Abstract
Purpose: Adding hyperthermia to chemotherapy improved the clinical outcome of patients with high risk soft tissue sarcoma. Further improvement might be possible if combined with vaccination strategies. As no sarcoma-associated antigens are known, the ectopic expression of a surrogate marker for which immune monitoring tools are available, is envisaged. We tested surrogate marker transfer into sarcoma cells in vitro using modified vaccinia virus Ankara (MVA), which has well established clinical safety. We examined its robustness against standard sarcoma treatment modalities, such as ifosfamide and hyperthermia.
Materials and methods: We transduced sarcoma cell lines and primary tumour cells from sarcoma patients with MVA encoding the human tyrosinase gene (MVA-hTyr). Kinetics of tyrosinase expression and the potency to activate tyrosinase-specific cytotoxic T cells were assessed. In addition cells were exposed to chemotherapy and heat, imitating the clinical setting.
Results: Tyrosinase was ectopically expressed in sarcoma cells. Infected cells presented tyrosinase epitopes for T cell recognition even if exposed to ifosfamide/heat.
Conclusions: As sarcoma patients receive surgery up front or after neoadjuvant systemic chemotherapy/hyperthermia, tumour material is generally available. Our data document that primary sarcoma cells can be infected with MVA-hTyr in vitro and antigen presentation is not affected by ifosfamide or heat treatment. Infected cells can serve as a source for vaccine preparation. MVA-hTyr infection of tumour cells lacking defined antigens is a feasible system to introduce a robust surrogate marker to provide an immune monitoring marker for assessing the induction of antigen-specific T cell activation.
Introduction
Immunotherapeutic strategies against malignant diseases utilise diverse strategies, including autologous and allogeneic tumour cells, peptides, proteins and dendritic cells modified to present antigenic fragments via MHC class I and II Citation1–5. They all aim at inducing tumour-associated antigen (TAA)-specific T cell responses (for review see Banchereau and Palucka Citation6 and Slingluff et al. Citation7. Clinical phase I and II trials could convincingly show that specific anti-tumour immune responses can be elicited with low toxicity and minimal side effects.
The need to know TAAs to establish a tumour-specific vaccine or to monitor antigen-specific CD8+ and/or CD4+ T cell responses and judge immunological efficacy of vaccination hampered the development of immunotherapies for tumour entities where useful TAAs have not been identified.
One group of the malignancies with sparse knowledge of specific TAAs is sarcomas; malignant mesenchymal neoplasms. Because of their unpredictable clinical behaviour and the lack of objective markers for its evaluation, progress in the management of these tumours has been minimal.
A recent phase III clinical European Organization for Research and Treatment of Cancer (EORTC) trial has provided strong evidence that applying regional hyperthermia simultaneously with chemotherapy, such as ifosfamide, significantly improves local progression-free survival of patients with high-risk sarcoma Citation8.Other innovative treatments such as vaccination trials in sarcoma have been ongoing since the early 1970 s Citation9. Due to the lack of knowledge of TAAs, the vaccines were based on autologous tumour cells and immune monitoring was limited to measurements of a delayed-type hypersensitivity response (DTH) and demonstration of lymphocytic infiltration at the vaccination site. A more detailed insight into the immunological response in patients could be gained if the vaccine could express a traceable surrogate marker antigen. This could be achieved by transferring a well-known surrogate antigen into primary tumour cells ex vivo. These engineered tumour cells containing the surrogate antigen in addition to the undefined tumour-associated antigens could be used for loading of dendritic cells (DCs).
For antigen transfer, recombinant modified vaccinia virus Ankara (MVA) is one of the most promising live viral vector systems. It has well-established safety, large packaging capacity for recombinant DNA and achieves high-level expression of heterologous genes Citation10, Citation11. Being a replication-defective form, it can be used under conditions of biosafety level 1. Drexler and colleagues showed in vitro and in vivo the efficient generation of HLA-restricted tyrosinase-specific cytotoxic T cells using autologous dendritic cells infected with MVA carrying the cDNA for human tyrosinase (MVA-hTyr) Citation12. Tyrosinase is an attractive candidate for a surrogate antigen as it encodes for a number of peptide epitopes presented in the context of MHC class I and II molecules for recognition by CD8+ and CD4+ T cells Citation13, Citation14. In contrast to synthetic peptides, the MVA gene transfer system generates cells that encompass the whole spectrum of potential T cell epitopes not limited by HLA compatibility. Several clinical trials documented that MVA lacks the drawback of adenoviral vectors. In detail MVA did not did not cause strong de novo activation of adaptive responses nor did it activate pre-existing memory responses to the vector itself. MVA-induced activation of innate immunity was observed and found to be supportive for induction of antigen-specific immunity. Regarding tyrosinase as a surrogate antigen of choice, its ectopic expression can stimulate antigen-specific T cell responses. These caused forms of vitiligo in some cases, which, however, were therapeutically manageable Citation15.
Altogether, several clinical trials attested the feasibility, safety and low toxicity of MVA-hTyr in clinical vaccination Citation16. MVA-hTyr-infected tumour cells can be used as antigen source to introduce tyrosinase as a marker to measure CD4 and CD8 adaptive T cell immunity.
We explored the MVA vector system to introduce human tyrosinase in sarcoma cell lines and primary cells. We evaluated whether the “genetically engineered” sarcoma cells allowed the presentation of the ectopically expressed tyrosinase to the immune system. HTyr-transfected HLA-A2-positive sarcoma cells activated TyrF8, a HLA-A*0201-restricted tyrosinase peptide (tyr368-376)-specific cytotoxic T cell clone, demonstrating that sarcoma cells efficiently processed and presented the tyrosinase epitope. As we have previously observed that hyperthermia can reduce antigen presentation Citation17, we also tested expression and presentation of the MVA-tyrosinase in combined treatment conditions. Antigen presentation and T cell stimulation was maintained after in vitro hyperthermia or combined hyperthermic and ifosfamide treatment.
Our data demonstrate that MVA-hTyr is a robust system with clinically approved status that achieves high transfection efficiency with low toxicity and stable protein expression and presentation even in cells that had been exposed to various treatment conditions. Introducing tyrosinase to tyrosinase-negative cells such as sarcoma cells endows these cells with a surrogate marker and opens the possibility to monitor the induction of an immune response without the requirement to know sarcoma-specific antigens.
Material and methods
Tumour and T cell lines
Ewing's sarcoma cell line RD-ES and human osteosarcoma cell line TC71 were obtained from ATCC (LGC Promochem, Wesel, Germany).
A375 is an HLA-A2 positive tyrosinase-negative human melanoma cell line and SK23-Mel is an HLA-A2 positive tyrosinase-positive human melanoma cell line (gift from M.C. Panelli, National Institutes of Health, Bethesda, MD, USA). RD-ES, A375 and SK23-Mel cells were cultured in Roswell Park Memorial Institute medium 1640 (RPMI) (Invitrogen, Karlsruhe, Germany) and TC71 cells in Iscove's Modified Dulbecco's medium (IMDM) (Invitrogen) each containing 10% FCS. The HLA-A*0201-restricted tyrosinase peptide tyr368-376 (YMNGTMSQV)-specific cytotoxic T-cell clone TyrF8 and the HLA-A2-alloreactive T cell clone JB4 were cultured as described (17). T cells were used on day 14 after the last stimulation. Cultures were periodically tested for mycoplasma by fluorescent DNA staining according to the manufacturer's instruction (Biochrom, Berlin, Germany). For infection experiments, tumour cells of passage 7 to 11 were used.
Primary cells
After informed consent of patients, appromximately 3 cm3 of resected tumour was used for preparation of primary cells. Tissue from five different patients was used. Patients were suffering from synovial sarcoma (2), liposarcoma (1), leiomyosaroma (1) and fibrosarcoma (1). Surgery of the tumour was performed for clinical reasons i.e. as a standard treatment of resectable sarcoma at first diagnosis. Tissue was minced into small pieces and then incubated with sterile serum-free medium containing hyaluronidase (10 mg/mL), collagenase (20 mg/mL) and deoxyribonuclease (40.000 IE/mL) for 4 h at room temperature. After incubation, cells were filtered through a sieve (approximately 1 mm pore size) to remove undigested tissue. The cell suspension was washed in RPMI medium and finally cells were resuspended in RPMI 1640 medium containing 10% fetal calf serum (FCS), sodium pyruvate and non-essential amino-acids (Invitrogen). After 48 h, cultured cells were washed with PBS to remove non-adherent cells. Adherent cells were further incubated until sufficient expansion (usually 3 to 5 passages). HLA-A2 expression was determined by staining with antibody HB82, specific for HLA-A2, and analysed by flow cytometry Citation17.
Heat treatment of cells
In complete medium containing 10% FCS, 2 × 106 cells each were seeded onto a 6-well dish. After 24 h of recovery at 37°C, cells were exposed to 37°C or 41.8°C for 2 h with or without chemotherapy (see below) using a water bath. After heat shock, cells had a recovery time for 6 h at 37°C. Thereafter, cells were infected with MVA or control vector, as described below. After MVA-infection, cells were washed and resuspended in fresh medium containing 2% FCS. After an additional 16 h of incubation, cells were harvested for western blot analysis and T cell assay.
Ifosfamide treatment
4-OH-ifosfamide was added to cells at a final concentration of 100 or 200 µM. 4-OH-ifosfamide treatment was performed over 2 h with or without concurrent heat treatment (see above). After 2 h of treatment, medium was changed and cells were further kept at 37°C for 6 h in order to recover. Thereafter, cells were infected with MVA-hTyr or MVA wild type (not encoding ectopic gene sequences) as indicated (see below). Cells were harvested 16 h after MVA infection.
Recombinant MVA virus
MVA expressing the human tyrosinase gene under control of the VACV synthetic early/late promoter SP (MVA-hTyr) has been described previously Citation18. MVA expressing GFP (MVA-GFP) was generated by homologous recombination as described previously Citation19, Citation20. DNA genomes of recombinant viruses were analysed by PCR. MVA were propagated and titrated following standard methodology.
Infection of cells
MVA infection was performed in cell line specific medium containing 2% FCS in a 6-well plate. Therefore, 2 × 106 cells were seeded per well and were allowed to recover for 6 h at 37°C. Thereafter, cells were co-incubated for 1 h with MVA wild-type, MVA-GFP or MVA-hTyr at a multiplicity of infection as indicated. After harvesting the cells and washing them with PBS, cells were resuspended in medium with 10% FCS. Cells were analysed right away (0 h time point) or kept in fresh medium at 37°C for 4 to 72 h as indicated. Unless indicated otherwise, cells were incubated for 16 h after 1 h of infection before analysis.
Assessment of viability
Cell viability was assessed by staining cells with 10 µg/mL propidium iodide (PI) and subsequent analysis by flow cytometry using a FACScan and CellQuest software (BD Biosciences, San Jose, CA). The number of non-viable cells is given as percentage of PI-positive cells of all cells. If not further specified, the number of non-viable cells was below 10%.
Quantification of tyrosinase protein expression by western blot analysis
1–2 × 106 cells were lysed in 300 µL of 2% CHAPS buffer (pH 7.5) in 200 mM NaCl and 50 mM HEPES containing a mixture of freshly added protease inhibitors pepstatin A, leupeptin A, aprotinin, antipain, chymostatin, PMSF (10 µg/mL) and phosphatase inhibitors sodium pyrophosphate (10 mM), sodium fluoride (50 mM) and sodium vanadate (0.2 mM). After 30 min on ice, samples were spun at 13,000 g for 30 min at 4°C. Supernatants were collected and protein levels were quantified using the TCA precipitation assay (Sigma Biotech, St Louis, MO, USA). Protein content was adjusted for each sample and probes were loaded onto a 10% SDS gel and run for 1.5 h at 150 V. Gels were blotted to a nitrocellulose membrane and incubated with a 1:500 dilution of T3.11 monoclonal IgG2a mouse anti-human antibody directed against the C-terminal peptide of human tyrosinase (Novocastra, Newcastle upon Tyne, UK). As a secondary antibody we used a 1:2000 dilution of goat anti-mouse antibody (Bio-Rad, Hercules, CA, USA). The enhanced chemiluminescence system (ECL) was used to detect bound first and secondary antibody.
Quantification of antigen presentation and T cell activation using IFN-γ
IFN-γ ELISA
Supernatants of stimulator/T cell co-cultures were collected after 24 h of co-culture and stored at −70°C until analysis. IFN-γ levels were quantified using the OptEIA ELISA kit (Becton Dickinson, Franklin Lakes, NJ, USA) according to manufacturer's protocol.
IFN-γ ELISPOT
For the detection of individual IFN-γ secreting cells, we used the IFN-γ ELISpot kit (Mabtech, Nacka Strand, Sweden) according to manufacturer's instruction. The coating antibody was the monoclonal mouse-anti-IFN-γ clone 1-D1K and the detection antibody was the biotinylated monoclonal mouse-anti-human IFN-γ clone 7-B6-1 (both from Mabtech). After coating the 96-well plates, wells were blocked in RPMI 1640 culture medium supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, penicillin/streptomycin (100 U/mL) and 10% human AB serum (Cambrex, Verviers, Belgium). Serial dilutions of effector T cells (TyrF8 or JB4 T cells) were co-incubated for 24 h with stimulator cells at an E:T ratio as indicated. After incubation, plates were washed and detection antibody was added for 2 h at 37°C. The enzyme-substrate reaction was performed according to protocol. The read-out was done by a computer-assisted video imaging analysis system (KS-ELISPOT, Zeiss, Jena, Germany). As positive control phytohaemagglutinin (1 µg/mL)-stimulated peripheral blood mononuclear cells (PBMC) were used.
Statistics
For calculation of p values we used the two-sided t-test for unpaired samples.
Results
GFP and tyrosinase expression levels after infection with MVA are dependent on the recipient cell line and multiplicity of infection (MOI).
To define the conditions for optimal MVA-hTyr infection we used MVA containing the cDNA-sequence of green fluorescence protein (GFP). 1 × 106 cells of the Ewing`s sarcoma cell lines RD-ES and TC71 and the melanoma cell line A375 were infected with MVA-GFP, as described in materials and methods, using different MOI as indicated. After 16 h, GFP expression was assessed by FACS analysis. For some experiments intracellular appearance of GFP was verified by fluorescence microcopy (data not shown). The percentage of GFP-positive cells of gated live cells (data not shown) as well as the mean fluorescence intensity () differed substantially, dependent on the cell line used. Percentage of positive cells increased with MOI reaching maximum values of around 68% for A375, 66% for TC71 and 83% for RD-ES at MOI of 15. Mean fluorescence intensity also increased with MOI reaching 382 for A375, 235 for TC71 and 138 for RD-ES.
Figure 1. Influence of MOI on GFP or tyrosinase expression by TC71, A375 and RD-ES cells. (A) Cells were infected using different MOI of MVA-GFP. Viable cells were evaluated 16 h after infection for mean fluorescence intensity of GFP-positive cells. Data are given as mean ± SEM of three independent experiments. (B) Infection with MVA-hTyr and assessment of tyrosinase protein expression by western blot analysis 16 h after infection. Control represents cells that were not infected.
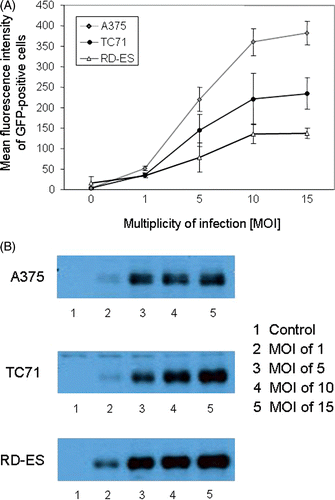
Tyrosinase protein expression after MVA-hTyr infection was analysed by western blot (). Similar to GFP-positivity, tyrosinase expression level increased with the MOI in all cell lines. In contrast to the low MFI after GFP transfection, RD-ES cells showed highest tyrosinase expression. The percentage of non-viable cells was below 10% for MOI up to 10 and increased to 20% when a MOI of 15 was used. Due to better viability without significantly less efficiency of infection, we chose an MOI of 10 for all further experiments.
Figure 2. Kinetics of GFP or tyrosinase expression of TC71, A375 and RD-ES cells after MVA infection. A375, TC71 and RD-ES cells, infected with MVA-GFP at an MOI of 10, were analyzed 0, 4, 8, 16, 24, 48 and 72 h post infection by flow cytometry and western blot analysis, respectively. (A) The percentage of GFP-positive cells. (B) Mean fluorescence intensity of MVA-GFP-infected cells and not infected control cells over time. Results of A and B are given as mean ± SEM of three independent experiments. (C) Tyrosinase expression of TC71 and RD-ES cells evaluated by western blot analysis. Control cells were not infected (lane 1), or infected with MVA wild-type not containing the cDNA for tyrosinase (lane 3).
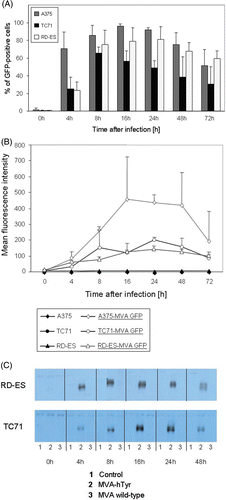
GFP and tyrosinase expression after infection with MVA is dependent on the time period after infection
To determine the best time point for the analysis of the T cell stimulatory capacity of MVA-hTyr-infected cells, we first investigated the kinetics of GFP and tyrosinase expression at different time points (0, 4, 8, 16, 24, 48 and 72 h) after infection. A375 cells and RD-ES cells showed the highest percentage of GFP-positive cells at almost all assessed time points (). Highest MFI of GFP expression was found 16 h after infection (). TC71 cells had an overall lower GFP expression. Highest percentages of positive cells were observed between 8 and 24 h after infection. Using MVA-hTyr and analysing tyrosinase protein levels by western blot, we observed strong expression between 8 and 24 h after transfection with variation of the optimal time point depending on the cell line used for infection (). Comparable results were obtained for A375 cells (data not shown).
Tyrosinase-negative cells infected with MVA-hTyr induce specific activation of T cell clone TyrF8
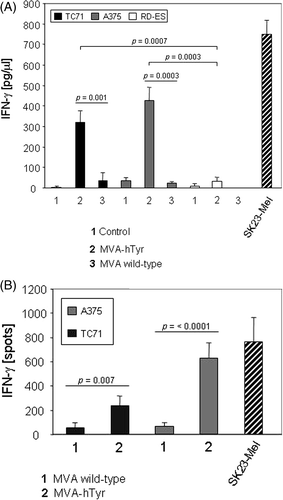
To investigate whether the tyrosinase protein is processed into T cell epitopes in sarcoma cells, tyrosinase-negative sarcoma cell lines RD-ES (HLA-A2-negative) and the HLA-A2 positive TC71 were infected with MVA-hTyr. As a control, the tyrosinase-negative and HLA-A2-positive melanoma cell line A375 was also transfected. Based on the observation that tyrosinase expression is highest 16 h after transfection, the HLA-A2-restricted tyrosinase-specific T cell clone TyrF8 was added at that time point. After 24 h of co-culture, supernatants were harvested and the amount of IFN-γ, as an indicator of T cell activation, was quantified by ELISA. As shown in , HLA-A2-positive sarcoma (TC71) and melanoma (A375) cell lines induced IFN-γ secretion of TyrF8 with higher levels observed in co-cultures with the melanoma cell line. Highest IFN-γ levels were detected in co-cultures with HLA-A2-positive and endogenously tyrosinase-positive SK23-Mel cells. These data were confirmed on a single-cell level using the IFN-γ ELISPOT assay as described in materials and methods ().
Figure 3. IFN-γ secretion of TryF8 T cells induced by cell lines infected with MVA-hTyr. (A) MVA-hTyr-infected A375, TC71 and RD-ES cells and melanoma cell line SK23-Mel, which expresses tyrosinase endogenously, were co-cultured with CTL clone TyrF8 at an E:T ratio of 1:7 for 24 h. Background IFN-γ production by T cells was below 10 pg/mL. IFN-γ content in supernatants was quantified by ELISA. Control cells were either not infected (bar 1) or infected with MVA-wild type not containing the tyrosinase cDNA (bar 3). There was no significant difference in IFN-γ production comparing hTyr-transfected A375 and TC71 cells (p = 0.087). (B) MVA-hTyr-infected A375 and TC71 cells were co-cultured with TyrF8 T cells at an E:T ratio of 1:5 and ELISPOT analysis was performed. Control cells were infected with MVA wild-type. TyrF8 alone produced less than 10 spots (data not shown). Results are given as mean ± SEM of three independent experiments.
Tyrosinase-negative primary cells of sarcoma patients produce tyrosinase and induce specific activation of T cell clone TyrF8 after infection with MVA-hTyr
We further investigated whether primary sarcoma cells would be accessible to MVA-hTyr infection making them capable of activating tyrosinase-specific T cells. Primary sarcoma cells were isolated using sarcoma tissue of therapy-naive patients undergoing surgery for diagnostic reasons. Viability of cells before and after infection was high with non-viable cells below 7% (data not shown).
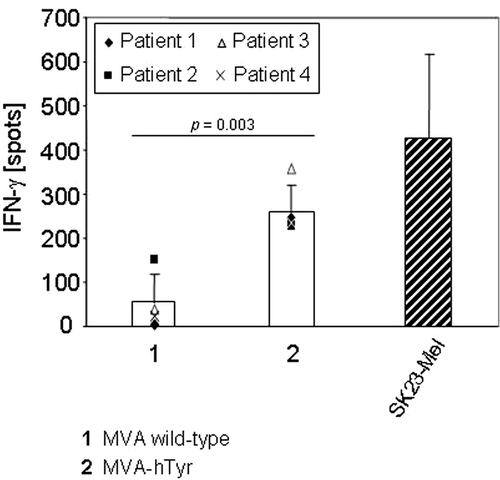
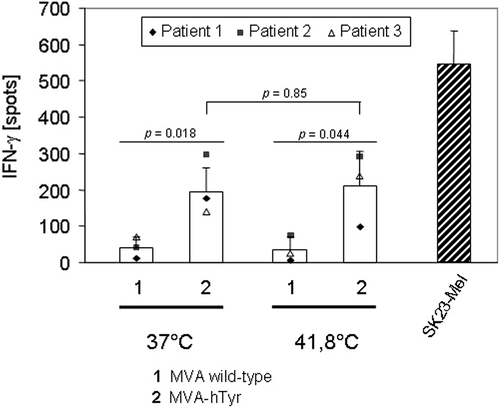
Four patients were HLA-A2-positive and all four samples of primary cells were capable of stimulating TyrF8 cytotoxic T lymphocyte (CTLs) after MVA-hTyr infection (mean number of IFN-γ-positive spots of the four patients: 265 ± 62 with MVA-hTyr; 55 ± 65 with MVA-WT; ). One patient was HLA-A2-negative and thus, as expected, was not able to induce IFN-γ synthesis by TyrF8 after MVA-hTyr infection despite tyrosinase protein expression (data not shown).
Figure 4. IFN-γ secretion of TyrF8 T cells achieved by primary sarcoma cells infected with MVA-hTyr. Primary HLA-A2+ sarcoma cells (n = 4 patients) were infected with MVA wild-type (bar 1) or MVA-hTyr (bar 2) as indicated and co-incubated with TyrF8 CTLs at an E:T ratio of 1:5 for 24 h. Control cell line SK23-Mel (HLA-A2 positive with endogenous tyrosinase expression) was used at the same E:T ratio. IFN-γ ELISPOT was performed. Results are shown as mean ± SEM of four independent experiments. P values indicate statistical significance between MVA wild-type control (1) and MVA-hTyr infected (2) cells.
Heat shock does not influence the capacity of tyrosinase-transfected primary cells to activate T cell clone TyrF8
In model systems, the cellular response to heat stress was linked to a response of the immune system to cancer Citation21. Clinically, hyperthermia is recognised as an adjuvant treatment to improve the management of sarcoma Citation8, and local recurrences of breast cancer and cervical cancer Citation22.
We have previously shown that endogenous tyrosinase presentation of peptides to T cells undergoes temporary changes during the heat shock response Citation17. We therefore investigated here whether heat-induced changes in tyrosinase presentation also occurred in cells that expressed tyrosinase under the control of the exogenous MVA promoter.
MVA-hTyr-infected primary cells that were exposed to heat shock (41.8°C for 2 h) showed similar viability than non-heat-treated cells (data not shown). When tested for the tyrosinase epitope presentation similar numbers of IFN-γ producing T cells were observed (195 ± 65 spots without heat shock versus 209 ± 99 spots with heat shock; ). The results were confirmed by IFN-γ-ELISA (data not shown).
Figure 5. IFN-γ secretion of TyrF8 T cells by infected primary sarcoma cells after heat treatment. Primary sarcoma cells were either exposed to 37°C or 41.8°C for 2 h. After 6 h of recovery at 37°C, cells were infected with MVA wild-type (bar 1) or MVA-hTyr (bar 2). Thereafter TyrF8 cells were added at an E:T ratio of 1:5 for 24 h. HLA-A2- and tyrosinase-positive SK23-Mel cells served as control. Results of the IFN-γ ELISPOT of three independent experiments are given. P values indicate statistical significance between MVA wild-type control (1) and MVA-hTyr infected (2) cells in the 37°C and heated samples.
Pretreatment of primary tumour lines with heat shock or ifosfamide alone or in combination does not influence infection with MVA-hTyr nor does it reduce the capacity to express and present the tyrosinase epitope for T cell activation
The combination of heat treatment and chemotherapy is a known therapeutic strategy in multiple solid tumours Citation8, Citation23, Citation24. Recently, the combination of chemotherapy such as ifosfamide, with hyperthermia in patients suffering from soft tissue sarcoma has proven to significantly improve local time to progression Citation8. Primary tumour cells harvested for intended vaccine development are therefore likely pretreated. Here, we tested the stability of ectopic tyrosinase expression by infecting sarcoma cells and control cells that had been pretreated with ifosfamide and heat shock.
Sarcoma and melanoma cells were exposed to ifosfamide for 2 h. After 6 h of recovery, cells were transfected with MVA-hTyr or MVA-wildtype and their potential of specific T cell activation was assessed 16 h later.
Admittedly the stimulation of tyrosinase specific CTL by MVA-hTyr infected TC71 sarcoma cell line was low and showed some variation in the different experiments (). Though ifosfamide treatment reduced the capacity of tumour cells to activate IFN-γ secretion by tyrosinase-specific CTL, this reduction did not reach significance. MVA-hTyr infected melanoma A375 induced strong IFN-γ production (). Subjecting the MVA-hTyr infected melanoma cell line A375 to the same treatment conditions, ifosfamide did not significantly alter the induced IFN-γ levels. Moreover, cells exposed to either ifosfamide or heat alone (data not shown) or to the combined treatment of ifosfamide with heat and subsequent infection with MVA-hTyr, were equally efficient to stimulate IFN-γ production of antigen-specific T cells than untreated cells ( and ). Taken together, MVA-mediated antigen expression, antigen presentation and stimulation of antigen-specific T cell activation as measured by IFN-γ synthesis was not impaired by exposure of tumour cells to ifosfamide, alone or in combination with heat, which are treatment schemes used in clinical settings Citation8.
Figure 6. IFN-γ secretion of TyrF8 T cells by infected TC71 sarcoma cell line and A375 melanoma cell line after exposure to 4-OH-ifosfamide. TC71 sarcoma cells (A), or A375 melanoma cells (B), were treated with ifosfamide and infected with MVA-hTyr (2). Cells treated with ifosfamide (100 or 200 µM) but either not infected (0) or infected with MVA wild-type (1) served as controls for background IFN-γ. IFN-γ production of TyrF8 after stimulation with SK23-Mel cells is depicted as reference value for T cell activity. IFN-γ content in 4-h co-culture supernatants was quantified by ELISA. Results are shown as mean ± SEM of three independent experiments.
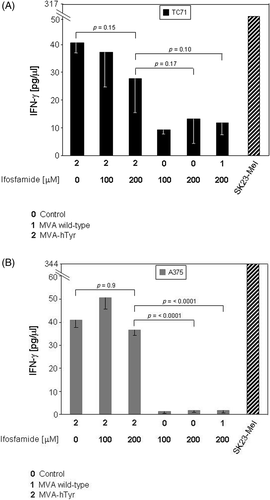
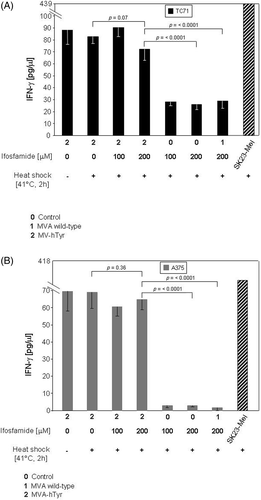
Figure 7. IFN-γ secretion of TyrF8 T cells by infected TC71 sarcoma cell line and A375 melanoma cell line after exposure to 4-OH-ifosfamide and heat. TC71 sarcoma cells (A), or A375 melanoma cells (B), were simultaneously treated with ifosfamide and heat shock (41.8°C, 2 h) and thereafter infected with MVA-hTyr (2). As controls, similarly treated cells but not infected (0) or cells infected with the MVA wild-type (1) were used. IFN-γ production of TyrF8 after stimulation with tyrosinase-expressing SK23-Mel cells is depicted as reference value for T cell activity. IFN-γ content in 4-h co-culture supernatants was quantified by ELISA. Results are shown as mean ± SEM of three independent experiments. Only the results of cells with concomitant heat shock treatment are shown, because cells without heat shock showed comparable values. For both cell lines p values indicate statistical significance as to whether ifosfamide- and heat-treated cells were MVA-hTyr-infected or not.
Discussion
Clinical hyperthermia is thought to be a way of immune stimulation Citation25. Results of an EORTC multicentre randomised clinical trial comparing the combination with systemic chemotherapy treatment and regional hyperthermia against chemotherapy alone in patients with high grade soft tissue sarcoma showed a significantly prolonged progression-free survival and overall disease-free survival in the combination arm Citation8. These results suggest that the management of sarcoma could be further improved by additional immune modulatory strategies.
A variety of immuno-modulatory strategies against malignant diseases have been established, including the administration of anti-tumour antibodies Citation26 or anti-tumour-reactive T cells and active vaccinations, in which tumour cells or DCs pulsed with tumour-specific peptides, tumour cell lysates, or tumour-derived RNA are used as vaccines for immune stimulation Citation27. Monitoring the induction of a cellular immune response in the patient is one parameter to estimate the efficacy of a vaccination approach. The ability to detect such responses is based on the knowledge of immunogenic peptides and peptide-specific T cell clones, which allow specific screening methods and the construction of monitoring tools, such as peptide-MHC multimers. The knowledge of TAAs, however, is limited to few tumour entities, such as melanoma, breast cancer or ovarian cancer.
Sarcoma, representing 1% of all solid tumours, is one of the tumour entities where TAAs are sparse and sufficient tools for immune monitoring are not available. Clinical trials investigating immunotherapeutic modalities in sarcoma are limited to the assessment of clinical response and the occurrence of a delayed-type hypersensitivity response Citation28–30.
In a recent study, Meyers and colleagues tested the effect of a muramyl tripeptide, which is a non-specific immune stimulant, in combination with chemotherapy in patients with osteogenic sarcomas Citation31. In this randomised study the clinical improvement of patients receiving ifosfamide combined with muramyl tripeptide was thought to result from a decrease in the frequency of regulatory T cells. Whether antigen-specific T cell responses were also induced could not be monitored due to the lack of immune monitoring tools. To allow the assessment of immune modulatory strategies in the treatment of sarcoma, immune monitoring tools need to be developed.
We envisaged the transfection of sarcoma cells with MVA encoding the human tyrosinase gene to endow them with a surrogate marker for immune monitoring. The human tyrosinase is an attractive candidate surrogate marker as it encodes for several well characterised antigenic peptides known to induce specific T cell responses (for review see Citation32). Moreover, the monitoring tools developed for tyrosinase could be used to monitor vaccination efficacy in such a setting.
MVA is a preferable delivery system for the surrogate marker as it achieves a high level of transfection efficacy and protein expression in primary non-dividing cells. Moreover, MVA has not shown the drawback of adenoviral vectors as it did not lead to strong de novo activation of adaptive responses, nor did it activate pre-existing memory responses to the vector itself. Observed activation of innate immunity, such as DC activation, was found to be supportive for induction of antigen-specific immunity.
The MVA-hTyr vector system has been used in several clinical trials and showed feasibility, safety and low toxicity Citation15, Citation16, Citation33–35.
Using the MVA-hTyr we document efficient infection and robust protein expression in primary fresh sarcoma cell suspensions. The infected sarcoma cells efficiently presented the tyrosinase epitopes for antigen-specific T cell stimulation. The efficacy of infection and antigen presentation was stable in ifosfamide- and/or hyperthermia-treated cells in vitro, treatment conditions that mimic the protocols applied in clinical settings. This documents that the MVA-based surrogate marker delivery system is applicable to tumours collected from treatment naïve patients or patients that had received thermo-/chemotherapy.
The MVA-hTyr infected tumour cells can be used as an antigen source to be presented by DCs for the induction of T cell-mediated immunity. Tyrosinase would serve as a marker to measure the extent of vaccine-induced immune activation within both the CD4 and CD8 arms of adaptive T cell immunity while unknown sarcoma-associated antigens, delivered by the sarcoma cells, would stimulate the clinically relevant immune responses.
Conclusion
MVA-hTyr infection can be used to reproducibly introduce tyrosinase into primary sarcoma cells. Tyrosinase expression was robust and resulted in antigen presentation and T cell activation, even after treatment regimens such as hyperthermia, which has been shown to reduce endogenous antigen expression in some situations Citation17. Subjecting cells to the clinical protocol of simultaneous ifosfamide and hyperthermia did not change their capacity to express and present the tyrosinase for T cell activation. The observation that primary material can be efficiently infected using the MVA system opens the possibility to develop autologous vaccines from primary tumour material within a short time frame. Endowing tumour cells where endogenous TAAs are not known with a surrogate marker, such as tyrosinase, allows monitoring the activation status of the immune system in patients enrolled in clinical trials using the tumour vaccine. The MVA-hTyr system may not be restricted to sarcomas, but should be applicable to tumours of other histologies.
Acknowledgements
The authors thank the editor and reviewers for their helpful suggestions to improve the manuscript.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med 1999, 190: 1669–1782
- Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res 2001, 61: 6451–6458
- Wolchok JD, Livingston PO. Vaccines for melanoma: Translating basic immunology into new therapies. Lancet Oncol 2001, 2: 205–211
- Marten A, Renoth S, Heinicke T, Albers P, Pauli A, Mey U, et al. Allogeneic dendritic cells fused with tumor cells: Preclinical results and outcome of a clinical phase I/II trial in patients with metastatic renal cell carcinoma. Hum Gene Ther 2003, 14: 483–494
- Finn OJ. Cancer vaccines: Between the idea and the reality. Nat Rev Immunol 2003, 3: 630–641
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005, 5: 296–306
- Slingluff CL, Jr, Engelhard VH, Ferrone S. Peptide and dendritic cell vaccines. Clin Cancer Res 2006, 12: 2342 s–2345 s
- Issels RD, Lindner LH, Verweij J, Wust P, Reichardt P, Schem BC, et al. Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: A randomised phase 3 multicentre study. Lancet Oncol 2010, 11: 561–570
- Marcove RC, Southam CM, Levin A, Mike V, Huvos A. A clinical trial of autogenous vaccine in osteogenic sarcoma in patients under the age of 25. Surg Forum 1971, 22: 434–435
- Moss B. Genetically engineered pox viruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci USA. 1996, 93: 11341–11348
- Sutter G, Staib C. Vaccinia vectors as candidate vaccines: The development of modified vaccinia virus Ankara for antigen delivery. Curr Drug Targets Infect Disord 2003, 3: 263–271
- Drexler I, Antunes E, Schmitz M, Wolfel T, Huber C, Erfle V, et al. Modified vaccinia virus Ankara for delivery of human tyrosinase as melanoma-associated antigen: Induction of tyrosinase- and melanoma-specific human leukocyte antigen A*0201-restricted cytotoxic T cells in vitro and in vivo. Cancer Res 1999, 59: 4955–4963
- Kawakami Y, Robbins PF, Wang X, Tupesis JP, Parkhurst MR, Kang X, et al. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, -A2, and -A3 alleles. J Immunol 1998, 161: 6985–6992
- Yee C, Gilbert MJ, Riddell SR, Brichard VG, Fefer A, Thompson JA, et al. Isolation of tyrosinase-specific CD8+ and CD4+ T cell clones from the peripheral blood of melanoma patients following in vitro stimulation with recombinant vaccinia virus. J Immunol 1996, 157: 4079–4086
- Di Nicola M, Carlo-Stella C, Mortarini R, Baldassari P, Guidetti A, Gallino GF, et al. Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD34(+)-derived dendritic cell vaccination: A phase I trial in metastatic melanoma. Clin Cancer Res 2004, 10: 5381–5390
- Meyer RG, Britten CM, Siepmann U, Petzold B, Sagban TA, Lehr HA, et al. A phase I vaccination study with tyrosinase in patients with stage II melanoma using recombinant modified vaccinia virus Ankara (MVA-hTyr). Cancer Immunol Immunother 2005, 54: 453–467
- Milani V, Frankenberger B, Heinz O, Brandl A, Ruhland S, Issels RD, et al. Melanoma-associated antigen tyrosinase but not Melan-A/MART-1 expression and presentation dissociate during the heat shock response. Int Immunol 2005, 17: 257–268
- Boulanger D, Baier R, Erfle V, Sutter G. Generation of recombinant fowlpox virus using the non-essential F11L orthologue as insertion site and a rapid transient selection strategy. J Virol Methods 2002, 106: 141–151
- Gasteiger G, Kastenmuller W, Ljapoci R, Sutter G, Drexler I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J Virol 2007, 81: 11925–11936
- Staib C, Drexler I, Ohlmann M, Wintersperger S, Erfle V, Sutter G. Transient host range selection for genetic engineering of modified vaccinia virus Ankara. Biotechniques 2000, 28: 1137–1142, 44–46, 48
- Hatzfeld-Charbonnier AS, Lasek A, Castera L, Gosset P, Velu T, Formstecher P, et al. Influence of heat stress on human monocyte-derived dendritic cell functions with immunotherapeutic potential for antitumor vaccines. J Leukoc Biol 2007, 81: 1179–1187
- Westermann AM, Jones EL, Schem BC, van der Steen-Banasik EM, Koper P, Mella O, et al. First results of triple-modality treatment combining radiotherapy, chemotherapy, and hyperthermia for the treatment of patients with stage IIB, III, and IVA cervical carcinoma. Cancer 2005, 104: 763–770
- Van Bree C, Beumer C, Rodermond HM, Haveman J, Bakker PJ. Effectiveness of 2′,2′difluorodeoxycytidine (gemcitabine) combined with hyperthermia in rat R-1 rhabdomyosarcoma in vitro and in vivo. Int J Hyperthermia 1999, 15: 549–556
- Miller RC, Richards M, Baird C, Martin S, Hall EJ. Interaction of hyperthermia and chemotherapy agents: Cell lethality and oncogenic potential. Int J Hyperthermia 1994, 10: 89–99
- Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins?. Int J Hyperthermia 2005, 21: 713–716
- Weiner GJ. Monoclonal antibody mechanisms of action in cancer. Immunol Res 2007, 39: 271–278
- Godelaine D, Carrasco J, Lucas S, Karanikas V, Schuler-Thurner B, Coulie PG, et al. Polyclonal CTL responses observed in melanoma patients vaccinated with dendritic cells pulsed with a MAGE-3.A1 peptide. J Immunol 2003, 171: 4893–4897
- Dillman R, Barth N, Selvan S, Beutel L, de Leon C, DePriest C, et al. Phase I/II trial of autologous tumor cell line-derived vaccines for recurrent or metastatic sarcomas. Cancer Biother Radiopharm 2004, 19: 581–588
- Dillman RO, Wiemann M, Nayak SK, DeLeon C, Hood K, DePriest C. Interferon-gamma or granulocyte-macrophage colony-stimulating factor administered as adjuvants with a vaccine of irradiated autologous tumor cells from short-term cell line cultures: A randomized phase 2 trial of the cancer biotherapy research group. J Immunother 2003, 26: 367–373
- Mahvi DM, Shi FS, Yang NS, Weber S, Hank J, Albertini M, et al. Immunization by particle-mediated transfer of the granulocyte-macrophage colony-stimulating factor gene into autologous tumor cells in melanoma or sarcoma patients: Report of a phase I/IB study. Hum Gene Ther 2002, 13: 1711–1721
- Meyers PA, Schwartz CL, Krailo M, Kleinerman ES, Betcher D, Bernstein ML, et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 2005, 23: 2004–2011
- Rosenberg SA. Progress in the development of immunotherapy for the treatment of patients with cancer. J Intern Med 2001, 250: 462–475
- Mayr A, Stickl H, Muller HK, Danner K, Singer H. [The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author's translation from German)]. Zentralbl Bakteriol (B). 1978 Dec, 167: 375–390
- Smith CL, Dunbar PR, Mirza F, Palmowski MJ, Sheperd D, Gilbert SC, et al. Recombinant modified vaccinia Ankara primes functionally activated CTL specific for melanoma tumor antigen epitope in melanoma patients with a high risk of disease recurrence. Int J Cancer 2005, 113: 159–166
- DiNicola M, Carlo-Stella C, Anichini A, Mortarini R, Guidetti A, Tragni G, et al. Clinical protocol. Immunization of patients with malignant melanoma with outologous CD34(+) cell-derived dendritic cells transduced ex vivo with a recombinant replication-deficient vaccinia vector encoding the human tyrosinase gene: Phase I trial. Hum Gene Ther 2003, 14: 1347–1360