–322
- Schaft N, Dorrie J, Thumann P, Beck VE, Muller I, Schultz ES, Kampgen E, Dieckmann D, Schuler G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J Immunol 2005; 174: 3087–3097
- Gilboa E. DC-based cancer vaccines. J Clin Invest 2007; 117: 1195–1203
- Kikuchi T. Genetically modified dendritic cells for therapeutic immunity. Tohoku J Exp Med 2006; 208: 1–8
- Melief CJ, Toes RE, Medema JP, van der Burg SH, Ossendorp F, Offringa R. Strategies for immunotherapy of cancer. Adv Immunol 2000; 75: 235–282
- Melief CJ, van der Burg SH, Toes RE, Ossendorp F, Offringa R. Effective therapeutic anticancer vaccines based on precision guiding of cytolytic T lymphocytes. Immunol Rev 2002; 188: 177–182
- Melief CJ. Mini-review: Regulation of cytotoxic T lymphocyte responses by dendritic cells: Peaceful coexistence of cross-priming and direct priming?. Eur J Immunol 2003; 33: 2645–2654
- Bachleitner-Hofmann T, Strohschneider M, Krieger P, Sachet M, Dubsky P, Hayden H, Schoppmann SF, Pfragner R, Gnant M, Friedl J, Stift A. Heat shock treatment of tumor lysate-pulsed dendritic cells enhances their capacity to elicit antitumor T cell responses against medullary thyroid carcinoma. J Clin Endocrinol Metab 2006; 91: 4571–4577
- Ostberg JR, Repasky EA. Emerging evidence indicates that physiologically relevant thermal stress regulates dendritic cell function. Cancer Immunol Immunother 2006; 55: 292–298
- Basu S, Srivastava PK. Fever-like temperature induces maturation of dendritic cells through induction of hsp90. Int Immunol 2003; 15: 1053–1061
- DeFillipo AM, Dai J, Li Z. Heat shock-induced dendritic cell maturation is coupled by transient aggregation of ubiquitinated proteins independently of heat shock factor 1 or inducible heat shock protein 70. Mol Immunol 2004; 41: 785–792
- Zheng H, Benjamin IJ, Basu S, Li Z. Heat shock factor 1-independent activation of dendritic cells by heat shock: Implication for the uncoupling of heat-mediated immunoregulation from the heat shock response. Eur J Immunol 2003; 33: 1754–1762
- Liao X, Li Y, Bonini C, Nair S, Gilboa E, Greenberg PD, Yee C. Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. Mol Ther 2004; 9: 757–764
- Ostberg JR, Patel R, Repasky EA. Regulation of immune activity by mild (fever-range) whole body hyperthermia: Effects on epidermal Langerhans cells. Cell Stress Chaperones 2000; 5: 458–461
- Hatzfeld-Charbonnier AS, Lasek A, Castera L, Gosset P, Velu T, Formstecher P, Mortier L, Marchetti P. Influence of heat stress on human monocyte-derived dendritic cell functions with immunotherapeutic potential for antitumor vaccines. J Leukoc Biol 2007; 81: 1179–1187
- Ostberg JR, Kabingu E, Repasky EA. Thermal regulation of dendritic cell activation and migration from skin explants. Int J Hyperthermia 2003; 19: 520–533
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 2000; 6: 435–442
- Lehner T, Wang Y, Whittall T, McGowan E, Kelly CG, Singh M. Functional domains of HSP70 stimulate generation of cytokines and chemokines, maturation of dendritic cells and adjuvanticity. Biochem Soc Trans 2004; 32: 629–632
- Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, Lehner T. Stimulation of Th1-polarizing cytokines, C-C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein 70. J Immunol 2002; 169: 2422–2429
- Clark GJ, Angel N, Kato M, Lopez JA, MacDonald K, Vuckovic S, Hart DN. The role of dendritic cells in the innate immune system. Microbes Infect 2000; 2: 257–272
- Yanagihara S, Komura E, Nagafune J, Watarai H, Yamaguchi Y. EBI1/CCR7 is a new member of dendritic cell chemokine receptor that is up-regulated upon maturation. J Immunol 1998; 161: 3096–3102
- Tournier JN, Hellmann AQ, Lesca G, Jouan A, Drouet E, Mathieu J. Fever-like thermal conditions regulate the activation of maturing dendritic cells. J Leukoc Biol 2003; 73: 493–501
- Chen T, Cao X. Stress for maintaining memory: HSP70 as a mobile messenger for innate and adaptive immunity. Eur J Immunol 2010; 40: 1541–1544
- Goloubinoff P, De Los RP. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends Biochem Sci 2007; 32: 372–380
- Massa C, Melani C, Colombo MP. Chaperon and adjuvant activity of hsp70: Different natural killer requirement for cross-priming of chaperoned and bystander antigens. Cancer Res 2005; 65: 7942–7949
- Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2002; 2: 185–194
- Guo J, Zhu J, Sheng X, Wang X, Qu L, Han Y, Liu Y, Zhang H, Huo L, Zhang S, Lin B, Yang Z. Intratumoral injection of dendritic cells in combination with local hyperthermia induces systemic antitumor effect in patients with advanced melanoma. Int J Cancer 2007; 120: 2418–2425