Abstract
Purpose: We sought to assess whether heat-induced autophagy, apoptosis and cell damage in H9c2 cells can be affected by pre-inducing HSP70 (heat shock protein 70).
Materials and methods: Cell viability was determined using 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide staining and a lactate dehydrogenase assay. Apoptosis was evidenced using both flow cytometry and counting caspase-3 positive cells, whereas autophagy was evidenced by the increased LC3-II expression and lysosomal activity.
Results: The viability of H9c2 cells was temperature-dependently (40–44 °C) and time-dependently (90–180 min) significantly (p < 0.05) reduced by severe heat, which caused cell damage, apoptosis and autophagy. Heat-induced cell injury could be attenuated by pretreatment with 3-methylademine (an autophagy inhibitor) or Z-DEVD-FMK (a caspase-3 inhibitor). Neither apoptosis nor autophagy over the levels found in normothermic controls was induced in heat-shock preconditioned controls (no subsequent heat injury). The beneficial effects of mild heat preconditioning (preventing heat-induced cell damage, apoptosis and autophagy) were significantly attenuated by inhibiting HSP70 overexpression with triptolide (Tripterygium wilfordii) pretreatment.
Conclusion: We conclude that pre-inducing HSP70 attenuates heat-stimulated cell autophagy, apoptosis and damage in the heart. However, this requires in vivo confirmation.
Introduction
Heat-stress-induced cardiovascular dysfunction is accompanied by severe cardiomyocyte injury [Citation1]. Patients with heatstroke display a predominant S wave and a depressed ST segment [Citation2]. Electron microscopic examination also shows that subcellular changes in the myocardium in response to heat stress are characterised by mitochondrial hypertrophy, intracellular oedema, destruction of the myofibrils, dilatation of the intercalated discs, and some abnormalities in the capillaries [Citation3]. Heat stress time- and dose-dependently induces apoptosis and necrosis of rat cardiomyocytes [Citation4]. The destruction of mitochondrial structure and function not only impairs the physiological function of cardiomyocytes under heat stress, but it may also lead to severe cellular injury and even cell death [Citation4].
Heat shock protein (HSP)-70 is one of the most extensively studied groups inducible by an increase in temperature as well as other environmental stresses [Citation5]. The HSP40 and HSP70 families work both independently and together to help prevent the misfolding of nascent polypeptide chains on cytoplasmic and endoplasmic reticulum-associated ribosomes [Citation6]. A knockout (KO) mouse brain has been recently developed that lacks functional HSPa 1a and HSPa 1b genes [Citation7,Citation8], both of which encode the HSP70 proteins, and differ by only one amino acid. Deleting the inducible HSP70 protein genes in mice damages the calcium handling and contractility of cardiac myocytes [Citation9]. Therefore, before heat shock and chemical stress, both of which induce HSP70 expression, it is necessary to increase the resistance of the heart to subsequent ischaemia and non-ischaemic injury [Citation10]. The acquisition and decay of the enhanced post-ischaemic ventricular recovery and the hyperthermic induction of the heat-shock response indicated by the accumulation of HSP70 and the increase in catalase activity has been strongly associated in rats [Citation11]. HSP70 expression in transgenic mice also protects against post-ischaemic myocardial dysfunction [Citation12]. Infarct studies [Citation13] confirm that HSP70 is protective after ischaemic preconditioning, and the transgenic expression of porcine HSP70 protected mice from hypothalamic ischaemic and oxidative damage in a mouse model of heat stroke [Citation14].
The present study had three purposes. First, to assess whether prior heat shock protects against heat-induced autophagy, apoptosis, or cell damage in H9c2 cells; second, to assess whether the pre-induction of HSP72 in H9c2 is essential for the beneficial effects of prior heat shock in preventing cell autophagy, apoptosis and damage; third, to assess whether heat-induced cell injury can be affected by preconditioning with 3-methylademine (3-MA) (an autophagy antagonist) [Citation15], z-DEVD-FMK (a caspase inhibitor) [Citation16], or triptolide (a human heat-shock response inhibitor) [Citation17] in H9c2 cells.
Materials and methods
Cell culture
The embryonic rat heart myoblast cell line H9c2 was purchased from the American Tissue Type Collection (catalogue no. CRL-1446, Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, and 4 mM L-glutamine supplemented with 10% heat-inactivated foetal bovine serum, 50 U/mL of penicillin and 50 µg/mL of streptomycin. The cells were grown on tissue culture dishes and subcultured to about 90% confluence before experiments.
Simulated hyperthermic injury model
The H9c2 cells were put in a circulatory 43 °C water bath for 120 min to induce hyperthermic injury, while the normothermic controls were kept in a 37 °C incubator for the equivalent time.
Heat shock preconditioning
Eight hours before the start of hyperthermic injury, the H9c2 cells were kept in a circulating 42 °C water bath for 30 min to induce HSP-70 expression [Citation18]. Triptolide (Calbiochem, San Diego, CA, USA) was dissolved in dimethyl sulphate at a concentration of 10 mM and added to the cells at the indicated concentrations [Citation18].
Experimental groups
Four groups of H9c2 cells were used in the present study (): (1) normothermic control (NC) group: the cells were kept in a 37 °C incubator for the entire experiment; (2) heat shock preconditioning (HSPre) group: the cells were kept in a circulating 42 °C water for 30 min and then allowed to recover in a 37 °C incubator for 8 h; (3) hyperthermic injury (HI) group: the cells were kept in a circulating 43 °C water bath for 120 min; (4) HSPre + HI group: 8 h before the start of HI, the cells were kept in a circulating 42 °C water bath for 30 min and then allowed to recover in a 37 °C incubator for 8 h; (5) (HSPre + triptolide + HI) group: 1 h before the start of HSPre + HI, the cells were exposed to triptolide for 1 h; (6) (3-MA + HI) group: 1 h before the start of HI, the cells were exposed to 3-MA (10 mM) for 1 h; and (7) (z-DEVD-FMK + HI) group: 3 h before the start of HI, the cells were exposed to z-DEVD-FMK (60 µM) for 3 h.
Figure 1. Experimental procedures. NC, normothermic controls; HI, hyperthermic injury; HSPre, heat shock preconditioning; HSPre + triptolide, heat shock preconditioning plus triptolide.
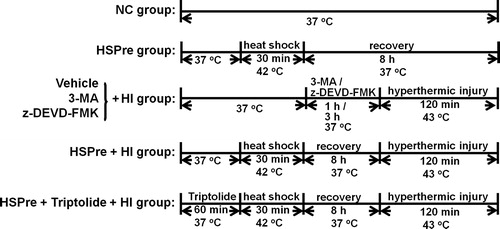
Cell viability
Cell viability was determined using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay (Invitrogen, Carlsbad, CA) in which the yellow tetrazolium salt is catalysed by nicotinamide adenine dinucleotide (NAD)-dependent dehydrogenase (in active mitochondria) to form purple formazan salt crystals [Citation18,Citation19]. Cells were seeded into a 96-well plate. After severe hyperthermic injury, the MTT reagent (5 mg/mL MTT in PBS) was added to each well and then incubated for 4 h. After the assay reagent had been removed, dimethyl sulphoxide (DMSO) was added to each well to solubilise the purple crystals. Plates were then immediately analysed using an ELISA plate reader (BioTek Instruments, Winooski, VT, USA) at 580 nm. Cell viability was defined relative to the ratio of normothermic control cells as follows: cell viability (%) = (value of optical density of treated sample)/(value of optical density of control × 100).
Western blotting
Cells were harvested for protein analysis using standard western blotting techniques. Briefly, the cells were washed twice with cold PBS, lysed with cold RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). Protein concentrations were assessed in duplicate in each sample using Bradford analysis with beef serum albumin (BSA) as a standard. Samples were thawed, and each lane was loaded with an equal amount of protein and subjected to 12% (or 15%) SDS-polyacrylamide gel electrophoresis (PAGE) with molecular weight standards, transferred from gels onto polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA), and then blocked in 5% non-fat milk for 1 h at room temperature (RT). The membranes were then probed with mouse monoclonal antibody to HSP-70 (1:200 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit polyclonal antibody to LC3B (1:5000 dilution) (Abcam, Cambridge, UK) for 1 h at RT or overnight at 4 °C, and then incubated with horseradish-peroxidase-conjugated secondary antibody for 1 h at RT. Bound antibodies were detected using a chemiluminescence reagent (Amersham Biosciences, Uppsala, Sweden), exposed to an X-ray film, and quantified by scanning the film using image software (ImageMaster TM2D Platinum, Amersham).
LDH release
Lactate dehydrogenase (LDH), which exists in all cell types, is a stable enzyme and rapidly released into the cell culture medium when plasma membranes are damaged. The LDH cytotoxicity assay (Lactate Assay Kit II, Biovision, Milpitas, CA) uses the advanced WST (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) substrate mix as a fast and sensitive detector for LDH release from damaged cells. WST substrate mix provides an indirect measurement of cellular reductive capacity correlating with reduced nicotinamide adenine dinucleotide (NADH) levels, and causes the formation of a highly light-absorbent formazan product [Citation20]. After hyperthermic injury, conditioned medium containing an LDH reagent with WST substrate was processed and immediately analysed using an ELISA plate reader at 450 nm for 30 min. This reaction was processed by the LDH, which oxidises lactate to generate NADH, which then reacts with WST and turns it yellow. The kinetic speed of the generated colour correlates directly with the quantity of damaged cells.
Flow cytometry
Apoptosis was analysed using a kit (Annexin V-FITC kit, Beckman Coulter, Fullerton, CA) that measures the binding of a fluorescein-labelled annexin V to phosphatidylserine residues translocated on the cell surface soon after the induction of apoptosis and before nuclear breakdown occurs. After hyperthermic injury, cells were collected by trypsinisation and were suspended at a concentration of 1 × 106/mL in PBS. All subsequent steps were done on ice. The cells were resuspended in binding buffer and incubated with annexin V-FITC for 15 min in the dark. The cells were then re-pelleted and re-suspended in binding buffer, propidium iodide was added, a flow cytometer (Beckman Coulter, Miami, FL) was used for analysis: (1) viable cells do not stain with either reagent, (2) necrotic cells stain with both reagents, and (3) apoptotic cells stain with annexin V-FITC only.
Caspase-3 activity assay
Measuring caspase-3 activity is based on the hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-p-NA) by the active form of caspase-3, which causes the release of the p-nitroaniline (pNA) moiety. Equal concentrations (100 µg) of sample protein were added to a 96-well plate and reacted with 10 µL of 20-mM peptide substrate for 1.5 h at 37 °C. The level of pNA release from the peptide substrate was detected using an ELISA plate reader at 405 nm and was calculated using the p-NA standard curve to determine the level of caspase-3 activity.
Immunocytochemistry
Cells were cultured in chambered cover glasses (Lab-Tek®II no. 154461; Nalge Nunc International, Rochester, NY) and fixed for immunocytochemistry analyses by removing culture medium, washing in PBS and adding 4% paraformaldehyde in PBS (pH 7.4) for 20 min at RT. Cells were permeabilised 0.1% Triton-X in PBS for 15 min and then blocked with 10% goat serum in PBS for 1 h at PT. Primary antibodies such as rabbit polyclonal cathepsin-B, -D, -L or -G antibody (all 1:50 dilution) (Santa Cruz Biotechnology) are added into blocking solution and incubated overnight at 4 °C. Cells were incubated with FITC-labelled secondary antibodies (1:1000/blocking solution) (Invitrogen) for 1 h at RT, and then mounted with coverslips onto microscope slides with anti-fade Gelmount (Biomedia, Foster City, CA). Fluorescence images are analysed using an Olympus BX41 fluorescence microscope.
Lysosomal activity
Cells were cultured in chambered cover glasses. After they had been subjected to hyperthermic injury, the cells were incubated with 50 nM probes (LysoTracker® Green DND-26, Invitrogen) at 37 °C for 15 min. The probes freely permeate cell membranes and typically concentrate in spherical organelles. Analysis of lysosomal morphology was photographed live using the fluorescence microscope.
Statistical analysis
All assays were independently performed three times. The data are means ± standard deviation (SD). Comparisons between groups were made using analysis of variance (ANOVA) F tests. Bonferroni adjusted t-tests were used for multiple-group comparisons, and an unpaired t-test was used for single comparisons. Significance was set at p < 0.05 (two-tailed).
Results
The cell viability of H9c2 cells was temperature- (40–44 °C) and time- (90–180 min) dependently significantly reduced (). In the following experiments a hyperthermia regimen (43 °C for 120 min) was used to induce cell death.
Figure 2. Cell viability of different degrees of temperature and different treatment time in the hyperthermic injury (HI). Cells were immersed in a circulating water bath at indicated periods of temperature (A) and time (B). After hyperthermic injury, cells were immediately incubated in MTT reagent for 4 h at 37 °C, and then analysed by optic density (OD) 450 nm. Data were presented as the means ± SD of three independent experiments and were compared among groups with ANOVA (F test) following by Bonferroni adjusted t-tests (p < 0.05).
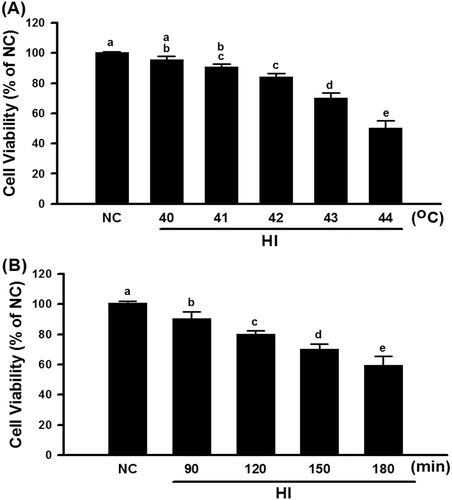
H9c2 cells were exposed to 42 °C for 30 min and then allowed to recover at 37 °C for 1, 4, 8, 12, 24, 48, or 72 h, after which HSP70 expression was determined. HSP70 expression was significantly higher in the group of H9c2 cells that recovered for 8 h (). Therefore, in the following experiments, the H9c2 cells that recovered 8 h after the termination of 42 °C exposure preconditioning were chosen for heat shock preconditioning. In addition, the heat shock preconditioning-induced HSP70 overexpression in H9c2 cells was significantly attenuated by triptolide preconditioning ().
Figure 3. HSPre induced the expression of HSP70 and had a spike at recovery 8 h. Cells suffered from a sub-lethal heat shock at 42 °C for 30 min in a circulating water bath followed by recovery of the indicated period time to express the HSP70 at 37 °C. Data were presented as the means ± SD of three independent experiments and were compared among groups with ANOVA (F test) followed by Bonferroni’s adjusted t-test (p < 0.05).
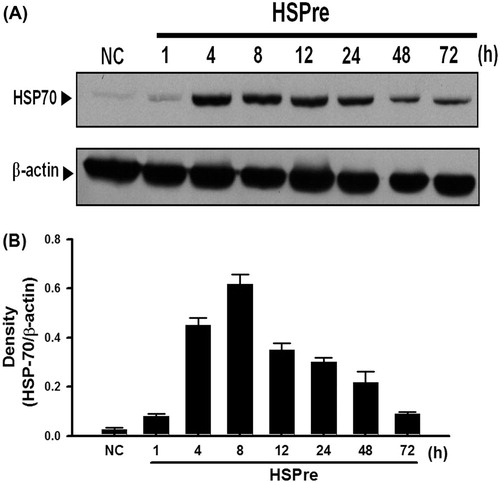
Figure 4. The inhibitor triptolide blocked the effect of HSPre-induced HSP70 expression. Immunoblotting analysed the HSP70 after HSPre with/without triptolide pretreatment. Data were presented as the means ± SD of three independent experiments. †p < 0.05 in comparison with NC group. ‡p < 0.05 in comparison with the HSPre group. Please see the legends of for group abbreviations.
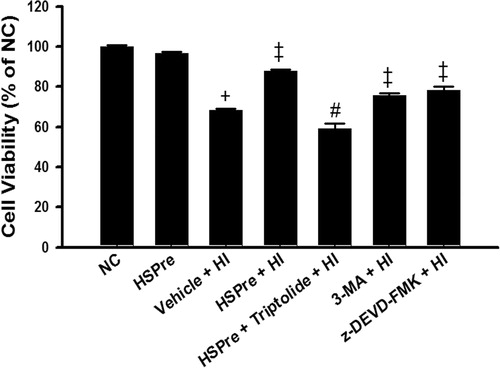
Cell viability was significantly (p < 0.05) lower in H9c2 cells exposed to 43 °C hyperthermia for 120 min (vehicle + HI) than in normothermic (37 °C) controls (). Hyperthermia-induced cell death was significantly attenuated by heat shock preconditioning (HSPre) (42 °C for 30 min, 8 h before 43 °C heat exposure), 3-MA preconditioning, or z-DEVD-FMK preconditioning (). HSPre-treated cells without HI showed no significant reduction in cell viability. Triptolide preconditioning (60 min before HSPre) significantly attenuated the benefits of HSPre.
Figure 5. The protective effect of HSPre, 3-MA preconditioning or z-DEVD-FMK preconditioning significantly attenuated HI-induced cell death. Cells were processed by hyperthermic injury in a 43 °C circulating water bath for 120 min; however, its damage was significantly attenuated by HSPre. Data were presented as the means ± SD of three independent experiments. †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI. #p < 0.05 in comparison with the HSPre + HI group. Please see the legends of for group abbreviations.
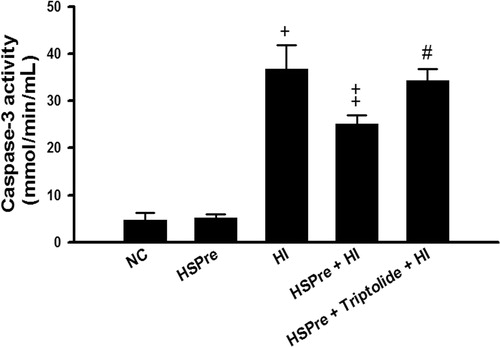
There were no significant differences between the normothermic controls and HSPre-treated cells in increased LDH activity in the medium (an indicator of cellular damage) (), apoptosis (), or caspase-3 activity (), but the differences between the HI-treated cells and the control and HSPre cells were all significant (p < 0.05–0.001). Again, triptolide preconditioning (60 min before HSPre) significantly attenuated the benefits of HSPre.
Figure 6. The LDH release that increased in HI was attenuated by HSPre. Data were presented as the means ± SD of three independent experiments. †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI. #p < 0.05 in comparison with HSPre + HI. Please see the legends of for group abbreviations.
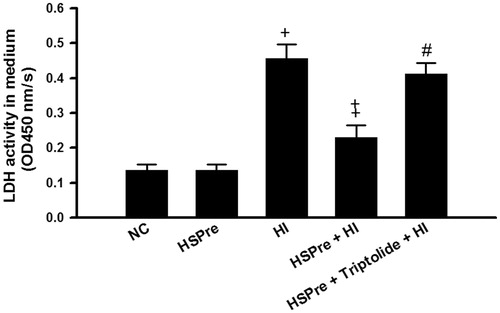
Figure 7. The level of apoptosis that increased in HI was attenuated by HSPre. Apoptosis was analysed according to the commercially available protocol (Annexin V-FITC kit; Beckman Coulter). Cells were analysed by flow cytometry using a Beckman Flow Cytometer, with results (1) viable cells that did not stain with either reagent (B3 area), (2) necrotic cells that stained with both reagent (B2 area), and (3) apoptotic cells that stained with annexin V-FITC only (B4 area). Data were presented as the means ± SD of three independent experiments. †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI group. #p < 0.05 in comparison with HSPre + HI group. Please see the legends of for group abbreviations.
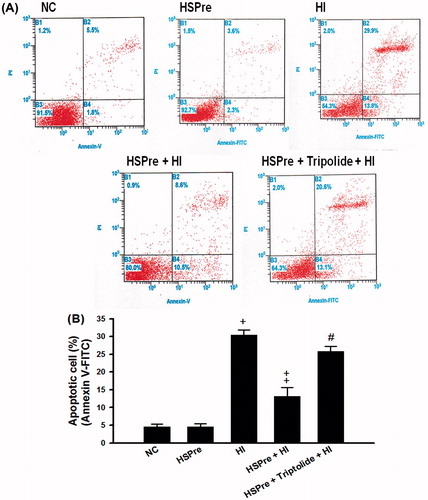
Figure 8. The level of caspase-3 activity that increased in HI was attenuated by HSPre. Caspase-3 activity was analysed according to the commercially available protocol (Sigma). Equal concentration of extract protein was reacted with caspase-3 substrate (Ac-DEVD-p-NA), and then the cleaved p-NA was detected at OD 405 nm and was calculated from the p-NA standard curve to gain intensity of caspase-3 activity. †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI. #p < 0.05 in comparison with HSPre + HI. Data were presented as the means ± SD of three independent experiments. Please see the legends of for group abbreviations.
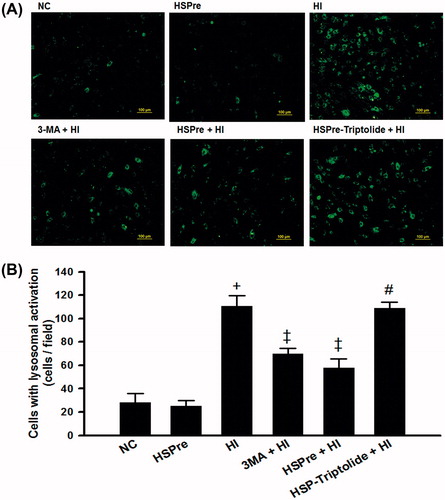
The levels of apoptosis () and autophagy (e.g. increased light chain 3 of microtubule-associated protein 2 (LC3-II) expression and increased lysosomal activity) () induced in controls with HSPre-only treatment (no subsequent heat injury) were not significantly different from those in normothermic controls. However, HI-treated and triptolide-preconditioned cells had significantly higher levels.
Figure 9. Autophagosome marker LC3-II that increased in HI was attenuated by HSPre. In cytosolic soluble form of LC3-I converted into the autophagic vesicle-associated form of LC-3 II that was used as a marker of autophagosome formation. Immunoblotting was used to analyse the LC-II expression and LC-I conversion into LC-II ratio (LC-II/total LC-3 (LC3-I + LC3-II)). Data were independently performed three times and presented at quality (A), quantity (B) and LC3-II/total LC3 ratio (C). †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI. #p < 0.05 in comparison with HSPre + HI group. Please see the legends of for group abbreviation.
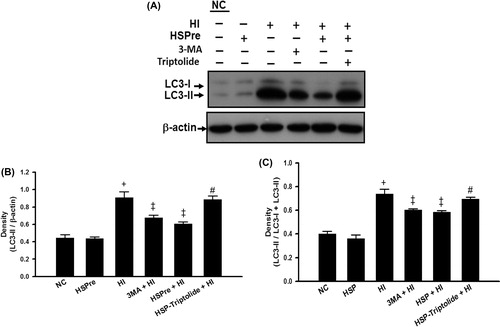
Figure 10. The lysosomal activity that increased in HI was attenuated by HSPre. Cells were incubated with LysoTracker® Green DND-26 (Invitrogen) probes that were fluorescent acidotropic probes for labelling and tracking acidic organelles (lysosome) in live cells. The lysosomal activity was photographed live on an Olympus BX41 fluorescent microscope. images were taken at 100 × magnification and scale bar represented 100 µm (A). Cells with lysosomal activation per field were counted for at least three fields/sample, and the average number of cells per field is shown in (B). Data were independently performed three times. †p < 0.05 in comparison with NC. ‡p < 0.05 in comparison with HI. #p < 0.05 in comparison with HSPre + HI. Please see the legends of for group abbreviation.
Discussion
We showed that exposure to heat time- (90–180 min) and temperature- (40–44 °C) dependently and significantly reduced the viability of and caused apoptosis, autophagy and necrosis in H9c2 myocytes. Although HSPre-treated cells without HI did not cause low-level induction of apoptosis, autophagy and necrosis, HSPre caused HSP70 overexpression and significantly attenuated heat-induced apoptosis, autophagy and necrosis. The beneficial effects of HSPre in preventing apoptosis, autophagy and necrosis were significantly attenuated by triptolide preconditioning, which inhibited HSP70 overexpression. These observations prompted us to hypothesise that pre-induction of HSP70 attenuates H9c2 myocyte death in the heat by inhibiting apoptosis, autophagy and necrosis. Our data are in part consistent with our previous results [Citation21]. In our previous study, effects of heatstroke on mean arterial pressure (MAP) were assayed in rats 0, 16, or 48 h after heat shock (42 °C for 15 min) or chemical stress (5 mg/kg sodium arsenite intraperitoneally) preconditioning. Heatstroke was induced by exposing the rats to a high ambient temperature (43 °C for 70 min); the moment at which MAP decreased from their peak values was taken as the time of heatstroke onset. Prior heat shock or chemical stress was significantly protective against heatstroke-induced hyperthermia, hypotension and bradycardia, and it correlated with HSP72 expression at 16 h. However, at 48 h, when HSP72 expression returned to basal values, the above parameters were indistinguishable between the two groups (0 h versus 48 h). Our previous and present results lead to the hypothesis that the heart can be preconditioned by thermal or chemical stress, that this preconditioning will induce HSP70 expression, and that HSP70 expression will protect cardiomyocytes from heatstroke injury.
This study has some limitations. The most important is that this was an in vitro study that used an isolated immortal cell line. It is not ethically possible to do an in vivo study on humans; therefore, it is possible that our findings will not be mirrored in the intact heart.
There is also a controversial issue: Is autophagy detrimental or protective? Our present data provide novel evidence of a positive association between heatstroke up-regulated autophagy and the decreased resistance of H9c2 myocytes to severe heat stress injury. Additionally, heat-induced cell injury can be attenuated by 3-MA preconditioning. Our results suggest that autophagy is detrimental during heatstroke, which is inconsistent with the findings of a recent study [Citation22]. These investigators showed that rats with heatstroke displayed neuronal shrinkage and pyknosis of the nucleus in the cerebral cortex and increased autophagy in the cerebral cortex. Pretreating rats with 3-MA, an autophagy inhibitor, aggravated neurodegeneration. They promote heatstroke-induced autophagy as a protection mechanism against neurodegeneration. Our present results are in part supported by the findings of another study [Citation23] on heat-induced autophagy in several cell lines without nutrient depletion. Heat treatment increased the light chain 3 of microtubule-associated protein 1 (LC3-I) and LC3-II autophagy markers at the protein levels. Interestingly, expression of a constitutively active heat shock factor 1 (HSF1) mutant, the key transcription factor that mediates the increased expression of HSP genes in cells exposed to heat, suppressed the basal LC3-II protein level and heat-induced increase of LC3-II. These findings provide evidence that heat is a potent inducer of autophagy in mammalian cells and imply the negative role of active HSF 1 in this process.
A similar controversial issue exists about autophagy’s role in determining the fate of ischaemic-reperfused cardiomyocytes: Is autophagy detrimental or protective? [Citation24]. Evidence obtained in isolated cardiomyocytes and rodent models has revealed that acute, pre-ischaemic induction of autophagy can confer a cardioprotective phenotype [Citation25,Citation26]. Also, data obtained in a clinically relevant in vivo swine model of acute myocardial infarction or heart attack [Citation24] provide novel evidence of a positive association between pharmacological up-regulation of autophagy and increased resistance to myocardial ischaemia-reperfusion injury. On the other hand, autophagy reportedly plays a detrimental role in the heart. For example, the failing myocardium caused by ischaemic heart disease displayed autophagy [Citation27–29]. The myocardium from patients with ischaemic cardiomyopathy also showed autophagy [Citation30]. In human hibernating myocardium, degenerated cardiomyocytes with autophagy were also observed [Citation28]. In animal models, dead and dying cardiomyocytes showing characteristics of autophagy were observed [Citation31,Citation32].
Conclusions
In summary, our data showed that heat injury in H9c2 involved a complex biochemical cascade that was characterised by autophagy, apoptosis and necrosis. Hyperthermia treatment of H9c2 cells caused autophagy, apoptosis and necrosis, all of which were significantly attenuated by heat shock preconditioning-induced HSP70 expression. The beneficial effects of HSP70 preconditioning in reducing the damage to H9c2 cells was reversed by triptolide preconditioning. These findings suggest that, in the heart, heat stimulates autophagy, apoptosis and necrosis, which can be attenuated by HSP70 preconditioning.
Declaration of interest
This work was supported by grants NSC100-2314-B-218-001, NSC99-2314-B-38, 4-004-MY3 and NSC99-2314-B-218-006-MY2 from the Taiwan National Science Council and a grant DOH99-TD-B-111-003 from the Taiwan Department of Health. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
We thank Professor Ching-Ping Chang (Department of Biotechnology, Southern Taiwan University of Science and Technology) for advice and support during the study.
References
- Chen SH, Niu KC, Lin MT. Cerebrovascular dysfunction is an attractive target for therapy in heat stroke. Clin Expt Pharmacol Physiol 2006;33:663–72
- Akhtar MJ, al-Nozha M, al-Harthi S, Nouh MS. Electrocardiographic abnormalities in patients with heat stroke. Chest 1993;104:411–14
- Lin MT, Chai CY, Sun SC, Kau SL. Myocardial lesions produced by external heat or cold exposure in rats. Chin J Physiol 1977;22:115–25
- Qian L, Song X, Ren H, Gong J, Cheng S. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones 2004;9:281–93
- Nollen EA, Morimoto RI. Chaperoning signaling pathways: Molecular chaperones as stress-sensing ‘heat shock' proteins. J Cell Sci 2002;115:2809–16
- Borges JC, Ramos CH. Protein folding assisted by chaperones. Protein Pept Lett 2005;12:257–61
- Hampton CR, Shimamoto A, Rothnie CL, Griscavage-Ennis J, Chong A, Dix DJ, et al. HSP70.1 and -70.3 are required for late-phase protection induced by ischemic preconditioning of mouse hearts. Am J Physiol Heart Circ Physiol 2003;285:H866–74
- Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, et al. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol 2004;24:899–11
- Kim YK, Suarez J, Hu Y, McDonough PM, Boer C, Dix DJ, et al. Deletion of the inducible 70-kDa heat shock protein genes in mice impairs cardiac contractile function and calcium handling associated with hypertrophy. Circulation 2006;113:2589–97
- Yellon DM, Marber MS. Hsp70 in myocardial ischemia. Experientia 1994;50:1075–83
- Karmazyn M, Mailer K, Currie RW. Acquisition and decay of heat-shock-enhanced postischemic ventricular recovery. Am J Physiol 1990;259:H424–31
- Trost SU, Omens JH, Karlon WJ, Meyer M, Mestril R, Covell JW, et al. Protection against myocardial dysfunction after a brief ischemic period in transgenic mice expressing inducible heat shock protein 70. J Clin Invest 1998;101:855–62
- Tranter M, Ren X, Forde T, Wilhide ME, Chen J, Sartor MA, et al. NF-kappaB driven cardioprotective gene programs: Hsp70.3 and cardioprotection after late ischemic preconditioning. J Mol Cell Cardiol 2010;49:664–72
- Chen ZC, Wu WS, Lin MT, Hsu CC. Protective effect of transgenic expression of porcine heat shock protein 70 on hypothalamic ischemic and oxidative damage in a mouse model of heatstroke. BMC Neurosci 2009;10:111–9
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007;100:914–22
- Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-FMK attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab 2004;24:1119–32
- Westerheide SD, Kawahara TLA, Orton K, Morimoto RI. Triptolide, as an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem 2006;281:9616–22
- Widestrand J, Lundh T, Pettersson H, Lindberg JE. Cytotoxity of four trichothecenes evaluated by three colorimetric bioassays. Mycopathologia 1999;147:149–55
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol Annu Rev 2005;11:127–52
- Yang YL, Lin MT. Heat shock protein expression protects against cerebral ischemia and monoamine overload in rat heatstroke. Am J Physiol 1999;276:H1961–7
- Liu TT, Hu CH, Tsai CD, Li CW, Lin YF, Wang JY. Heat stroke induces autophagy as a protection mechanism against neurodegeneration in the brain. Shock 2010;34:643–8
- Zhao Y, Gong S, Shunmei E, Zou J. Induction of macroautophagy by heat. Mol Biol Rep 2009;36:2323–27
- Przyklenk K, Undyala VVR, Wider J, Sala-Mercado JA, Gottlieb RA, Mentzer RM Jr. Acute induction of autophagy as a novel strategy for cardioprotection. Getting to the heart of the matter. Autophagy 2011;7:432–3
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004;304:1500–2
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004;6:1221–8
- Yan L, Vatner DE, Kim SJ, Ge H, Masureker M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci USA 2005;102:13807–12
- Elsässer A, Vogt AM, Nef H, Kostin S, Möllmann H, Skwara W, et al. Human hibernating myocardium is jeopardized by apoptosis and autophagic cell death. J Am Cell Cardiol 2004;43:2191–9
- Knaapen MW, Davies MJ, De Bie M, Haven AJ, Martinet W, Kockx MM. Apoptotic versus autophagic cell death in heart failure. Cardiovasc Res 2001;51:304–12
- Shewan LG, Coats AJ. Ethics in the authorship and publishing of scientific articles. Int J Cardiol 2010;144:1–2
- Miyata S, Takemura G, Kawase Y, Li Y, Okada H, Maruyama R, et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony stimulating factor. Am J Pathol 2006;168:386–97
- Akazawa H, Komazaki S, Shimomura H, Terasaki F, Zou Y, Takano H, et al. Diphtheria toxin-induced autophagic cardiomyocyte death plays a pathogenic role in mouse model of heart failure. J Biol Chem 2004;279:41095–103