Abstract
Purpose: HER-2 is in the EGF tyrosine kinase receptor family, overexpressed by many human cancers and minimally expressed by normal adult tissues. HER-2 expression in human cancers is correlated with reduced survival, increased metastasis, reduced apoptosis and increased proliferation. Herceptin is a humanised mouse antibody that targets and inactivates HER-2. In the present study, Herceptin was used to deliver ferric oxide-enriched nanoparticles to HER-2+ cancer cells. If exposed to alternating magnetic field (AMF), the nanoparticles heat. We tested the ability of AMF-activated Herceptin-directed nanoparticles to selectively kill HER-2+ human cancer cells.
Methods: Herceptin-conjugated nanoparticles were incubated with normal human mammary epithelial cells (HMEC)(HER-2-) or malignant human mammary epithelial cells (SK-BR-3)(HER-2+). Cells were stained to detect Herceptin or iron and the kinetics of binding quantified. Once conditions were optimised for binding, cells were exposed to either antibody-directed or non-antibody-conjugated nanoparticles, washed and sham-treated or exposed to AMF and cell death quantified.
Results: SK-BR-3 cells bound Herceptin-directed nanoparticles in increasing amounts over 3 h but did not retain non-antibody conjugated nanoparticles. HMECs did not retain either nanoparticles. SK-BR-3 cells with bound Herceptin-directed-nanoparticles, exposed to AMF, died by apoptosis, quantifiable by Live/Dead and nuclear morphology assays and released LDH. Sham-treated SK-BR-3 cells with Herceptin-directed nanoparticles, HMECs with either nanoparticles, with or without AMF treatment, exhibited no increase in toxicity above baseline cell death using these three assays.
Conclusions: These studies demonstrate Herceptin-directed nanoparticles can selectively kill HER-2+ cancer cells via hyperthermia after AMF activation.
Introduction
Trastuzumab, or Herceptin, is a high affinity engineered mouse-human-chimeric IgG1 monoclonal antibody that was raised against the extracellular domain of human epidermal growth factor receptor 2 (HER-2) Citation[1]. HER-2 belongs to a family of receptor tyrosine protein kinase receptors that are overexpressed in 20–30% of breast, lung, ovarian and gastric adenocarcinomas [Citation1–3]. Herceptin was approved by the FDA for the treatment of patients with HER-2 overexpressing breast cancer in 1998 Citation[4], Citation[5]. However, only 25–30% of patients who overexpress HER-2 exhibited tumour regression in response to treatment with the antibody alone Citation[5]. Recently, Herceptin has been tried as a vehicle to deliver dyes, drugs or nanoparticles to malignant cells [Citation2–7]. These approaches include the use of polypyrrole nanospheres with a hyaluronic acid coating that was functionalised using Herceptin and shown to be rapidly endocytosed by human cancer cells and used for targeting Citation[6]. And magnetite-containing immuno-liposomes functionalised using Herceptin that were direct injected into subcutaneous human cancers in nude mice, were activated by AMF and caused tumour regression Citation[7]. Beginning with Gilchrist in the 1950s and with interest steadily increasing in tandem with the research community's understanding of the importance of nanotechnology and the utility of hyperthermia, the use of magnetic material to induce hyperthermia has emerged as a possible cancer therapy [Citation8–10]. Directly injected iron-containing nanoparticles which respond to alternating magnetic field (AMF) by generating magnetic heat have been shown to destroy a proportion of the cancer cells or sensitise them so they are more responsive to chemotherapy or radiation [Citation9–12]. Some pre-clinical and clinical studies have reported the use of magnetic material for the treatment of brain glioblastomas Citation[13], prostate cancer Citation[14] and breast cancer Citation[15]. Thermotherapy using non-directed nanoparticles has not received widespread support for clinical development. Reasons include the non-specific distribution of nanoparticles in the tumour mass, poor penetration of the nanoparticles in the tumour mass, insufficient tumour killing and adverse effects to the surrounding normal tissue.
The purpose of our study was to demonstrate selective killing of HER-2 positive human cancer cells using Herceptin conjugated magnetic nanoparticles. We evaluated the binding and uptake of Herceptin conjugated magnetic nanoparticles by a HER-2 overexpressing breast cancer cell line vs. HER-2 negative human normal mammary cells, and evaluated the cytotoxicity of nanoparticles alone verrsus antibody conjugated nanoparticles, with and without AMF. Because we are interested in the potential for a cancer treatment localised at or close to the cellular level, we conducted our experiments so that during the course of AMF-driven particle excitation, the macroscopic temperature, that is the temperature measured in the incubation wells, did not rise into the hyperthermia region.
Materials and methods
Cell Lines and Cell Culture
SK-BR-3 (human breast adenocarcinoma) cells from American Type Tissue Culture Collection (ATCC, Manassas, VA, USA) and HMEC (normal human mammary epithelial) cells (Lonza, Basel, Switzerland) were grown in the media recommended by the manufacturers, at 37°C in a humidity controlled incubator with 5% CO2. SK-BR-3 cells were grown in McCoy's 5A media (Sigma, Saint Louis, MI) with the added ingredients of 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), 20 mM L-glutamine, 1 K IU penicillin, 1 K mg/mL streptomycin sulfate (GPS; Sigma), and amphotericin B (FGZ; Lonza) supplementation. HMECs were grown in mammary epithelial growth medium (MEGM) (Lonza) with bullet kit without FBS.
Bionised nanoferrite (BNF) nanoparticles
The bionised nanoferrite (BNF) nanoparticles were of magnetite crystals (Fe3O4) coated with dextran [Citation16–19]. Briefly, the magnetic particle manufacturing process consisted of synthesising iron oxide (IO) nano-crystal particles, coating these crystals with dextran and fractionating the material to specific sizes by magnetic separation. The particles are approximately 50 to 60% weight iron in the form of iron oxide, with each particle containing one or more iron oxide crystals. The particles have an average size of approximately 100 nm (Z average weighted hydrodynamic diameter) with a polydisperity index of less than 0.2 as measured in aqueous solution by dynamic light scattering. These particles efficiently produce heat upon application of AMF; for instance, a specific absorption rate (SAR) of 250 W/g iron is produced with application of 35,828A/M peak amplitude at 150 kHz. To facilitate use as targeted particles, the dextran coatings were cross-linked to provide stability and functionalised at the surface with amines to facilitate attachment of targeting agents as discussed below. Cross-linking and amination were accomplished using epichlorohydrin and ammonia. The preparation of the functionalised, dextran-coated nanoparticles was carried out by Micromod Partikel-Technologie (Rostock, Germany).
These ‘base particles’ were then functionalised with targeting agents at Aduro BioTech (Berkeley, CA). For the preparation of Herceptin particle conjugates, amines on the surface were converted to thiols using 2-iminothiolane. Then, a total of 1 to 2 mL of ∼2 × 10-5 M thiolated Herceptin (Genentech, San Francisco, CA) in degassed phosphate buffered saline (PBS) was added to 20 mL (400 mg) of BNF-maleimide nanoparticles in degassed PBS. The reaction mixture was shaken for at least 1 h at room temperature. Sufficient N-ethymaleimide (NEM) was then added to achieve a final concentration of 10 mM. The mixture was shaken for 40 min and then subjected to magnetic separation for 30 min. The supernatant was removed and fresh buffer added for the next magnetic separation. Washing was repeated two or more times and the conjugates were resuspended in 2 mM 2-mercaptoethanol. The suspension was then shaken and the washing sequence repeated for two or more washes at which point the antibody-functionalised particle conjugates were resuspended in PBS. Control nanoparticles have functionalised sites blocked using N-ethylmaleimide and then all particles undergo magnetic size separation in parallel. After Herceptin-conjugated nanoparticles are isolated by magnetic separation, they are tested for binding efficiency in a microELISA using a pure HER-2 standard. The number of antibodies per nanoparticle are determined by monitoring the disappearance of a secondary antibody in the titration microELISA as previously reported Citation[20], Citation[21] and nanoparticle conjugate batches used in the present study range from 30–40 Ab per particle. In pilot studies we used nanoparticles in a range of 0.01, 0.1 and 1 µg/mL to 80,000 cells to optimise the binding reaction.
Immunohistochemical staining of her-2 associated with living cells after incubation with nanoparticles
To determine the availability of surface HER-2 on cells, the cells were grown to confluency in 75cm2 rectangular canted neck cell culture flasks with vent caps (Corning Life Science, Lowell, MA). Cells were trypsinised with 0.05% trypsin, with EDTA (Invitrogen) and plated in 8-well Lab-Teks™ II (Nalge Nunc, Rochester, NY) at 20,000 cells/well and grown in full media. Once cells were 70–80% confluent, the media were changed to 10% heat inactivated FBS medium before staining the cells. Cells were incubated in 3% heat inactivated goat serum (Sigma) in Tris-buffered saline solution (TBS) for 0.5 h for blocking, then without washing the cells, the primary antibody, mouse-human chimeric Herceptin conjugated to nanoparticles, was added at room temperature at various concentrations. Control wells received the blocking solution but the primary antibody was omitted. After rinsing with 1% heat inactivated goat serum in TBS, the cells were then incubated with the secondary antibody Alexa Fluor 555 goat anti-human IgG1 (H&L) (1:1000, Invitrogen) in TBS 1% heat inactivated serum at room temperature. After washing with PBS, the cells in Lab-Teks were fixed by incubation with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS for 30 min at room temperature. The cells were counter stained with a DNA stain, 4′,6-diamidino-2-phenylindole (DAPI). DAPI staining was performed by removing the PBS and replacing it with a 0.2 µg/mL solution of DAPI in methanol and incubating the cells at room temperature for 30 min. The cells were then rinsed with PBS, air dried, mounted with Fluoromount G (Electron Microscopy Sciences) and a cover slip applied. Images were obtained using fluorescence microscopy (Olympus, Center Valley, PA).
Prussian blue staining and quantification of iron
To investigate the cell-specific binding of Herceptin conjugated nanoparticles compared to non-antibody directed control nanoparticles, total binding by SK-BR-3 and HMECs was determined by iron visualisation and quantification using a Prussian blue iron stain kit (Polysciences, Warrington, PA) and spectroscopy. Cells were plated in 8-well Lab-Tek IIs at 6 × 104/well in full growth media. The media were replaced 24 h before treatment with 10% heat inactivated FBS (Invitrogen) culture medium. Then cells were blocked with 10% heat inactivated FBS medium for 0.5 h, and then bionised nanoferrite nanoparticles (BNF) alone or BNF Herceptin-directed nanoparticles were added to the cell culture medium at various concentrations. In control wells, the cells were plated at the same cell density and grown and treated without nanoparticle addition. After 1, 2 or 3 h incubation at 37°C and 5% CO2, the cells were washed with PBS containing 1% heat inactivated FBS. For quantification of the uptake of the nanoparticles, washes were collected and pooled and quantified for iron using the Prussian blue assay. To estimate 100% Prussian blue reaction product, wells without cells were blocked in parallel, and received an identical amount of nanoparticles, and washed and collected in parallel. The iron concentration of the sample was calculated from the calibration curve obtained using standard Fe particle solutions of various concentrations Citation[22]. Using six known concentrations of standard Fe, the Prussian blue reaction product can be used to generate a linear curve which can be used as a standard. A modified Prussian blue reaction has already been used as a well accepted quantitative assay for cyanidins in foods Citation[22] and can be used as a quantitative assay for iron. Colour was allowed to develop for 60 min and measured at 535 nm. Four sets of duplicates were done for each condition and the results were averaged.
For viewing nanoparticles bound to the cells, the cells were fixed in 10% neutral buffered formalin in PBS for 30 min at room temperature. After washing with PBS, equal volumes of 4% potassium ferrocyanide and 4% hydrochloric acid were added for two series of 10 min each. The cells were counterstained with 1% nuclear fast red for 4 min at room temperature. Then after final PBS washes, the Lab-Teks were air dried, then cover-slipped using mounting medium, viewed under the light microscope, and photographed.
In vitro AMF studies
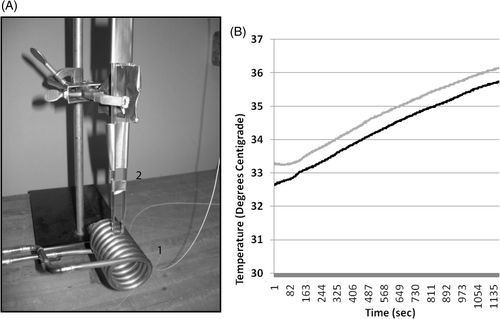
A system was designed and built to provide high-amplitude AMF to cells in Lab-Teks (). The system consists of three main parts: the induction coil made of a copper tube (, part 1), the recirculating chiller (Thermo Electron, Newington, NH), which maintained temperature by closed loop circulating of cold water through the selenoid (not shown); and a 10-kW AMF power supply (not shown, Huttinger TIG 10/300). Two temperature probes (Fiso, Quebec, Canada) were mounted above the coil and could be lowered into individual Lab-Tek chambers via pipette catheters (, part 2). The temperature probes measured media temperatures at 1-s intervals for each probe during nanoparticle activation using AMF and were plotted in real time in wells that contained SK-BR-3 that had been incubated with Herceptin-directed (black line) and non-antibody directed nanoparticles (grey line) and then washed prior to AMF (). In pilot experiments, wells with media alone not containing cells or treated with either nanoparticle were tested. Coil contact with the sides of the Lab-Tek chambers results in a 3°C rise of temperature during a 20-min AMF exposure. Only experiments where global media temperatures did not rise above 37°C were used. Cold water was passed through the coil and controlled to ensure that the coil did not itself heat the media during AMF exposures in excess of 37°C. In vitro AMF studies were done under three different conditions with: (1) BNF-Herceptin directed particles, (2) non-antibody directed BNF-control particles, or (3) using cell culture medium without particles added to cells. Each condition was conducted in parallel in duplicate Lab-Teks wells per experiment and each experiment was replicated at least three times. AMF at 150 kHz was applied having a peak amplitude of 35,828A/M (Gauss) and the recirculating chiller (Thermo Electron) was set at 35°C with a total treatment time of 20 min. The capacitance was designed so that in parallel with the inductor, the LC resonant circuit produced a stable oscillation frequency of 150 kHz. Sham controls were handled exactly the same way but were not AMF exposed. A pulse-timer circuit (manufactured by Giltron, Medway, MA) was included to allow 0.5- to 9,999-s pulses at duty ranging from 0–100% as previously described [Citation16–21].
Figure 1. The experimental apparatus used to deliver a focused homogeneous electromagnetic field to growth chambers containing living cells and nanoferrite nanoparticles while monitoring temperature in the cell growth media. A 10 kW AMF power supply was connected to a copper solenoid coil (A1) and used to expose living cells, in the presence or absence of nanoferrite nanoparticles, to an alternating magnetic field. The solenoid coil diameter was designed to accommodate two stacked eight-chamber Lab-Tek devices to bring the cell growth areas into a uniform aspect of magnetic field. The power source was operated at a 150 kHz frequency for 20 min during irradiations. Two temperature probes were inserted through pipette catheters into two growth chamber wells (A2) for all treatments, and temperature of the media in two wells were monitored continuously (B) during and following AMF exposures. Shown are readings for SK-BR-3 cells with Her-2 nanoparticles (black) versus non-directed nanoparticles (grey) collected in parallel under the same AMF exposure. The chiller was set to 33°C and after 20 min of AMF exposure, the media above cells only rose 3°C. This rise in temperature is due to induction coil contact and transference of heat to the Lab-Tek slides. It is observed in wells with medium alone without cells that never received nanoparticles. No cell death is observed using either HMEC or SK-BR-3 cells in control wells without nanoparticles exposed to AMF in parallel and experiencing these temperature ranges during AMF.
Quantification of live and dead cells using viability dyes
The death of HMEC or SK-BR-3 cells after AMF was measured with the Live/Dead® Viability/Cytotoxicity assay for mammalian cells (Invitrogen) and the cells were processed according to the manufacturer's recommendations. Briefly, the Live/Dead reagent stock solutions were thawed and allowed to warm to room temperature. After gently removing the medium from the live cells, the ethidium homodimer-1 (EthD-1)-calcein AM mixture was added and incubated with the live cells for 30 min at room temperature. After gently washing with PBS, the cells were then fixed with 2% formaldehyde solution in PBS (200 µL) for 30 min at room temperature. The cells were then rinsed with PBS, air dried, mounted with Fluoromount G (Electron Microscopy Sciences) and a cover slip applied. Live cell cultures were compared to those killed using 70% methanol exposure to assure calcein AM stained only the cytoplasm of live SK-BR-3 and HMEC cells green and EthD-1 only stained the nucleus of killed SK-BR-3 and HMEC cells red and to determine optimised exposure times to photograph each dye reaction.
Images were obtained using fluorescence microscopy (Olympus, Center Valley, PA). The calcein AM (green) images were taken using an excitation wavelength filter of 480 nm and the EthD-1 cell (red) images were taken using an excitation wavelength filter of 545 nm. To create a composite, the two images obtained were overlaid using DP manager 3.1 software (Olympus). Exposure time was adjusted using 100% dead and live cell cultures prepared in parallel as controls. Percentages of living (green-stained) and dead (red-stained) cells were determined by counting a total of at least 1000 cells in 5–10 fields chosen randomly at 40× magnification.
Quantification of live/dead cells using lactate dehydrogenase cell release
Lactate dehydrogenase enzyme (LDH) activity in cell supernatants was measured with the TOX-7 assay kit (Sigma Aldrich) as previously described by us Citation[23], Citation[24]. The calibration standard curve was generated using known numbers of cells representing at least six points from 1,000–50,000 that were exposed to 100% lysis solution. SK-BR-3 and HMECs were assayed separately as the total LDH pool per cell type can vary and different standard curves have to be used. For experiment treatments, after 6 or 24 h post sham or post-AMF, the supernatants were collected into microcentrifuge tubes, centrifuged at 250 g for 5 min at room temperature. Supernatants were transferred to microcentrifuge tubes for storage until the LDH assay. Lactate dehydrogenase assay mixtures were freshly prepared for each experiment. To perform the assay, equal amounts of lactate dehydrogenase assay substrate, assay dye, and 1′ × LDH assay cofactor were added to each sample. The samples from each well were analysed in triplicate. In 96-well microplates, 30 µL of the test sample and 60 µL of the substrate/dye/cofactor mixture solution were added to each well and mixed, covered with an opaque material to protect from light and incubated at room temperature for 30 min. Ten µL (1/10 volume) of 1 M HCl was added to each well to stop the reaction. Blank controls were prepared by similar procedures as the LDH assay except unused media was used instead of cell supernatant. Spectrometric absorbances were measured by a Synergy HT Multi-Mode Microplate Reader (Bio-Tek, Winooski, VT). Absorbance at the wavelength of 490 nm was minus the absorbance at wavelength of 690 nm. The optical density values were then normalised to cell number using calibration curves.
LDH release and the Live/Dead assay are used in distinct ways. The two-dye Live/Dead viability cytotoxic assay can only be used to quantify live and damaged cells that remain attached to the plates not cells that have detached or lysed. The LDH assay measures LDH release from totally lysed or leaked from damaged cells, still attached to plates or detached and floating in media. The Live/Dead assay is based on live cells having intracellular esterases that convert the non-fluorescent, cell-permeable calcein acetoxymethyl (calcein AM) to the green fluorescent calcein. Cleaved calcein is retained within live but not dead cells. In contrast, dead or damaged cells that remain attached have leaky or damaged membranes allowing ethidium homodimer-1 (EthD-1), the second distinct dye, to enter damaged cells and fluoresce when it becomes bound to nucleic acids. If cells are completely lysed, this can be accounted for only by counting a change in field number required to count 100 cells on average or by the detection of LDH in the media. Therefore these two assays taken together can account for all of the cells.
The means of cell death can affect when LDH is seen. Necrotic death results in immediate LDH release and no cells can be seen or counted in the media. In contrast, apoptotic death can result in cell detachment but not leakage or breakage of cells and no detection of LDH in the media. There is a well-known phenomenon of late or delayed necrosis secondary to apoptosis in that the detached apoptotic cells eventually begin to leak and/or lose membrane integrity but this can take 48–72 h post the appearance of apoptotic bodies via nuclear morphology studies or DNA laddering by gel analysis Citation[25], Citation[26].
Quantification of apoptosis using nuclear morphology
Cells were fixed by adding 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 30 min at room temperature. Lab-Teks with fixed cells were stored at 4°C until staining with 4′,6-diamidino-2-phenylindole (DAPI), which binds to the A-T-rich regions of DNA. All subsequent staining procedures were performed in the dark. DAPI solution was prepared in methanol to a final concentration of 0.2 µg/mL. Fixed cells were washed once with DAPI solution, then incubated in DAPI solution at 37°C for 30 min. The cells were then rinsed with PBS, air dried, mounted with Fluoromount G (Electron Microscopy Sciences) and a cover slip applied. Images were obtained using fluorescence microscopy (Olympus). In microscopic studies, a cell was considered to be apoptotic if at least three distinct clusters of fragmented DNA of different size were observed as previously described by us [Citation23–26].
Statistical analysis
All experiments were repeated at least three times under the same conditions, and the data were pooled. Statistical analysis was carried out using student's t-test and Excel software. A P value of less than 0.05 was accepted as statistically significant.
Results
Detection of live cell selective binding and/or uptake of Herceptin-directed nanoparticles using indirect immunofluorescence
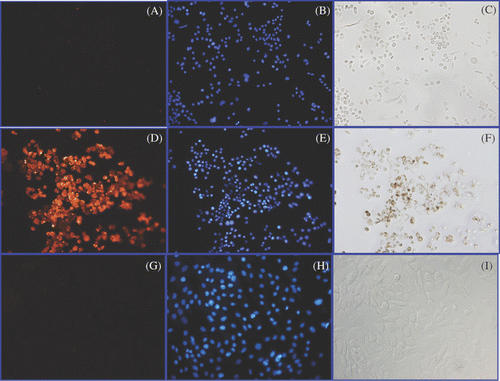
We first wanted to determine the ability of a Herceptin-directed nanoparticle to bind and be retained selectively by HER-2 positive human cancer cells. The same number of live SK-BR-3, human breast adenocarcinoma cells, or HMEC, normal human mammary epithelial cells, were incubated with Herceptin-conjugated nanoparticles. After 1 h, cells were stained using an Alexa Fluor labelled secondary antibody to human IgG1 (, , ) or counterstained with a DNA stain (DAPI) to reveal nuclear morphology and cell locations (, , ) determined using fluorescent microscopy as described in Methods. Cells could also be viewed under light microscopy to detect cell-associated iron seen as brown without any counter-stain (, , ). In the upper panels, identical fields of SK-BR-3 cells are shown when the nanoparticles were omitted during the first step. This serves as a control to indicate the secondary antibody (A) does not bind the cells and there is no staining of the numerous cells revealed by DAPI staining (B), and by light microscopy no endogenous iron deposition is seen (C). However, if the same number of live SK-BR-3 cells were incubated first with Herceptin-directed nanoparticles, washed and then the humanised IgG1 Herceptin antibody is localised with the secondary antibody (D), significant cell associated Herceptin was detected in essentially every cell revealed by DAPI staining of the identical field (E), and with a corresponding cell-associated iron deposition (F). In contrast, the same number of DAPI stained HMEC cells (H) that received Herceptin-directed nanoparticles in parallel demonstrated no retained cell associated Herceptin (G) and no residual cell-associated iron (I), indicating the Herceptin directed nanoparticles did not bind to these normal cells. Only the live SK-BR-3 cells incubated with Herceptin-directed nanoparticles after washing demonstrated retention of both Herceptin and iron material. The DAPI staining revealed not only location of all cells, but also normal nuclear morphology and no evidence of apoptosis in any of the cell cultures following this 1-h incubation as live cells with or without Herceptin-directed nanoparticles.
Figure 2. Detection of Herceptin, nuclear morphology and iron in cultures of live SK-BR-3 and HMEC cells. Live SK-BR-3 cells were incubated in medium alone (A–C) (control) or were incubated with Herceptin direct nanoparticles for 1 h (D–F) in parallel with live HMEC cells (G–I). Cells were then washed and fixed and examined using light microscopy for the detection of iron (C, F, I); or subjected to indirect immunostaining to detect retention of Herceptin antibody associated with the cells (A, D, G) or stained with DAPI to reveal nuclear morphology (B, E, H). Only SK-BR-3 cells (D) retained Herceptin-directed nanoparticles, whereas normal mammary cells did not (G). Iron was only found in association with SK-BR-3 cells incubated with Herceptin directed nanoparticles (F). All cells exhibited normal nuclear morphology with no indication of apoptotic bodies occurring in these cultures (B, E, H).
Time course and detection of Herceptin-directed nanoparticle binding to SK-BR-3 cells using Prussian blue staining of iron
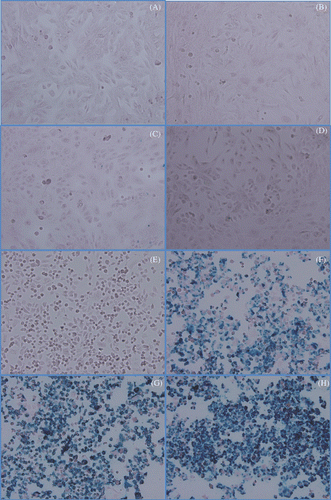
We wanted to define the optimal incubation time for human cancer cells to retain Herceptin-directed nanoparticles while still exhibiting specificity of this binding. We incubated either live HMEC cells () or live SK-BR-3 cells (panels E–H) in parallel with Herceptin-directed nanoparticles for 0 (, ), 1 (, ), 2 (, ) or 3 (, ) h, and then washed, fixed and processed all the cells immediately for Prussian blue staining of iron (). Regardless of incubation time, negligible amounts of iron containing nanoparticles were retained after washing when using live HMEC cells (). In contrast, with increasing incubation time ( versus 3F versus 3G versus 3H), more Prussian blue reaction product was apparent as a function of time when using live SK-BR-3 cells, indicative of higher amounts of iron containing nanomaterial retained by the cells (). We also noted by 2 and 3 h that iron nanomaterial was associated with every SK-BR-3 cells in the field and some cells appeared to be capping the iron-rich material at one pole. Capping is a term to indicate that when bivalent antibodies become bound to a membrane-associated antigen, this can trigger movement and coalescence of the Ab:Ag complexes within the plasma membrane to a patch or pole of cell. This can lead to internalisation of the nanoparticle attached to the antibody in the complex, an event that can enhance significantly nanoparticle retention, aggregation and heating within the targeted cells when AMF is applied. Longer incubation times (4 and 5 h) for binding did not appear to increase the cell-associated nanomaterial that was SK-BR-3 associated (data not shown). We estimate iron bound per cell based on the addition of 0.1 µg/mL nanoparticles to be 80,000 cells in 0.5 mL and a 30% binding efficiency at the conclusion of the binding incubation time to be a mean average of ∼28 pg iron/per cell for the SK-BR-3 cells using HER-2 directed nanoparticles.
Figure 3. Prussian blue localisation of iron in cultures of HMECs or SK-BR-3 cells treated with Herceptin-directed nanoparticles as a function of time. Live HMEC cells (A–D) and SK-BR-3 cells (E–H) were incubated for either 0 (A, E), 1 (B, F) 2 (C, G) or 3 (D, H) h with Herceptin-directed iron oxide crystal-containing nanoparticles increase. Cells were then fixed and stained to localise iron using Prussian blue. No iron was retained by HMEC cells over the time course, but increasing amounts of iron could be visualised in association with Her-2 positive malignant mammary cancer cells in parallel.
Herceptin directs binding of nanomaterial to human cancer cells and triggers internalisation and capping of the material
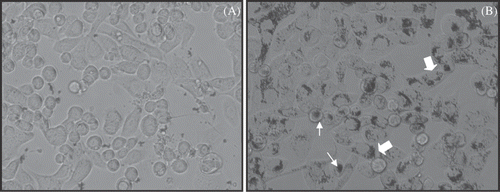
To study the role of the Herceptin antibody in the specific binding and cellular localisation of the iron nanoparticles in exclusive association with the human breast cancer cells we conducted binding studies using live SK-BR-3 cells at 2–3 h with the Herceptin-directed () versus the non-antibody directed control nanoparticles (). In living cells, after washing with PBS and under phase microscopy without fixation or staining, little nanomaterial is associated with the SK-BR-3 cells when using non-antibody directed nanoparticles (). In contrast, the same cell type, SK-BR-3, appears to internalise (large arrows) and cap (small arrows) the same nanoparticles if they are conjugated to a Herceptin antibody (). It is clear from these studies that HER-2 positive cells bind, cap and internalise Herceptin-directed nanoparticles after 2–3 h of incubation, and without the antibody this does not occur. We did not quantify the relative amounts bound to the surface versus internalised, a subject for future studies. Another observation of the studies is the lack of non-specific uptake by the cancer cells when using non-directed nanoparticles.
Figure 4. Microscopic study of binding and internalisation of iron oxide crystal containing nanoparticles with or without Herceptin antibody delivery to SK-BR-3 cells. Live SK-BR-3 cells were incubated for 2 h with either non-antibody-directed nanoparticles or the same amount of nanoparticles with Herceptin conjugated to the surface. After washing with PBS and without any fixation or staining, the highly malignant human breast cancer cells could be observed capping (small arrows) and internalising (large arrows) nanomaterial if directed by Herceptin (B), but retained no observable nanomaterial without the benefit of the HER-2 specific antibody (A).
Quantification of the time dependent specific binding and retention of Herceptin-directed nanoparticles to human cancer cells
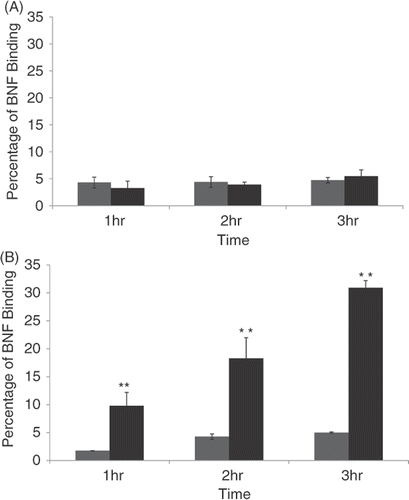
Using spectroscopy, we analysed the percentage of nanomaterial that remained bound to cells after various incubation times when a Herceptin-directed (black bars) versus minus antibody, non-directed control nanoparticles (grey bars) were used with either SK-BR-3 () or normal human mammary cells (HMEC) (). By quantifying iron that remained associated with the cells we found a steady increase up to 30% of nanoparticles were retained by SK-BR-3 cells when the Herceptin-directed nanoparticles were used (). If the nanoparticles were not conjugated to the Herceptin antibody (grey bars), but conjugated to non-specific IgG1 antibody, less than 4% of the nanoparticles were retained in the wells, and this amount did not statistically change with incubation time in SK-BR-3 cell cultures. When using HMEC cells, less than 4% of the nanomaterial was retained in the wells whether the nanoparticles were conjugated with Herceptin or not and regardless of incubation time (). Upon microscopic examination, the small amount of nanomaterial that was in HMEC cultures with either type of nanoparticles, or SK-BR-3 cell cultures when using non-antibody directed nanoparticles, was associated with the glass of the Lab-Tek plates and not cell associated. These studies demonstrated that a statistically significant amount of nanoparticle binding and cell retention occurred, but was dependent on the cells expressing HER-2 on their surface and the nanoparticles being directed to the cell surface marker using Herceptin. The binding increased with time and by 2–3 h was maximal, resulting in 30% of the nanoparticles added to the human cancer cells being retained. We had defined the conditions under which we could now test effective and selective killing of human cancer cells using Herceptin-directed nanoparticles activated by alternating magnetic field energies.
Figure 5. Quantification of non-antibody versus antibody-directed nanoparticle retention and binding to normal versus malignant human mammary cells over time. HMEC cells (A) or SK-BR-3 cells (B) were incubated for 1, 2 or 3 h with either non-antibody directed nanoparticles (grey bars) or Herceptin-directed nanoparticles (black bars) and then washed and stained with Prussian blue. Spectroscopy was used to analyse the amount of iron retained by the cells over time compared to a starting sample of nanoparticles not used with cells processed in parallel to represent 100% binding. These studies reveal a time-dependent increase in binding of Herceptin-directed nanoparticles only to HER-2 positive human cancer cells (lower panel, black bars). Minimal binding of directed or non-directed nanoparticles occurs in response to normal mammary epithelial cells (upper panel) or when non-directed nanoparticles are added to the same Her-2 positive human breast cancer cells (lower panel, grey bars).
Herceptin-directed nanoparticles activated by alternating magnetic field energies induce selective cytotoxicity of human cancer cells via hyperthermia
First, live HMEC cells () or SK-BR-3 cells () were incubated with either media alone, non-antibody directed nanoparticles (BNF-control) or Herceptin-directed nanoparticles (BNF-Herceptin) and placed in the incubator for 2 h. The cells were then washed. The wells were then relocated to the AMF room, temperature probes inserted and chilling regulated. Cells were exposed to either sham treatments or treatments with AMF for 20 min. The total set up time for washing AMF, temperature probe insertion into the wells, cooling regulation and AMF exposure added an additional hour for a total cell binding time of three hours prior to cell replacement in the incubator. All the cells were washed, fixed and stained in parallel using the Live/Dead cell viability assay (upper images) 6 h later, and in a blind study the number of live (green) or dead (red) cells in five randomly selected fields were quantified ( and , lower graphs). HMEC cells remained alive and stained green regardless of receiving AMF (lower panels of microscopic images) and the inclusion of either nanoparticle type during the binding to incubation time resulted in no increase in cytotoxicity (, upper panels). In a blind study, baseline cell death as measured by counting the number of red cells in HMEC cultures remained constant in all treatments, with less than 5% of cells staining with the death detection dye (, graph).
Figure 6. Quantification of HMEC killing before and after AMF exposure following incubation with non-antibody versus antibody-directed nanoparticles using the Live/Dead assay. Live HMECs were incubated in media alone, or with non-antibody-directed nanoparticles (BNF-control) or with Herceptin-directed nanoparticles (BNF-Herceptin) for 2 h. The cells were then washed and treated with a 163 kHz frequency AMF using a steady amplitude of 35,828 A/M for 20 min. Temperature monitoring of the media showed no rise in temperature above 37°C in wells that had been treated with the two types of nanoparticles. Cells were fixed and stained at 6 h using the Live/Dead assay and photographed (upper panels: 40×; calcein (green live) 1/100 s; EthD-1 (red dead) 1/200 s) and cells counted to determine the live/dead cell numbers (lower graphs). Data were pooled for at least duplicate wells and student t-tests used to test the null hypothesis. No killing of normal mammary epithelial cells was seen and cell viability remained at over 90% in all treatment groups. Statistical differences of p < 0.05* or p < 0.01** were not observed.
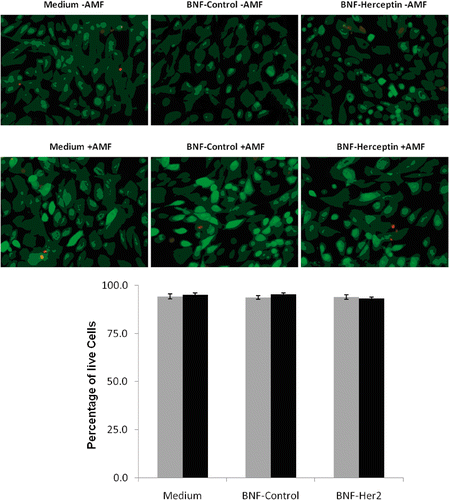
Figure 7. Quantification of SK-BR-3 cell killing before and after AMF exposure following incubation with non-antibody- versus antibody-directed nanoparticles using the Live/Dead assay. Live SK-BR-3 cells were incubated in medium alone, or with non-antibody-directed nanoparticles (BNF-control) or with Herceptin-directed nanoparticles (BNF-Herceptin) for 2 h. The cells were then washed and treated with a 163 kHz frequency AMF using a steady amplitude of 450 Orsteds for 20 min. Temperature monitoring of the media showed no rise in temperature above 37°C in wells that had been treated with the two types of nanoparticles. Cells were fixed and stained at 6 h using the Live/Dead assay and photographed (upper panels: 40×; calcein (green live)1/200 s; EthD-1 (red dead) 1/250 s) and cells were counted to determine the live/dead cell numbers (lower graphs). Data were pooled for at least duplicate wells and student t-tests used to test the null hypothesis. Only the SK-BR-3 cells that had been incubated with Herceptin-directed nanoparticles and exposed to AMF exhibited a statistically increased incidence of cell toxicity in the Live/Dead assay (p < 0.01**).
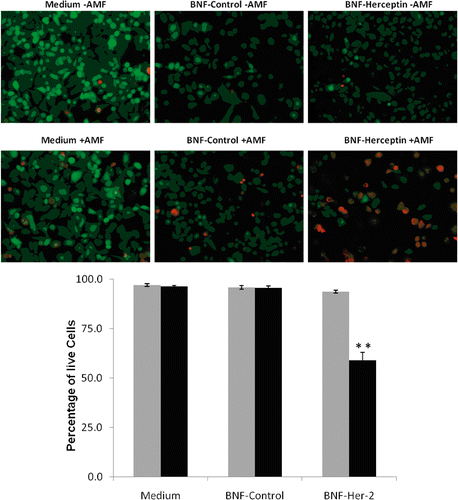
In contrast, AMF exposure of SK-BR-3 cells () that had received Herceptin-directed nanoparticles during the 2–3 h binding incubation time exhibited statistically significant cell death (red cells) (upper panel images), and at 6 h 40% of the cells were stained with a death detection dye, representing a statistically significant increase in cell death above all other treatment groups (, graph). Other treatments of SK-BR-3 cells did not result in any increase in cell death above baseline controls at 6 h using the Live/Dead cell viability assay.
Quantification of cell death by the lactate dehydrogenase release assay
A second method was used to quantify and document specific cell killing when AMF was used to activate and heat Herceptin-directed nanoparticles bound to HER-2 positive human cancer cells. Also, this assay captures an accounting of cells that have been lost from the plates and not seen in the Live/Dead assay described above. HMEC cells () or SK-BR-3 cells ( and ) were incubated with media alone, non-antibody directed control nanoparticles (BNF-C) (grey bars) or Herceptin-directed nanoparticles (BNF-Herceptin) (black bars) for 2–3 h in the incubator. As above, all the cells were then washed and exposed to sham or AMF (shown) treatments for 20 mins. After sham or AMF treatments, the cells were placed in the incubator for 6 () or 24 h ( and ) and then the medias were collected, centrifuged to remove nanomaterial and debris, and assayed for LDH. Two parallel cultures of each cell type that had received no treatment were lysed in total and LDH quantified to represent 100% LDH for each cell type. In the absence of AMF treatment (grey bars), little to no LDH release was detected in the HMEC cultures at 6 (not shown) or 24 h () or in SK-BR-3 cultures at 6 h () or 24 h (). With AMF exposure (black bars), no significant LDH release was seen in any of the cultures at 24 h, with one exception (). At 24 h, a statistically significant release of LDH was measured in cultures of SK-BR-3 cells that had been treated with AMF after incubation and binding/retention of Herceptin-directed nanoparticles. These studies confirm the observations of the Live/Dead cell viability assay, that AMF is required to achieve cell killing by heating of nanoparticles retained or internalised after antibody recognition and binding to HER-2 by Herceptin-directing antibodies on the nanoparticles. Human cancer cells are killed via this focused hyperthermia on the surface or within intracellular vesicles of the cells. The Live/Dead cell viability assay was predictive of an increase in cell toxicity by 6 h in these cultures, but we did not observe an increase in the release of LDH from these cells until 24 h. At 24 h we began to observe cells with partitioned nuclei resembling apoptotic bodies in the treatment group of SK-BR-3 cells that received Herceptin-directed nanoparticles and AMF. To examine this more closely, we treated SK-BR-3 cells with Herceptin-directed nanoparticles as above and then sham () or AMF treated them () as above. At 24, 48 and 72 h, we stained the cells to examine their nuclear morphology.
Figure 8. Quantification of lactate dehydrogenase (LDH) release into the media of HMECs or SK-BR-3 cells after incubation with media alone, non-antibody-directed nanoparticles or Herceptin-directed nanoparticles followed by sham or AMF exposures. Lactate dehydrogenase (LDH) was measured in the growth media of HMECs incubated with medium alone, BNF-control and BNF-Herceptin after 24 h (A) and compared to the LDH release by SK-BR-3 cells under parallel conditions at 6 (B) or 24 h (C) after sham (grey) or AMF irradiations (black bars). Total cell lysis standard curves had to be generated in parallel with known numbers of cells. Values are mean ± SE. Statistically significant differences from control are indicated with **; p < 0.01.
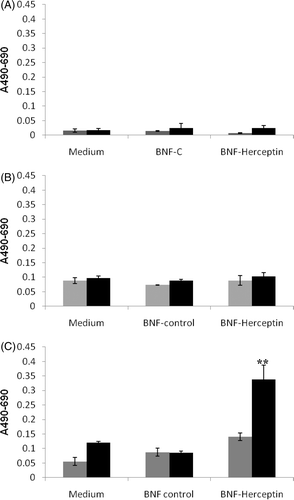
Figure 9. Detection of apoptosis using nuclear morphology in SK-BR-3 cells incubated with Herceptin-directed nanoparticles before and after AMF irradiation. Live SK-BR-3 were incubated with Herceptin-directed nanoparticles for 3 h and then washed and sham treated (A) or AMF treated (B) as above. Cells were fixed and stained 24, 48 (not shown) and 72 h later, using DAPI to stain DNA. Less than 1% of apoptotic cells were seen in sham exposed cultures, but cells that were AMF irradiated had significant apoptosis noted at 72 h.
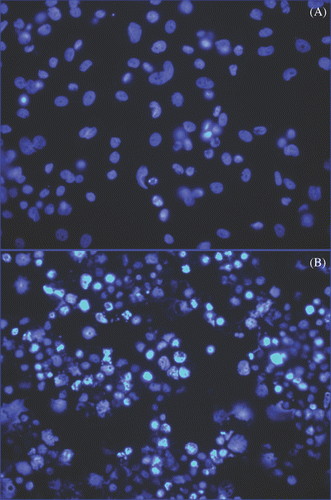
Herceptin-directed nanoparticles activated by AMF cause focused hyperthermia of human cancer cells and trigger apoptosis in these cells
Live SK-BR-3 cultures incubated with Herceptin-directed nanoparticles and exposed to AMF as described above, exhibited significant apoptosis at 72 h (p < 0.01) after treatment as evidenced by numerous apoptotic bodies (). At 24 and 48 h, increased numbers of apoptotic cells above controls were observed but did not reach statistically significant numbers when they were counted using student T-test analysis until 72 h. Without AMF treatment, apoptosis did not occur in SK-BR-3 cultures treated with the same Herceptin-directed nanoparticles in parallel (). No other treatment group exhibited statistically significant apoptosis in the cultures above baseline apoptosis. These studies indicate that HER-2 positive human cancer cells will selectively bind and internalise Herceptin-directed nanoparticles. This material had no cytotoxic effect until induced to exhibit Joule heating by an alternating magnetic field. When the nanoparticles heat in the microdomains of the cells or on their surface, global heating of the media does not occur. In pilot experiments using Lab-Teks without cells or nanoparticles, we observed a rise in temperature of approximately 3°C regardless of the starting point. As mentioned above, the temperature change is due to induction coil contact and transference of heat to the Lab-Tek slides. These pilot experiments were therefore used to determine the starting chiller temperature of 33°C to accommodate the anticipated rise in temperature during the AMF treatment. We then conducted Live/Dead assay studies using HMEC and SK-BR-3 cells without nanoparticles under these conditions: 33°C chiller and 20 min AMF treatments. We documented that no cell death was observed using either HMEC or SK-BR-3 cells in control wells without nanoparticles but exposed to AMF as cells experienced these temperature shifts before and then during AMF treatments, well outside the ranges of hypo- or hyperthermia. These pilot studies defined the experimental conditions used for all subsequent studies using nanoparticles. For the data shown, in every subsequent experiment, the probes were placed in wells that contained cells and either non-directed nanoparticles, after washing (grey line), or in wells containing cells and HER-2 nanoparticles, after washing (black line)(shown). No additional heating was ever detected even in wells containing HER-2-directed nanoparticles bound to SK-BR-3 cells. Yet at 6 h we can detect statistically significant increases in cytotoxicity using a Live/Dead cell viability assay; an increase in LDH release by 24 h and an increase in apoptotic cells at 72 h in these cultures. Normal mammary cells do not bind or internalise these nanoparticles and do not exhibit any signs of cell death when treated with AMF in parallel using these three independent assays of cytotoxicity.
Discussion
Herceptin received FDA approval in 1998 and is currently used in the treatment of human breast cancer after or in combination with other chemotherapeutics such as taxanes, doxorubicin and/or cyclophosphamides [Citation27–29]. It is thought that when Herceptin has efficacy for breast cancer cell growth it is via Herceptin binding and interfering with the mitogen receptor HER-2, overexpressed and chronically active in many human cancer cells Citation[29], Citation[30]. Herceptin has been shown to slow the growth of residual or resistant tumour cells and prevent or delay recurrence after patients have received one or more regimens of chemotherapy following surgery [Citation28–30]. Herceptin is approved for use in early stage, node negative and positive breast cancers that are HER-2 positive and is approved for follow-up therapy in metastatic breast cancer. The finding of HER-2 positivity with human cancer cells is a prognostic indicator of an aggressive tumour with high metastatic potential. As a short-term therapy, few side effects occur, but with longer treatments there are risks of cardiotoxicity and of reduced white blood cell numbers Citation[31], Citation[32]. In recent years, Herceptin has also been investigated as an agent to image HER-2 positive tumours, localise metastatic foci and deliver chemotherapeutic agents to the tumour. The chimeric antibody has been conjugated to radionuclides, dyes, or electron dense contrast molecules, and the tumour cells that bind these agents via membrane Her-2 can be visualised using single photon emission, optical tomography or magnetic resonance imaging, depending on the payload agent [Citation2–7, Citation[34].
In the current study we describe the use of Herceptin to deliver magnetite crystal composite nanoparticles to HER-2 positive highly malignant human mammary cells which then undergo apoptosis and then cell lysis if the cell-associated nanoparticles are activated to heat via exposure to high power alternating magnetic field energy. These nanoparticles were chosen due to their small size (100 nm hydrodynamic diameter), their low spontaneous aggregation in serum, their low intrinsic toxicity for cells, their rapid clearance rates in vivo and their magnetic hyperthermia properties [Citation9–15, Citation[33]. Herceptin was chosen as the antibody to direct the targeted delivery of the nanoparticles due to its widespread clinical use, low side effects in short term applications and its relevance to many types of human cancers, including breast cancer, and to metastatic disease. This therapeutic approach could be taken using the same nanomaterial but with different targeting antibodies or agents. In the study we describe methods to characterise binding kinetics, quantify iron and measure efficacy by three different cytotoxicity measures in pre-clinical studies.
First we demonstrated that Herceptin conjugated nanoparticles can be visualised in association with the cells using either indirect immunofluorescence to localise the human Ig G1 portion of the Herceptin antibody or by using Prussian blue histological stains to localise iron. Using these methods, we describe the time course for optimal cell binding using living cells. We compared the retention of Herceptin-directed nanoparticles in association with HER-2 positive cancer cells when compared to their HER-2 negative normal counterpart. We showed that in the first hour, Herceptin-directed nanoparticles only bound to HER-2 positive malignant cells and resisted washing removal. Non-directed nanoparticles were not retained by malignant cells and normal human mammary cells did not retain either form of nanoparticle. With increasing time, we observed that malignant HER-2 positive cells concentrated the bound Herceptin-directed nanoparticles and by 2–3 h internalised the material. This is similar to the finds by Wuang et al. Citation[6], where SK-BR-3 cells internalised and retained hyaluronic acid-coated Herceptin-directed polypyrrole nanospheres.
We then developed an experimental apparatus to expose cells to AMF without heating of the surrounding media during magnetic field irradiation. Lab-Teks used to grow the cells were positioned within a copper coil inductor designed to direct the magnetic flux to the solenoid interior, creating a uniform high amplitude AMF zone corresponding to the growth area of the cells. The coil was cooled with circulating water from a chiller during and following AMF, and fibre-optic temperature probes were inserted into treated wells to monitor media temperature continuously and ensure global temperature changes of the 500 µL media and cells within the wells above 37°C did not occur. Using this system, we tested the ability of the nanoparticles to induce specific killing of human cancer cells using AMF.
Hyperthermia is traditionally applied directly to tissue and tumour cell killing achieved when temperatures reach the 40–47°C range Citation[35]. A distinct advantage for the use of nanoparticles containing iron oxide to deliver heat to cell surfaces and interiors is the fast rate of heating within seconds of AMF Citation[36] to achieve hyperthermia at a multi-cellular level not a gross tissue level Citation[37]. Rabin has suggested intracellular loading of single tumour cells will not be sufficient to achieve hyperthermia but a combination of intracellular and extra-cellular nanoparticles retained in a region of at least 1.1 mm diameter would reach killing levels of heat Citation[38]. Our observations using Herceptin-directed nanoparticles indicate this targeting agent binds to all the HER-2 positive cells with an intracellular and membrane distribution. Although cell killing effects of hyperthermia are thermal dose dependent, a broad range of temperatures from 41–47°C can achieve similar effects and have been shown to be influenced by rate of temperature change, duration and the means by which the heat is delivered. This is particularly true when the materials used to deliver the heat are in the nano size range. Where they heat, at the subcellular level, at the membrane, or at subcellular locations, as single molecules or aggregates within vesicles, will influence the mechanism of cell killing. At this time, a thermal dose calculation as cumulative equivalent minutes is difficult to estimate in our studies. One piece of evidence suggests that the cellular temperatures we are achieving are above 41°C and less than 46°C, and that is because we primarily see an apoptotic response Citation[25], Citation[39–41]].
Normal human mammary cells exposed to AMF following incubation with either Herceptin-directed nanoparticles or control nanoparticles lacking antibodies exhibited no increase above basal cell death by the Live/Dead assay at 6 h, LDH release quantification at 6 or 24 h and nuclear morphology assay to detect apoptosis at 24, 48 or 72 h. In contrast, highly malignant human breast cancer cell cultures that did express HER-2, exhibited statistically significant cell toxicity at 6 h when measured using the Live/Dead assay, at 24 h using the LDH quantitative release assay and at 72 h using the apoptosis assay after incubation with Herceptin directed nanoparticles followed by AMF irradiation. If the nanoparticles were not directed via Herceptin (control nanoparticles) or if AMF irradiation was not applied, cell death in these HER-2 positive malignant human mammary cell cultures did not occur. These studies provide proof of principle in vitro evidence that Her-2 directed magnetite composite nanoparticles could be used to target Her-2 positive malignant cells and achieve a high therapeutic ratio.
We propose, if introduced in vivo, these nanoparticles can be imaged, for instance using MRI, to estimate their concentration in tumours and then the tumour containing the nanoparticles can be exposed to localised AMF to activate them to undergo hysteretic heating. If the concentration is in a certain range, particle excitation and heating will occur proximal to the excited nanoparticles and thus selective killing of the tumour cells to which they are attached and internalised via hyperthermia is possible. The diameter of the nanoparticles, the ferric oxide content, the concentration of the nanoparticles, the selective coatings, the AMF amplitude, pulse duty and duration, can be used to control and focus the hyperthermia to tumour tissue and minimise damage to surrounding normal tissues [Citation35–38]. While the possibility of a cell-localised temperature rise – sometimes referred to as intracellular hyperthermia – has been discounted on the basis of heat transport calculations [Citation35–38], energised magnetic particles have been shown to achieve cancer cell killing via a nano-localised temperature rise or other mechanism. Following treatment, the nanoparticles, the core and coating materials, appear to be metabolised, cleared and excreted with little toxicity in vivo, as observed with similar formulations in several mouse model studies Citation[17], Citation[19].
Besides antibodies, antibody fragments, various engineered protein and peptides, as well as aptamers and other agents may be used to target particles. For example, an exciting development in recent years is the production of ‘Affibodies’ incorporating Herceptin specific binding to carry imaging agents or therapeutics to the tumour by exploiting the Her-2 extracellular domain epitope Citation[42], Citation[43]. Affibodies are small, ∼6 kDa, proteins consisting of three helices that retain binding and recognition specificity of the monoclonal antibody their sequence is based on. Unlike antibodies, these recombination protein scaffolds bind covalently to the epitope with the specificity of antibody Fab sites. Since they lack the disulphide bonds of traditional antibodies, these Herceptin-based Affibodies are highly stable, tolerating exposure to very high temperatures (90°C), acid (ph 2.5) or alkaline (pH 11) treatments. These antibody mimetics have several potential advantages over antibodies and antibody fragments in that they are so small they are not deposited non-specifically in the liver or kidney, are quickly cleared from blood, and can be produced in large quantities using bacterial expression systems Citation[42], Citation[43]. They retain structural elements that allow conjugation methods developed with antibodies or Fab fragments to be used to bind nanomaterials to them in much the same way. In future, these nanoparticles could be used with Affibodies or monoclonal antibodies to tumour antigens other than HER-2, thus the technology represents a multi-platform therapeutic approach.
The ability to deliver an agent to tumour cells in close proximity to critical tissues such as nerves, capillaries and normal epithelial cells, and then activate that agent using focused AMF to target hyperthermia and deliver a killing blow limited to the tumour cells would be of great value in oncology. The in vitro approaches described in this paper can be used to characterise the conditions for such a treatment by examining and comparing nanoparticles of different diameters and core material, targeting agent surface functionalisation level, coatings, kinetics of uptake and internalisation by cells, and AMF strength, frequency, duty and pulse duration. While there are many challenges to the development of magnetic particle therapeutics Citation[44], the refinement of particle structures as described in this paper and AMF parameters for use in vivo may lead to clinical applications using the antibody-directed, magnetically energised nanoparticle therapeutic approach described in this paper.
Acknowledgements
The authors thank Patricia Arnold for her assistance with the manuscript.
Declaration of interest: We thank Triton Systems for partial funding of the Triton Biosystems and University of Massachusetts Lowell Collaborative Project TSI-4014-04-003 and The US Army Medical Research Acquisition (W81XWH-05-1-0496; W81XWH-04-C-0142). The authors alone are responsible for the content and writing of the paper.
References
- Park JW, Neve RM, Szollosi J, Benz CC. Unraveling the biologic and clinical complexities of HER2. Clin Breast Cancer 2008; 8: 392–401
- Mukai H. Treatment strategy for HER2-positive breast cancer. Int J Clin Oncol 2010; 15: 335–340
- Mannocci A, De Feo E, de Waure C, Specchia ML, Gualano MR, Barone C, Ricciardi W, La Torre G. Use of trastuzumab in HER2-positive metastatic breast cancer beyond disease progression: A systematic review of published studies. Tumori 2010; 96: 385–391
- Capala J, Bouchelouche K. Molecular imaging of HER2-positive breast cancer: A step toward an individualized ‘image and treat’ strategy. Curr Opin Oncol 2010; 22: 559–566
- Smith TA. Towards detecting the HER-2 receptor and metabolic changes induced by HER-2-targeted therapies using medical imaging. Br J Radiol 2010; 83: 638–644
- Wuang SC, Neoh KG, Kang E, Pack DW, Leckband D. HER-2-mediated endocytosis of magnetic nanospheres and the implications in cell targeting and particle magnetization. Biomaterials 2009; 29: 2270–2279
- Kikumori T, Kobayashi T, Sawaki M, Imai T. Anti-cancer effect of hyperthermia on breast cancer by magnetite nanoparticle-loaded anti-HER2 immunoliposomes. Breast Cancer Res Treat 2009; 113: 435–441
- Gilchrist RK, Medal R, Shorrey WD, Hanselman RC, Parrott JC, Taylor CB. Selective inductive heating of lymph nodes. Ann Surg 1957; 146: 596–606
- Lacroix LM, Ho D, Sun S. Magnetic nanoparticles as both imaging probes and therapeutic agents. Curr Top Med Chem 2010; 10: 1184–1197
- Santra S, Kaittanis C, Grimm J, Perez JM. Drug/dye-loaded, multifunctional iron oxide nanoparticles for combined targeted cancer therapy and dual optical/magnetic resonance imaging. Small 2009; 5: 1862–1868
- Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr Opin Chem Biol 2010; 14: 529–537
- Hu JCM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Current Drug Metabolism 2009; 10: 836–841
- Mohs AM, Provenzale JM. Applications of nanotechnology to imaging and therapy of brain tumors. Neuroimaging Clin N Am 2010; 20: 283–292
- Krishnan S, Diagaradjane P, Cho SH. Nanoparticle-mediated thermal therapy: Evolving strategies for prostate cancer therapy. Int J Hyperthermia 2010; 26: 775–789
- Hilger I, Hergt R, Kaiser WA. Use of magnetic nanoparticle heating in the treatment of breast cancer. IEE Proc Nanobiotechnol 2005; 152: 33–39
- DeNardo S, DeNardo G, Natarajan A, Miers L, Foreman A, Gruettner C, Adamson G, Ivkov R. Thermal dosimetry predictive of efficacy of 111 in-ChL6 nanoparticle AMF-induced thermoablative therapy for human breast cancer in mice. J Nucl Med 2007; 48: 437–444
- Ivkov R, DeNardo S, Daum W, Foreman A, Goldstein R, Nemkov V, DeNardo G. Application of high amplitude alternating magnetic fields for heat induction of nanoparticles localized in cancer. Clin Cancer Res 2005; 11: 7093–7103
- Lehmann J, Natarajan A, Denardo GL, Ivkov R, Foreman AR, Catapano C, Mirick G, Quang T, Gruettner C, Denardo SJ. Nanoparticle thermotherapy and external beam radiation therapy for human prostate cancer cells. Cancer Biother Radiopharm 2008; 23: 265–271
- Dennis CL, Jackson AJ, Borchers JA, Hoopes PJ, Strawbridge R, Foreman AR, van Lierop J, Grüttner C, Ivkov R. Nearly complete regression of tumors via collective behavior of magnetic nanoparticles in hyperthermia. Nanotechnology 2009; 20: 395103 doi: 10.1088/0957-4484/20/39/395103
- Grüttner C, Müller K, Teller J, Westphal F, Foreman A, Ivkov R. Synthesis and antibody conjugation of magnetic nanoparticles with improved specific power absorption rates for alternating magnetic field cancer therapy. J Magn Magn Mat 2007; 311: 181–186
- Natarajan A, Gruettner C, Ivkov R, DeNardo GL, Mirick G, Yuan A, Foreman A, DeNardo SJ. NanoFerrite particle based radioimmunonanoparticles: Binding affinity and in vivo pharmacokinetics. Bioconjug Chem 2008; 19: 1211–1218
- Kelm MA, Hammerstone JF, Schmitz HH. Identification and quantitation of flavanols and proanthocyanidins in foods: How good are the data?. Clin Dev Imm 2005; 12: 35–41
- Wang G, Dewilde AH, Zhang J, Pal A, Vashist M, Bello D, Marx KA, Braunhut SJ, Therrien JM. A living cell quartz crystal microbalance biosensor for continuous monitoring of cytotoxic responses of macrophages to single-walled carbon nanotubes. Part Fibre Toxicol 2011; 8: 4
- Vorotnikova E, Rosenthal RA, Tries M, Doctrow SR, Braunhut SJ. Novel synthetic SOD/catalase mimetics can mitigate capillary endothelial cell apoptosis caused by ionizing radiation. Radiat Res 2010; 173: 748–759
- Vorotnikova E, Ivkov R, Foreman A, Tries M, Braunhut SJ. The magnitude and time-dependence of the apoptotic response of normal and malignant cells subjected to ionizing radiation versus hyperthermia. Int J Radiat Biol 2006; 82: 549–559
- Braunhut SJ, McIntosh D, Vorotnikova E, Zhou T, Marx KA. Detection of apoptosis and drug resistance of human breast cancer cells to taxane treatments using quartz crystal microbalance biosensor technology. Assay Drug Dev Technol 2005; 3: 77–88
- Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S. Neoadjuvant chemotherapy and concomitant trastuzumab in breast cancer: A pooled analysis of two randomized trials. Anticancer Drugs 2011; 22: 128–135
- Guarneri V, Barbieri E, Dieci MV, Piacentini F, Conte P. Anti-HER2 neoadjuvant and adjuvant therapies in HER2 positive breast cancer. Cancer Treat Rev 2010; 36: 62–66
- Saxena R, Dwivedi A, ErbB family receptor inhibitors as therapeutic agents in breast cancer: Current status and future clinical perspective. Med Res Rev 2010; DOI: 10.1002/med.20209
- Chu PY, Li TK, Ding ST, Lai IR, Shen TL. EGF-induced Grb7 recruits and promotes Ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells. J Biol Chem 2010; 285: 29279–29285
- Carver JR. Management of trastuzumab-related cardiac dysfunction. Prog Cardiovasc Dis 2010; 53: 130–139
- Metzger Filho O, Saini KS, Azim Jr HA, Awada A. Prevention and management of major side effects of targeted agents in breast cancer. Crit Rev Oncol Hematol 2011; Sep 1. [Epub]
- Schlorf T, Meincke M, Kossel E, Glüer C, Jansen O, Mentlein R. Biological properties of iron oxide nanoparticles for cellular and molecular magnetic resonance imaging. Int J Mol Sci 2011; 12: 12–23
- Tookman L, Roylance R. New drugs for breast cancer. Br Med Bull 2010; 96: 111–129
- Roti-Roti JL. Cellular responses to hyperthermia (40–46°C): Cell killing and molecular events. Int J Hyperthermia 2008; 24: 3–15
- Adraä W, d’Ambly CG, Hergt R, Hilger I, Kaiser WA. Temperature distribution as function of time around a small spherical heat source of local magnetic hyperthermia. J Magn Magn Mat 1999; 194: 197–203
- Kalambur VS, Longmire EK, Bischof JC. Cellular level loading and heating of superparamagnetic iron oxide nanoparticles. Langmuir 2007; 23: 12329–12336
- Rabin Y. Is intracellular hyperthermia superior to extracellular hyperthermia in the thermal sense?. Int J Hyperthermia 2002; 18: 194–202
- Vauthier C, Tsapis N, Couvreur P. Nanoparticles: Heating tumors to death?. Nanomedicine (Lond) 2011; 6: 99–109
- Yonezawa M, Otsuka T, Matsui N, Tsuji H, Kato KH, Moriyama A, Kato T. Hyperthermia induces apoptosis in malignant fibrous histiocytoma cells in vitro. Int J Cancer 1996; 66: 347–351
- Dewhirst MW, Prosnitz L, Thrall D, Prescott D, Clegg S, Charles C, MacFall J, Rosner G, Samulski T, Gillette E, et al. Hyperthermic treatment of malignant diseases: Current status and a view toward the future. Sem Oncol 1997; 24: 616–625
- Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett 2010; 584: 2670–2680
- Ahlgren S, Orlova A, Wållberg H, Hansson M, Sandström M, Lewsley R, Wennborg A, Abrahmsén L, Tolmachev V, Feldwisch J. Targeting of HER2-expressing tumours using 111In-ABY-025, a second-generation affibody molecule with a fundamentally reengineered scaffold. J Nucl Med 2010; 51: 1131–1138
- Barry SE. Challenges in the development of magnetic particles for therapeutic applications. Int J Hyperthermia 2008; 24: 451–466