Abstract
Purpose: Basal lamina is a major part of the microvascular wall and plays a critical role in the integrity of microvasculature. The aim of this study is to determine whether hyperthermia worsens the destruction of microvascular integrity in the ischaemic injured brain.
Materials and methods: Focal cerebral ischaemia was induced by embolising a pre-formed clot into the middle cerebral artery (MCA). Rats received either normothermic or hyperthermic treatment. Neurological score and infarct size were evaluated at 24 h after the MCA occlusion. Microvascular collagen type IV and laminin were measured with fluorescence microscopy. The activities of matrix metalloproteinases (MMP-2 and MMP-9) and plasminogen activators (tPA and uPA) were determined by zymography.
Results: Treatment with hyperthermia significantly increased infarct volume (p < 0.01), cortex swelling (p < 0.01), striatum swelling (p < 0.05) and neurologic score (p < 0.01) at 24 h after the MCA occlusion. Compared to the normothermic groups, hyperthermia significantly worsened the losses of microvascular basal lamina structure proteins, collagen type IV and laminin, at 6 h (p < 0.001) and 24 h (p < 0.01) after MCA occlusion. Hyperthermia increased the MMP-9 activity at 6 and 24 h after MCA occlusion compared with normothermia (p < 0.05), whereas increased the MMP-2 activity at 6 h only (p < 0.05). Hyperthermia also elevated uPA activity significantly at 6 and 24 h after MCA occlusion compared to normothermia (p < 0.05).
Conclusions: These results demonstrate that hyperthermia exacerbates the destruction of microvascular integrity possibly by increasing the activities of MMP-2, MMP-9 and uPA in the ischaemic cerebral tissues.
Introduction
Many common complications occurring in the initial phase of an ischaemic stroke adversely affect the final outcome. Hyperthermia is one such condition and is seen frequently in stroke patients and animal models of ischaemic brain injury. Hyperthermia negatively correlates with the outcome of stroke Citation1. Clinical studies have shown that mild hyperthermia in stroke patients can enlarge infarct size and worsen outcome of stroke Citation2–4. Animal studies demonstrated that hyperthermia markedly exacerbates neuronal injury in transient and permanent models of cerebral ischaemia Citation5–8.
Previously, we have studied the effects of hyperthermia on ischaemic brain injury in a focal embolic model of ischaemic stroke Citation9–10. We found that treatment with hyperthermia aggravated brain infarct size, worsened severity of neurological deficits and increased perfusion deficits, indicating hyperthermia exacerbates the ischaemic injury in this model. Furthermore, hyperthermia also antagonises the neuroprotective actions of thrombolytic therapy in this model Citation10. In addition, other groups have reported that hyperthermia even makes neuroprotective drugs deleterious Citation11.
Although it is well known that hyperthermia is detrimental for the injured brain, the mechanisms about how hyperthermia adversely affects injury processes are unknown. In focal ischaemia, the cerebral microvasculature rapidly displays multiple dynamic responses, including breakdown of the basal lamina, expression of several matrix-degrading protease families, e.g. matrix metalloproteinases (MMPs), plasminogen activators (PAs), which occurs simultaneously with neuronal injury Citation12. In the present study we have examined whether hyperthermia worsens ischaemic injury via damaging the microvascular basal lamina. We found that hyperthermia not only increased neurological deficits, enlarged infarct size, but also exacerbated the destruction of the microvasculature through the activation of digesting enzymes.
Materials and methods
Stroke model
Male Sprague-Dawley rats weighing 300 to 350 g were used for all experiments. Animal care and the general protocols for animal use were approved by the Animal Ethics Committee of the University of Alberta and Illinois State University. Focal cerebral ischaemia was induced by embolising a preformed clot into the MCA, as described previously Citation13. Briefly, rats were anaesthetised with halothane in a mixture of oxygen and nitrous oxide during surgery. A longitudinal incision of 1.5 cm in length was made in the midline of the ventral cervical skin. The right common carotid artery (CCA), right internal carotid artery (ICA) and right external carotid artery (ECA) were exposed. The distal portion of the ECA was ligated and cut. A modified PE-10 catheter, filled with bovine thrombin (Thrombostat™, Warner-Lambert, Scarborough, ON, Canada), was introduced into the lumen of the right ECA via a small puncture. An aliquot of 10 mL of blood were withdrawn into the catheter and retained for 15 min to allow formation of a clot. Once the clot formed, the catheter was advanced 17 mm in the ICA until its tip was 1–2 mm away from the origin of the MCA. The preformed clot in the catheter was then injected. In the sham group, the surgical procedure was the same as in the ischaemic rats except no clot was injected. Our unpublished data suggest that this sham procedure did not produce any neurological deficits and histological lesion in the brain. The possible explanation is that since the MCA was not blocked by the catheter placement, and blood from the contralateral arteries still can reach the MCA through anterior communication artery and ACA.
Physiological parameters were measured using an i-STAT analyser (Abaxis, Union City, CA). Briefly, blood from the CCA was collected into a syringe that was flushed with heparin lithium (500 IU/mL) (Sigma, St. Louis, MO). After removing air spaces, the blood was loaded into a cartridge and analysed.
Induction of hyperthermia
The body temperature was measured by placing a thermometer in rectum (Harvard Apparatus, Holliston, MA) as detailed previously Citation9–10, Citation14. The rectal temperature in the normothermic group was kept at 37.5°C, while the hyperthermic group at 39.5°C with a feed-back controlled YSI heating system (Yellow Springs Instrument, Yellow Springs, OH) Citation9–10, Citation14. The body temperature was raised to desired level in the anaesthetised rat immediately before the MCA occlusion, and maintained at this level for a period of 3 h after the occlusion. Thereafter, the rat was allowed to recover from the anaesthesia and housed in a room temperature.
Neurological deficits
Neurological deficits were recorded at 24 h after MCA occlusion using a modified Bederson's scoring system Citation13, Citation15, grade 0, no observable deficit; grade 1, forelimb flexion; grade 2, forelimb flexion plus decreased resistance to lateral push; grade 3, unidirectional circling; grade 4, unidirectional circling plus decreased level of consciousness.
Infarct volume and brain swelling
The methodology for quantification of infarct volume has been detailed previously Citation9, Citation13. Briefly, at 24 h after MCA occlusion the anaesthetised rats were sacrificed, and the brains were removed. For morphometric study, 2 mm-thick coronal sections were cut using a rat brain matrix. A total of eight coronal sections were collected and the sections were stained using a 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution. The infarct appears pale white on a background of red ‘normal’ brain. The stained brain sections were scanned with an HP ScanJet flatbed scanner (Palo Alto, CA). The infarct volume was calculated and expressed as a percentage of the total volume from the ipsilateral hemisphere. Cortex and striatum swelling was determined using the formula:The brain swelling was expressed as a percentage Citation16.
Immunohistochemistry
At 3, 6 and 24 h after MCA occlusion the rats were sacrificed and brains were removed. After sectioning, the brain sections were incubated with primary antibody (goat anti-type IV collagen (Southern Biotechnology, Birmingham, AL); rabbit anti-laminin (Sigma)), and then secondary antibody (Cy™3-conjugated donkey anti-goat (Jackson ImmuoResearch Laboratories, West Grove, PA); FITC-conjugated goat anti-rabbit (Sigma)), as described previously Citation17–18. The cyanine and FITC labelled microvessels were identified under fluorescent microscope and photographed. The changes of microvessel matrix constituents in cortex and striatum were automatically quantified by computerised video imaging microscopy (Improvision Image Analysis System, Coventry, UK). To identify microvascular targets, two criteria were defined. After optimal adjustment of contrast for each section, an intensity threshold and a minimum target length (10 µm) were set before automated quantification. Then, the numbers of target per field of view (vessel count) were determined by the software. Ratios were calculated between the ischaemic and non-ischaemic mirror areas Citation17. Four brain sections were collected serially from each rat starting at 0.7 mm anterior to the bregma with an interval of 1 mm. Data were acquired from an area of 1.16 mm2 at ×400. Five predetermined areas, three in the cortex and two in the striatum, were sampled from each brain section, as detailed previously Citation18. The procedure of this automated quantification was verified by manual counting the stained microvessels using a Fisher hand tally counter. The percentage changes of microvessels stained were similar between these two procedures.
Gel zymography
Gelatin gel zymography was used to determine MMP-9 and MMP-2 activities Citation19. In brief, the frontoparietal cortex and basal ganglia, areas of MCA supply, were dissected from the right MCA supply area. Brain tissue was homogenised in lysis buffer (50 mM Tris-HCl, pH 7.6; 150 mM NaCl; 5 mM CaCl2; 0.02% NaN3), and centrifuged. Following the supernatant collection, protein concentration was determined using Bio-Rad protein assay reagent. Equal amount of proteins, mixed with sample buffer (125 mM Tris-HCl pH 6.8, glycerol 10 mL, sodium dodecyl sulfate (SDS) 4 g), were loaded onto 8% acrylamide gel containing 0.1% gelatine as a substrate. Upon the completion of protein separation, the gels were incubated in 2.5% v/v Triton-X 100 for 1 h and then incubated in incubation buffer (50 mM Tris HCL;0.15 mM NaCl; 5 mM CaCl2) at 37°C until bands appeared. Gels were then stained with Coomassie blue R-250 for 1 h and de-stained accordingly. MMP activation appeared as transparent bands on blue background. For detection of plasminogen activators, casein and plasminogen were copolymerised into the gels instead of gelatine Citation20. Images of the gels were captured by scanning on an HP ScanJet flatbed scanner and analysed with National Institutes of Health image software.
Experimental protocol
Animals were randomly assigned to different experimental groups. In the study of neurologic deficit, infarct volume and brain swelling, there were two groups: normothermic (n = 8) and hyperthermic group (n = 8). The animals were sacrificed at 24 h after the MCA occlusion. In the study of collagen type IV and laminin immunoreactive microvessels, there were six groups: normothermic, animals were sacrificed at 3 h (n = 5), 6 h (n = 5) and 24 h (n = 5) after MCA occlusion; hyperthermic, animals were sacrificed at 3 h (n = 5), 6 h (n = 5) and 24 h (n = 5) after MCA occlusion. In the study of MMP activity, there were six groups: normothermic, animals were sacrificed at 3 h (n = 5), 6 h (n = 5) and 24 h (n = 5) after MCA occlusion; hyperthermic, animals were sacrificed at 3 h (n = 5), 6 h (n = 5) and 24 h (n = 5) after MCA occlusion. In the study of plasminogen activators, there were six groups: normothermic, animals were sacrificed at 3 h (n = 4), 6 h (n = 4) and 24 h (n = 4) after MCA occlusion; hyperthermic, animals were sacrificed at 3 h (n = 4), 6 h (n = 4) and 24 h (n = 4) after MCA occlusion. In the normothermia group, three rats died prematurely and a total of 53 rats were used to obtain 50 rats for assessment of neurologic deficit, infarct volume and brain swelling (n = 8), immunohistochemistry (n = 15), gel zymography (n = 27). In the hyperthermia group, 12 rats died prematurely and a total of 62 rats were used to obtain 50 rats for assessment of neurologic deficit, infarct volume and brain swelling (n = 8), immunohistochemistry (n = 15), gel zymography (n = 27).
Statistical analysis
Neurological deficit scores were expressed as medians and interquartile ranges, considering 25–75th percentiles. Other values were presented as mean ± standard deviation (SD). Neurological deficit was analysed with Mann-Whitney test. The infarct volume and brain swelling were analysed with student's t-test. The data of immunohistochemistry, densitometry and physiological parameters were analysed with one-way ANOVA followed by Tukey test. Differences were considered significant when p < 0.05. The experimenters were blinded to the treatment groups when performing analytic and outcome procedures.
Results
Physiological parameters
Physiological parameters were measured in the blood collected from CCA (). Statistical analyses showed that there were no significant differences in the parameters measured among the groups (p > 0.05).
Table I. Physiological parameters.a
Infarct size, brain swelling and neurological deficits
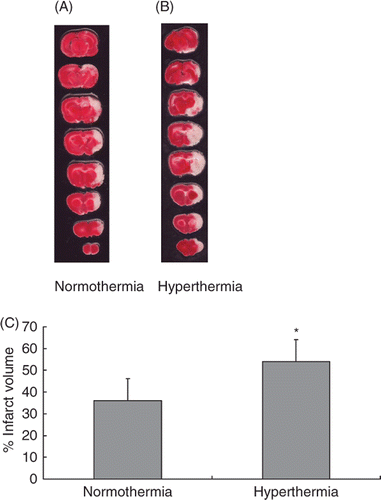
Representative samples of TTC-stained brain sections in the normothermic and hyperthermic groups are shown in Figure 1A and 1B. The mean infarct volume in the normothermic group was 36.2 ± 10.3% versus 54.0 ± 10.9% in the hyperthermia group at 24 h after the MCA occlusion. Hyperthermic treatment significantly increased the infarct volume as compared with the normothermic group (p < 0.01; ).
Figure 1. Hyperthermia enlarged the infarct in an embolic model of ischaemic brain injury. Representative TTC-stained brain sections from normothermic (A) and hyperthermic (B) rats, sacrificed at 24 h after the MCA occlusion. The infarct volume was measured in the TTC stained brain sections at 24 h after MCA occlusion (C). *p < 0.01 compared with the normothermic group.
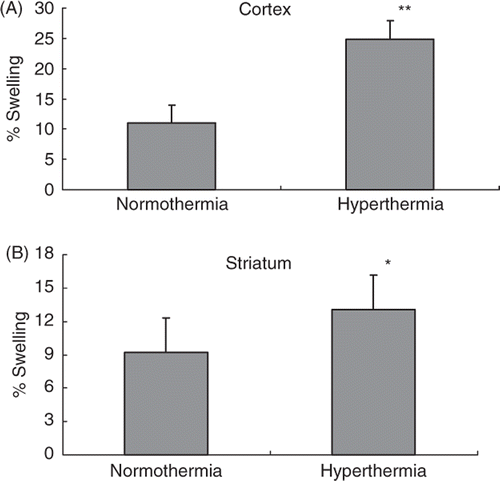
shows brain swelling in the normothermic and hyperthermic rats. Compared with normothermic rats (11 ± 3.1%), ischaemic swelling in the cortex increased significantly to 24.9 ± 3.7% in hyperthermic rats (p < 0.01; ). Ischaemic swelling in the striatum was also increased from 9.2 ± 2.8% in normothermic rats to 13.1 ± 3.6% in hyperthermic rats (p < 0.05; ).
Figure 2. Ischaemic swelling in the cortex (A) and striatum (B). The swelling was measured in the TTC-stained brain sections at 24 h after the MCA occlusion. *p < 0.05, **p < 0.01 compared with the normothermic group.
Neurological deficit score was 1 (0.75–2) in the normothermic group and 3 (2.75–3) in the hyperthermic group at 24 h after MCA occlusion. Hyperthermia treatment was found to increase the neurological scores significantly (p < 0.01).
Immunohistochemistry
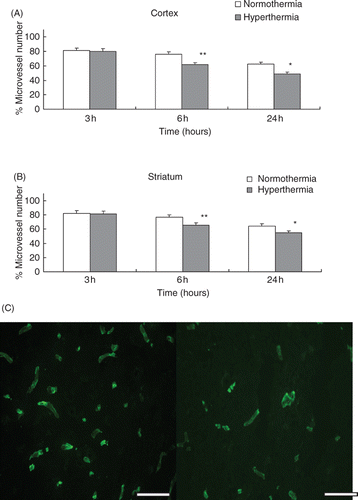
The decreases of the collagen type IV-containing microvascular structure are shown in the ischaemic cortex () and the striatum (). Hyperthermic treatment resulted in a significant reduction in the number of collagen type IV and laminin immunoreactive microvessels in the cortex and striatum by 6 h after the MCA occlusion compared with normothermic treatment (p < 0.001; , ; and ). Significant reduction of immunoreactive microvessels was also observed at 24 h after the MCA occlusion (p < 0.01; , ; and ). Figure 3C and 4C show representative microvessels stained by Cy™3-conjugated anti-type IV collagen and FITC-conjugated anti-laminin at 24 h after MCA occlusion in the cortex, respectively.
Figure 3. (A) The reduction of the number of collagen type IV positive stain microvessels in the cortex. The microvessel numbers are expressed as percentage changes between the ipsilateral and contralateral hemispheres to the occluded MCA. *p < 0.01, **p < 0.001 compared with the normothermic group. (B) Number of collagen type IV positive stain microvessels in the striatum. *p < 0.01, **p < 0.001 compared with the normothermic group. (C) Representative photomicrographs show microvessels stained by Cy™3-conjugated anti-type IV collagen at 24 h after MCA occlusion in the cortex. Left panel: normothermic and right panel: hyperthermic (scale bar = 200 µm).
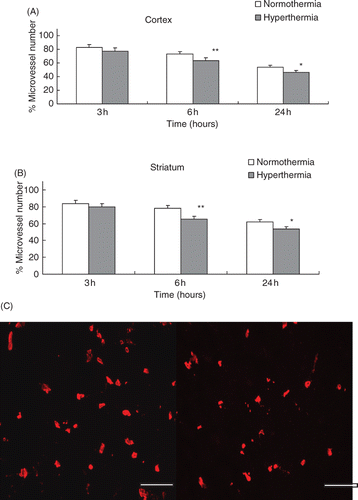
Figure 4. (A) The reduction of the number of laminin positive stain microvessels in the cortex. The microvessel numbers are expressed as percentage changes between the ipsilateral and contralateral hemispheres to the occluded MCA. *p < 0.01, **p < 0.001 compared with the normothermic group. (B) Number of laminin positive stain microvessels in the striatum. *p < 0.01, **p < 0.001, compared with the normothermic group. (C) Representative photomicrographs show microvessels stained by FITC-conjugated anti-laminin at 24 h after MCA occlusion in the cortex. Left panel: normothermic, right panel: hyperthermic (scale bar = 200 µm).
MMP activities
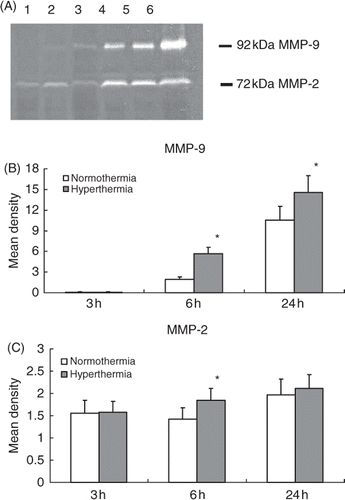
MMP-9 and MMP-2 activities were evaluated by gelatine gel zymography. Treatment with hyperthermia significantly elevated the MMP-9 activities at 6 and 24 h after the MCA occlusion compared to the normothermic group, respectively (p < 0.05; and ). Compared to the normothermic group treatment with hyperthermia significantly raised the MMP-2 activity only at 6 h after the MCA occlusion (p < 0.05; and ).
Figure 5. Gelatine zymogram shows the activities of MMP-2 (72 kDa) and MMP-9 (92 kDa) in the ischaemic injured brain collected from normothermic and hyperthermic rats. Lane 1: 3 h after normothermia; lane 2: 3 h after hyperthermia; lane 3: 6 h after normothermia; lane 4: 6 h after hyperthermia; lane 5: 24 h after normothermia; lane 6: 24 h after hyperthermia (A). Histograms show the densitometric analyses of MMP-2 (B) and MMP-9 (C) activities. *p < 0.05 compared with the normothermic group.
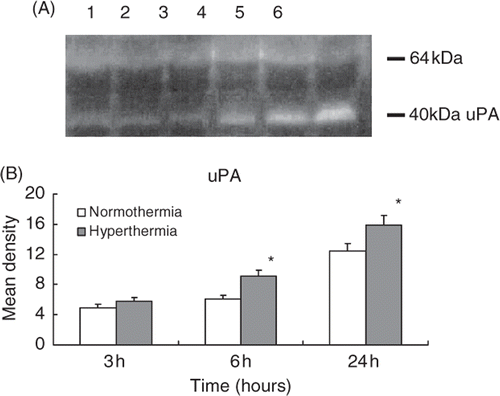
Plasminogen activators
depicts the casein/plasminogen zymogram for detection of urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA). The zymogram revealed that two bands were observed: the upper band represented the activity of tPA while the lower band was uPA (). When compared to the normothermic control, uPA activity markedly increased in the hyperthermia rats at 6 and 24 h after the MCA occlusion respectively (p < 0.05; ). No difference in tPA activity was observed between normothermic and hyperthermic groups at each time ().
Figure 6. Casein/plasminogen zymogram shows the activities of uPA and tPA in the ischaemic injured brain collected from normothermic and hyperthermic rats. Lane 1: 3 h after normothermia; lane 2: 3 h after hyperthermia; lane 3: 6 h after normothermia; lane 4: 6 h after hyperthermia; lane 5: 24 h after normothermia; lane 6: 24 h after hyperthermia. The upper band with a molecular weight of 64 kD represents tPA and the lower band with molecular weight of 40 kD represents uPA (A). Histogram shows the pooled data of uPA activities in the infarcted brain tissue (B). *p < 0.05 compared with the normothermic group.
Discussion
The present study demonstrated that hyperthermia treatment for 3 h following MCA occlusion significantly increased the infarct volume at 24 h after MCA occlusion. This treatment also increased brain swelling at 24 h after MCA occlusion, suggesting an aggravation of ischaemic oedema. In addition, this treatment worsened neurological score, which indicates that hyperthermia, in acute ischaemic stroke, is associated with a poor clinical outcome Citation21.
The microvascular basal lamina is a specialised part of the extracellular matrix that connects the endothelial cell compartment to the subjacent cell layers, end feet of astrocytes, and smooth muscle. In the microvascular basal lamina, collagen type IV chains form a covalently stabilised polygonal framework. Laminin self-assembles to form a second polymer network and it also serves to connect the basal lamina with the surrounding structures Citation22–23. These components of basal lamina are essential in the strength of microvascular walls and contribute to the integrity of the blood brain barrier (BBB) Citation17. Studies showed that the major basal lamina constituents, including laminin and collagen type IV, are degraded in very early stages in cerebral ischaemia Citation17, Citation24. This degradation is associated with microvascular haemorrhage Citation24 and oedema Citation25. In the present study, collagen type IV and laminin did not change significantly in sham operated animals (n = 3; data not shown). However, data from present study demonstrated for the first time that post-ischaemic hyperthermia significantly increases the loss of collagen type IV and laminin from the basal lamina of cerebral microvessels at 6 and 24 h after MCA occlusion. The results also showed the aggravation of brain swelling and neurologic deficit at 24 h in hyperthermia group. Previously, we demonstrated that hyperthermia worsens BBB damage in ischaemic brain injury Citation10, Citation14. Collectively, these findings suggest that microvascular basal lamina destruction may be a key factor in damage processes during ischaemic brain injury.
At least three pathways potentially lead to degradation of the microvascular basal lamina: (1) activation of plasminogen by endogenous PAs; (2) secretion of MMPs and (3) secretion of polymorphonuclear leukocyte specific granule enzymes Citation26. In the present study, analysis revealed that MMP-9 activity could not be detected whereas MMP-2 was constitutively expressed in the brain from the sham animals (n = 3; data not shown). Data also revealed that there was some discrepancy regarding the timing of the increased activities of MMP-2 and MMP-9. The time of MMP-2 activity increase was earlier than that of MMP-9, corroborating previous studies Citation27–28. MMPs are zinc-dependent endopeptidases that digest components of the basal lamina including collagen type IV, fibronectin and laminin Citation29–30. Collagen type IV is a substrate of MMP-2 and MMP-9, and laminin is a substrate of MMP-2 Citation31. Therefore, increased activities of these digesting enzymes likely contribute to basal lamina breakdown after focal cerebral ischaemia Citation31. In the present study, MMP activities are measured at 3, 6 and 24 h after ischaemic injury. These time points are chosen based on our previous findings that MMP-9 activities are significantly elevated in the model employed in a normothermic condition Citation18, Citation32. Importantly, the present study for the first time showed that hyperthermia further increased the activities of these digesting enzymes, which was accompanied by increased losses of collagen type IV and laminin. Thus, these data suggest that hyperthermia exacerbates microvascular lesion by regulating the digesting enzymes.
Evidence supports that the plasminogen/plasmin system contributes to the microvascular damage and BBB disruption Citation20, Citation31, Citation33. Our data showed that the activities of uPA and tPA were detected in sham operated animals (n = 3; data not shown). uPA activities were elevated significantly at 6 and 24 h within the ischaemic injured brain after MCA occlusion, which is similar to the previous studies Citation33–34. uPA mRNA assessed by RT-PCR in brain ischaemia did not increase, suggesting that increased uPA activity may be post-transcriptionally regulated Citation34. tPA did not change in the ischaemic injured brain which supports that the increase in endogenous plasminogen activators activity was mainly due to uPA. Plasminogen activators (tPA and uPA) convert plasminogen to plasmin which not only directly hydrolyses extracellular matrix proteins Citation31, but this plasminogen-plasmin system also contributes to the cleavage of collagen type IV through the activation of MMP-2 and MMP-9 Citation33, Citation35. The present study also demonstrated that uPA was significantly increased by hyperthermia, which may play a critical role in the brain after ischaemia Citation20, Citation33.
In the present study, an embolic model of ischaemic brain injury was used since in stroke patients 80–90% of cases are caused by thromboembolism Citation36–37, and the majority of ischaemic episodes occur as a result of MCA occlusion Citation38. Since Kudo et al. first introduced embolic injury model in rats, several groups have improved the technique for the injury induction Citation39. Our research has also further improved this model so that the lesion production is more reliable and consistent Citation13, Citation40. The microvessel patency in this model has also been characterised Citation40. Microvessels had reopened in the cortex 1 h after clot injection, but remained occluded in some portions of the striatum in most of the animals. By 3 h post-clot injection, microvessels in the cortex were patent in all animals, whereas in the striatum, microvessels were patent in 50% of the animals. At 24 h after clot injection, microvessels were patent in both the cortex and striatum of almost all animals. Further analyses revealed that the originally embolised clots dissolve over time and fragments formed from the clots obstruct more distal blood vessels Citation41.
In summary, the present study demonstrated that hyperthermia significantly increases infarct volume and brain swelling at 24 h after ischaemia in an embolic stroke model. The results also revealed that hyperthermia worsens the destruction of microvascular basal lamina structure proteins, type IV collagen and laminin at 6 and 24 h after MCA occlusion, possibly through the regulation of the digesting enzymes, MMP-2, MMP-9 and uPA. These findings thus contribute to our understanding the mechanisms of microvascular damage under hyperthermia condition after stroke.
Declaration of interest: This work was supported by Canadian Institutes of Health Research and National Institutes of health (R15NS072858). The authors alone are responsible for the content and writing of the paper.
References
- Zaremba J. Hyperthermia in ischemic stroke. Med Sci Monit 2004; 10: 148–153
- Reith J, Jorgensen HS, Pedersen PM, Nakayama H, Raaschou HO, Jeppesen LL, Olsen TS. Body temperature in acute stroke: Relation to stroke severity, infarct size, mortality and outcome. Lancet 1996; 347: 422–425
- Wang Y, Lim LL, Levi C, Heller RF, Fisher J. Influence of admission body temperature on stroke mortality. Stroke 2000; 31: 404–409
- Castillo J, Dávalos A, Marrugat J, Noya M. Timing for fever-related brain damage in acute ischemic stroke. Stroke 1998; 29: 2455–2460
- Reglodi D, Somogyvari-Vigh A, Maderdrut JL, Vigh S, Arimura A. Postischemic spontaneous hyperthermia and its effects in middle cerebral artery occlusion in the rat. Exp Neurol 2000; 163: 399–407
- Chen H, Chopp M, Welch KM. Effect of mild hyperthermia on the ischemic infarct volume after middle cerebral artery occlusion in the rat. Neurology 1991; 41: 1133–1135
- Baena RC, Busto R, Dietrich WD, Globus MY, Ginsberg MD. Hyperthermia delayed by 24 hours aggravates neuronal damage in rat hippocampus following global ischemia. Neurology 1997; 48: 768–773
- Minamisawa H, Smith ML, Siesjö BK. The effect of mild hyperthermia and hypothermia on brain damage following 5, 10, and 15 minutes of forebrain ischemia. Ann Neurol 1990; 28: 26–33
- Noor R, Wang CX, Shuaib A. Effects of hyperthermia on infarct volume in focal embolic model of cerebral ischemia in rats. Neurosci Lett 2003; 349: 130–132
- Noor R, Wang CX, Shuaib A. Hyperthermia masks the neuroprotective effects of tissue plasminogen activator. Stroke 2005; 36: 665–669
- Meden P, Overgaard K, Pedersen H, Boysen GBR. The influence of body temperature on infarct volume and thrombolytic therapy in a rat embolic stoke model. Brain Res 1994; 647: 131–138
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab 2003; 23: 879–894
- Wang CX, Yang T, Shuaib A. An improved version of embolic model of brain ischemic injury in the rat. J Neurosci Methods 2001; 109: 147–151
- Shabanzadeh AP, Shuaib A, Wang CX. Reduction of ischemic brain injury in rats with normothermic and hyperthermic conditions. J Neurosurg 2008; 109: 522–529
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986; 17: 472–476
- Shuaib A, Wang C, Yang T, Noor R. Effects of nonpeptide V(1) vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke 2002; 33: 3033–3037
- Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke 1995; 26: 2120–2126
- Wang CX, Ding X, Noor R, Pegg C, He C, Shuaib A. Rosiglitazone alone or in combination with tissue plasminogen activator improves ischemic brain injury in an embolic model in rats. J Cereb Blood Flow Metab 2009; 29: 1683–1694
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998; 29: 1020–1030
- Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab 1996; 16: 360–366
- Saini M, Saqqur M, Kamruzzaman A, Lees KR, Shuaib A. Effect of hyperthermia on prognosis after acute ischemic stroke. Stroke 2009; 40: 3051–3059
- Risau W, Esser S, Engelhardt B. Differentiation of blood–brain barrier endothelial cells. Pathol Biol 1998; 46: 171–175
- Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. FASEB J 1990; 4: 1577–1590
- Hamann GF, del Zoppo GJ, von Kummer R. Hemorrhagic transformation of cerebral infarction: Possible mechanisms. Thromb Haemost 1999; 82: S92–94
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: A possible role in blood–brain barrier dysfunction. J Cereb Blood Flow Metab 1999; 19: 1020–1028
- del Zoppo GJ, von Kummer R, Hamann GF. Ischaemic damage of brain microvessels: Inherent risks for thrombolytic treatment in stroke. J Neurol Neurosurg Psychiatry 1998; 65: 1–9
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab 1999; 19: 624–633
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 2007; 27: 697–709
- Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: Role for plasminogen activators and matrix metalloproteinases. J Neurosci Res 2002; 69: 1–9
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005; 50: 329–339
- Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke 2004; 35: 998–1004
- Noor R, Wang CX, Todd K, Elliott C, Wahr J, Shuaib A. Partial intra-aortic occlusion improves perfusion deficits and infarct size following focal cerebral ischemia. J Neuroimaging 2010; 20: 272–276
- Hosomi N, Lucero J, Heo JH, Koziol JA, Copeland BR, del Zoppo GJ. Rapid differential endogenous plasminogen activator expression after acute middle cerebral artery occlusion. Stroke 2001; 32: 1341–1348
- Ahn MY, Zhang ZG, Tsang W, Chopp M. Endogenous plasminogen activator expression after embolic focal cerebral ischemia in mice. Brain Res 1999; 837: 169–176
- Lee SR, Guo SZ, Scannevin RH, Magliaro BC, Rhodes KJ, Wang XY, Lo EH. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci Lett 2007; 417: 1–5
- Duncan GW, Pessin M, Mohr JP. Relevance of duration of transient ischaemic attacks in carotid territory. Br Med J 1978; 2: 1020
- Sloan MA. Thrombolysis and stroke. Past and future. Arch Neurol 1987; 44: 748–768
- Petty GW, Brust JC, Tatemichi TK, Barr ML. Embolic stroke after smoking ‘crack’ cocaine. Stroke 1990; 21: 1632–1635
- Kudo M, Aoyama A, Ichimori S, Fukunaga N. An animal model of cerebral infarction. Homologous blood clot emboli in rats. Stroke 1982; 13: 505–508
- Wang CX, Yang Y, Yang T, Shuaib A. A focal embolic model of cerebral ischemia in rats: Introduction and evaluation. Brain Res Brain Res Protoc 2001; 7: 115–120
- Wang CX, Yang T, Shuaib A. Clot fragments formed from original thrombus obstruct downstream arteries in the ischemic injured brain. Microcirculation 2006; 13(3)229–236