Abstract
Purpose: Previously we showed that mild thermal stress increased natural killer (NK) cell-mediated tumour cytotoxicity and that this could be blocked by anti-NKG2D or anti-MICA (major histolocompatability complex (MHC) class I related chain A) antibodies. Here, we investigated the role of the transcription factor heat shock factor 1 (HSF1) in thermal regulation of MICA expression in tumour cells in vitro and in vivo.
Materials and methods: Hyperthermia experiments were conducted in vitro and in mice using a target temperature of 39.5 °C. Apoptotic cells and NK cells in situ were visualised by use of the TUNEL assay or expression of NKp46 respectively. Using Colo205 cells, HSF1 message was blocked utilising siRNA while luciferase reporter assays were used to measure the activity of the MICA promoter in vitro. Cell surface MICA was measured by flow cytometry.
Results: Following whole body hyperthermia (WBH), tumour tissues showed an increase in NK cells and apoptosis. Mild thermal stress resulted in a transient increase in surface MICA and enhanced NK cytotoxicity of the Colo205 colon cancer cell line. Silencing (mRNA) HSF1 expression in Colo205 cells prevented the thermal enhancement of MICA message and surface protein levels, with partial loss of thermally enhanced NK cytotoxicity. Mutations of the HSF1 binding site on the MICA promoter implicated HSF1 in the thermal enhancement of MICA. Some, but not all, patient-derived colon tumour derived xenografts also exhibited an enhanced MICA message expression after WBH.
Conclusions: Up-regulation of MICA expression in Colo205 cells and enhanced sensitivity to NK cell killing following mild thermal stress is dependent upon HSF1.
Introduction
Natural killer (NK) cells are immune effector cells with the innate ability to target and kill major histocompatibility complex (MHC) class I negative target cells [Citation1,Citation2]. Therefore NK cells provide host surveillance against cells that exhibit loss or down-regulation of MHC Class I such as virally infected or transformed cells [Citation3–5], and they are therefore recognised as having an important role in controlling tumour growth and metastasis [Citation6,Citation7]. NK cells express two distinct classes of cell surface receptors, inhibitory and activating receptors, which enable target recognition and ensure the appropriate response. Inhibitory receptors, which protect target cells from cytotoxicity include killer inhibitory receptor family members (KIR) that recognise human leucocyte antigen (HLA) family members [Citation8], and CD94/NKG2A-B heterodimeric receptors that specifically recognise HLA-E [Citation9]. On the other hand, there are a variety of activating receptors which induce cell cytotoxicity and include natural cytotoxicity receptors (NCR) (i.e. NKp30, NKp44, NKp46 and NKp80) [Citation10], NKG2D [Citation11], KIR2DS [Citation12], 2B4 [Citation13], DNAM-I [Citation14], NTB-A [Citation15] and CD94/NKG2C heterodimer [Citation16]. NK cell-mediated killing cannot be triggered unless there is a dominance of activating receptor signals over inhibitory receptor signals for receptor ligand-mediated recognition of target cells [Citation17]. Therefore, identification of factors which can result in elevated levels of activating ligands on target cells may play an important role in enhancing regulating NK cell function.
A variety of hyperthermia protocols have been shown to have different effects on the cytotoxic activity of NK cells (for review see [Citation18]) but overall these studies indicate a general increase in NK-mediated cytotoxicity induced by hyperthermia in both pre-clinical and clinical studies. Our group has previously shown that the activity of NK cells against the human colon carcinoma cell line, Colo205 can be enhanced following treatment with mild (fever-range) hyperthermia. [Citation19]. However, little is known about potential mechanism(s) by which NK cell function is regulated by temperature.
In response to several different types of stress, cells increase expression of MHC class I related chain A (MICA) [Citation20–35] a ligand for NKG2D on NK cells, and we have previously speculated that this increase in response to thermal stress could act as an evolutionarily conserved danger signal that may play a role during febrile states [Citation19,Citation36]. The MICA core promoter contains heat shock elements that inducibly bind heat shock factor 1 (HSF1) [Citation21,Citation32]. Many previous studies have shown that HSF1 acts as a temperature sensitive transcription factor that regulates heat shock protein expression following heat shock and other stresses [Citation37]. Fionda et al. [Citation38] showed that inhibition of (mRNA) HSF1 with shRNA interference blocks MICA and MICB up-regulation in human myeloma cell lines and that HSF1 is recruited to MICA/B promoters by Hsp90 inhibitors. Moreover, HSF1 is increased in T lymphocytes following mild thermal stress temperatures of 39 °C [Citation39]. In line with these findings, heat shock treatment up-regulated MICA promoter activity in quiescent HCT116 colon tumour cells [Citation32]. However, there is little information on whether HSF1 could be a key regulator of MICA expression under physiological (fever-range) temperatures. Thus, in this study we tested whether MICA might be regulated by HSF1 following mild thermal stress which in turn, leads to enhanced recognition of target cells by NK cells.
Materials and methods
Cell lines, NK and human colon cell isolation and in vitro heating
Colo205 and HT29 human colon adenocarcinoma cell lines, CT26 colon and B16-F10 melanoma murine cell lines (ATCC) were propagated in RPMI-1640 medium with 2 mM L-glutamine and 10% FBS. For in vitro heating of cells, we incubated control cell culture plates at 37 °C, and experimental cell culture plates at 39.5 °C for 6 h in a controlled humidity CO2 incubator.
Human NK cells were isolated from healthy donor peripheral blood as described before [Citation19]. Briefly, NK cells were purified by depletion of non-NK cells from peripheral blood mononuclear cells with magnetic separation using a NK cell isolation kit (Miltenyi Biotech, Auburn, CA) according to the manufacturer’s protocol. Cell viability and purity were found to be over 90% with propidium iodide staining.
For isolation of human colonocytes, approximately 5 cm long (∼10 g) samples from normal regions of ascending colon were collected through Tissue Procurement from recently deceased patients at Roswell Park Cancer Institute using an approved protocol and processed within 18 h. Blood and luminal contents were removed by washing the section with cold tap water and then the sections were dissected longitudinally and placed in sterile ice-cold RPMI-1640 with L-glutamine, penicillin/streptomycin and amphotericin B. Visible fat, necrotic tissue/debris and mucus were removed. The mucosal layer (top layer, which contains epithelial cells) was separated from the connective tissue and membranes (bottom layer) and strips (approximately 4–5 mm) were cut and placed in Petri dishes and washed with warm Hanks balanced salt solution (HBSS) with 0.15% DTT to remove residual matter. Mucosal strips were transferred to a new container with 200 mL of RPMI-1640 with 1 mM EDTA, 10% fetal bovine serum (FBS) and antibiotic/antimycotic solution (RPMI-EDTA-FBS) with a stir bar and stirred at room temperature at 60 rpm for a minimum of 4 h to release cells from basal lamina. These isolated colon cells were cultured as previously described [Citation40], and propagated in epithelial growth media consisting of RPMI-1640 medium supplemented with 5% fibroblast conditioned media, antibiotic/antimycotic solution (penicillin, streptomycin, amphotericin B), 2 mM L-glutamine, 10% FBS, insulin (5 μg/mL) and transferrin (5 μg/mL).
Whole body hyperthermia
The systemic heating of mice was carried out in an incubator (Memmert Model BE500, East Troy, WI). Sterile cages were preheated to ∼38.5 °C in the incubator. Sentinel mice bearing tumours and implanted with glass-encased temperature transponders (BioMedic Data Systems, Seaford, DE) at least 2 weeks prior to heating were included in each cage in order to allow monitoring of temperature during the incubation period. Temperature readings were performed using a hand-held, non-contact temperature measurement device. (BMDS Pocket Scanner, Model DAS-5007, BioMedic Data Systems). Baseline measurements were taken, mice were injected intraperitoneally with 1 mL sterile saline and then placed in either a cabinet at room temperature (controls) or in pre-heated cages in the incubator (whole body hyperthermia (WBH)). Temperatures of sentinel mice were taken after 15–30 min and then every hour for the duration of the WBH. The temperature of the incubator was adjusted as necessary to maintain a mouse core temperature 39.5° ± 0.5 °C.
Cytotoxicity assays
Cytotoxicity assays were performed using the CytoTox 96 non-radioactive cytotoxicity assay (Promega, Madison, WI) according to the manufacturer’s instructions as described before [Citation19]. Briefly, human colon tumour cells were mixed with purified human NK cells at several dilutions and after brief centrifugation to bring them in contact, incubated for 6 h. The supernatant was analysed for lactate dehydrogenase (LDH) release from dead cells and compared to maximum LDH release from detergent-mediated lysis and spontaneous LDH release from untreated cells.
Monoclonal antibodies and flow cytometry
For flow cytometric staining, 106 cells were washed twice with PBS followed by incubation with anti-MICA monoclonal antibody (mAb) (Immatics Biotechnologies, Tubingen, Germany), anti-HLA-ABC mAb (BD Biosciences Pharmingen, San Jose, CA), or isotype control antibodies for 30 min. on ice. They were then washed twice with sterile PBS, and stained with PE conjugated secondary antibody (BD Biosciences Pharmingen) for 20 min on ice. Finally, cells were washed twice with PBS and fixed with freshly prepared 2% paraformaldehyde in PBS, and analysed with FACScan flow cytometer (BD Biosciences, San Jose, CA) and FCS Express 3 software (De Novo Software, Los Angeles, CA).
Immunohistochemistry
Frozen sections 6–8 μm thick of tumours isolated from mice were air-dried overnight and fixed in cold acetone for 10 min. Slides were washed with PBS/Tween 20 (0.5% v/v) and endogenous peroxidase and biotin was blocked with 0.3% hydrogen peroxide for avidin/biotin treatments. Sections were treated with 0.03% casein before treatment with goat anti-mouse NKp46/NCR1 antibody (AF2225, R&D Systems Minneapolis, MN) diluted 1:500 in Dako antibody diluent (Carpinteria, CA) for 1 h. After washing with PBS/Tween three times, and with PBS once, sections were treated with secondary antibody (1:250 dilution of biotinylated horse anti-goat antibody from Vector Laboratories, Burlingame, CA). Sections were washed three times with PBS-Triton X100 (0.2% v/v) and incubated with streptavidin-HRP followed by the ImmPACT DAB substrate (Vector Laboratories). Sections were counter-stained with Haematoxylin, dehydrated with gradual washes of ethanol and xylene and mounted with Permount mounting media (Fisher Scientific, Pittsburg, PA), examined and photographed with an Olympus BX40 microscope mounted with a Hitachi HV-C20 CCD camera.
RNA preparation and quantitative real-time PCR procedure
(mRNA) MICA. HSF1 and HSP70 were analysed by reverse transcription and PCR analysis as well as quantitative real time PCR. RNA from tumour tissue homogenised in lysis buffer was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA) and quantified by measuring absorbance at 260 and 280 nm using GeneQuant RNA/DNA Calculator (Pharmacia/Pfizer, New York). cDNA was prepared as described before [Citation19]. 18S ribosomal RNA (18S) was used as an internal control in quantitative PCR experiments where FastStart Universal SYBR Green Master mix was used (Roche Diagnostics, Mannheim, Germany). Briefly, SYBR Green Master Mix containing SYBR Green dye, FastStart Taq DNA polymerase, dNTPs, passive reference (ROX) and buffer components, were mixed with MICA reverse (5′-CATGGATCTCACAGACCCTAATCT-3′) and forward (5′-GGATGACCCTGGCTCATATCA-3′), HSF1 reverse (5′-GAGATCAGGAACTGAATGAGC-3′) and forward (5′-AAGTACTTCAAGCACAACAACAT -3′), or HSP70 reverse (5′-GCGATCTCCTTCATCTTGGT -3′), forward (5′-AAGGTGGAGATCATCGCCAA -3′) primers, cDNA, and water according to the manufacturer’s protocols. Amplifications were performed in 96-well QRT-PCR plates (Axygen Scientific, Union City, CA) using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Fold change in relative signal was determined using the 2−ΔΔCt formula. When necessary, PCR products were analysed with 1.5% agarose gel electrophoresis and ethidium bromide staining. Images of the gels were obtained with a BioDoc-It Transilluminator System (UVP, Upland, CA).
TUNEL assay
Apoptosis in tumour sections was detected by staining sections of frozen tissue in Tissue-Tek OCT blocks for TUNEL assay using MEB-STAIN Apoptosis Kit Direct (MBL, Woburn, MA) according to the manufacturer’s instructions. Briefly, 6-μm frozen sections were prepared, fixed in 4% paraformaldehyde and washed with PBS. Next, they were treated with TdT enzyme and FITC conjugated dUTP. Tissue sections were mounted on slides with Vectashield mounting media containing DAPI (Vector Laboratories). Positive control sections were treated with DNase I solution (Sigma-Aldrich, St Louis, MO). Sections were examined with a Zeiss Axioskop2 MOT fluorescent microscope mounted with a SPOT camera (Diagnostic Instruments, Sterling Heights, MI).
Silencing of HSF1
HSF1 in Colo205 cells was silenced using Silencer pre-designed siRNAs (Ambion, Austin, TX). The 21-mer siRNA sequences are as follows: 115674 sense 5′-GCAACAGAAAGUCGUCAACtt-3′, antisense 5′-GUUGACGACUUUCUGUUGCtg-3; 3134 sense 5′-AUAGUCAGAAUUGUAUUUUtt-3′, antisense 5'-AAAAUACAAUUCUGACUAUtg-3′. Five million cells were transfected with 5 μg HSF1 or control siRNA using a Nucleofector Kit T (Amaxa Biosystems, Gaithersburg, MD), according to the manufacturer’s instructions. The cells were harvested 72 h after transfection for further experiments.
Cycloheximide treatment
Colo205 cells were divided into four groups: the first group was treated with DMSO for the duration of 12 h at 37 °C. The second group was treated with DMSO for 6 h at 37 °C and with cycloheximide (5 μg/mL) for another 6 h at 37 °C. The third group was treated with DMSO for 6 h at 39.5 °C and then with cycloheximide (5 μg/mL) for another 6 h at 37 °C. The fourth group was treated with DMSO for 6 h at 39.5 °C and for another 6 h at 37 °C. Cell surface MICA was measured at 0, 6, 7, 8, 9 and 12 h with flow cytometry.
Luciferase reporter assays of MICA promoter
Luciferase reporter activity under the MICA promoter was determined following transfection into Colo205 cells using a Nucleofector Kit T, according to the manufacturer’s instructions. One million cells were transfected with 1 μg luciferase reporter plasmid and 0.1 μg of pRL-CMV Renilla vector, for standardisation of transfection efficiency. The cells were harvested 72 h after transfection. Luciferase assays were performed using a dual luciferase reporter assay system (Promega) according to the manufacturer’s protocol. Transcriptional activity was measured in a Fluoroskan Ascent FL luminometer (Thermo Electron Corporation, Vantaa, Finland).
MICA promoter plasmids were constructed by cloning the minimal MICA promoter into pGL3.basic vector (Promega), named pGL3.MICA. HSF1 binding site mutations were performed on the pGL3.MICA plasmid using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Mutations were confirmed by standard sequencing protocols. The mutated plasmid was designated pGL3.HSRE. A reporter plasmid carrying the Simian virus 40 (SV40) promoter and enhancer was used as positive control (pGL3.SV40).
Statistics
All data were analysed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA) statistical analysis software. ANOVA analysis was used in tests involving more than two variables. T-test with Welch`s correction was used for the analysis of significance between two unmatched experimental groups. Significance was accepted over 95% confidence.
Results
Tumour cell apoptosis and the presence of NK cells in the tumour microenvironment is increased after mild systemic thermal stress
Previous studies from our group demonstrated increased numbers of leucocytes, including NK cells, in the tumour microenvironment of tumour-bearing SCID mice treated with fever-range whole body hyperthermia (WBH) [Citation41]; moreover, in that study we also observed evidence of increased apoptosis which we hypothesised was due to the increased presence of NK cells in the tumour microenvironment. We have confirmed this information using other model systems here. Using a patient-derived colon tumour xenograft we again looked for changes in apoptosis following mild WBH. Tumours from unheated mice showed a low level of apoptosis (determined using the TUNEL assay [Citation42]) with only a few apoptotic cells visible at normothermic conditions (). However, within a day after WBH we observed evidence of increased TUNEL positive cells in tumour sections (). In another experiment, using CT26 cells grown as a tumour in BALB/c mice, we found that WBH resulted in an increase in the number of cells bearing a marker for NK cells, NKp46 [Citation10]. Representative photomicrographs from normothermic and hyperthermic BALB/c mice carrying CT26 tumours are shown in , respectively
Figure 1. Whole body hyperthermia (WBH) increases the numbers of NK cells and apoptotic cells in colon tumours. (A, B) Localisation of apoptotic cells by TUNEL assay in a patient tumour xenograft from control (A) or WBH treated (B) SCID mice (arrows, original mag. 40X). (C, D) Immunohistochemical localisation of NKp46+ cells in CT26 tumours from control (C) and WBH treated (D) mice at 24 h post-WBH (arrows, original magnification ×10).
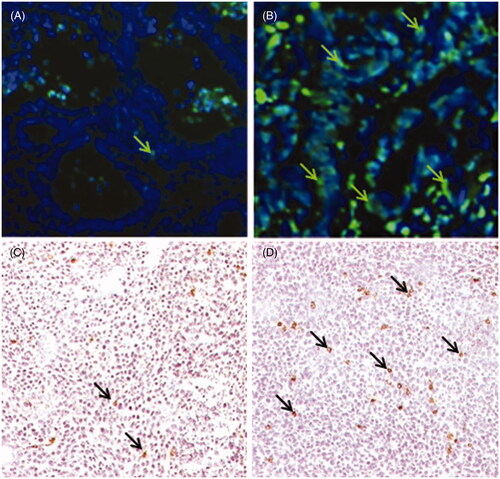
Since NK cells are the primary cytolytic effector cells in the SCID mouse model, these anatomical and immunohistochemistry data support our hypothesis that mild heating may, in some way, stimulate increased NK cell-mediated anti-tumour activity. Additionally, we and others have also shown previously that in vitro heat treatments can enhance NK cell cytotoxicity [Citation18,Citation19] and that this effect coincides with increased surface expression of MICA on the human colon adenocarcinoma cell line Colo205 [Citation19]. Collectively, these observations support the hypothesis that NK cells are sensitive to thermal stress and that by better understanding the mechanisms underlying this sensitivity, we may better target these cells for clinical hyperthermia applications. Thus, next we examined in greater molecular detail the mechanism by which MICA expression could be regulated by mild thermal stress and focused largely on the role of HSF1.
Thermal enhancement of NK cytotoxicity against colon carcinoma cells is dependent upon HSF1-dependent induction of MICA
We have previously shown that mild heating in vitro results in a significant increase in surface expression of MICA on cells from the human Colo205 cell line [Citation19]. To determine whether the increase in MICA following thermal stress was due to HSF1, Colo205 cells were transfected with two constructs of (mRNA) HSF1 siRNA to silence the transcription factor. Control transfectants included negative control siRNA at the highest concentration used for (mRNA) HSF1 silencing. HSF1 is endogenously present in Colo205 cells, and was suppressed to a quarter of the levels in controls by siRNA-mediated silencing as determined by western blotting (data not shown). Transfected Colo205 cells were exposed to mild (fever-range) thermal stress (39.5 °C) for 6 h or kept at normothermic (37 °C) temperature in vitro. Analysis for (mRNA) HSF1 carried out with quantitative RT-PCR at 48 h showed that silencing with (mRNA) HSF1 siRNA resulted in a five-fold decrease in thermal stress-inducible HSF1 message () correlating with the reduced protein expression.
Figure 2. Silencing of (mRNA) HSF1 in tumour cells results in decreased MICA expression and impaired thermally enhanced cytotoxicity by human NK cells. Colo205 cells were transfected with two different siRNA constructs against HSF1 (3134 and 115674) at two different concentrations 3 pg/cell (▪) and 5 pg/cell (□), or with control siRNA at 5 pg/cell ![]()
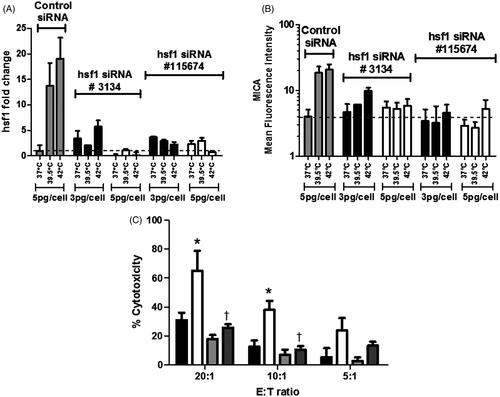
The effect of HSF1 reduction on surface MICA expression by Colo205 cells was examined with flow cytometry (). Colo205 cells were treated with thermal stress (39.5 °C) for 6 h and then analysed for MICA expression. We observed an overall reduction of thermally enhanced surface MICA expression on Colo205 cells after transfection with both (mRNA) HSF1 siRNA constructs at all concentrations.
We next asked whether silencing of HSF1 message would affect susceptibility of the Colo205 cells to NK cell killing. NK cells were isolated from three healthy human donors. Colo205 cells transfected with (mRNA) HSF1 siRNA were used as target cells in NK cell cytotoxicity assays. Transfected cells were either held at (37 °C) or treated with mild thermal stress (39.5 °C) for 6 h in vitro as above. Cytotoxicity assays showed that (mRNA) HSF1 silencing resulted in a significant decrease in NK cell-mediated cytotoxicity at higher NK cell to target ratios (). These findings confirm our earlier report that thermal stress enhances NK activity and up-regulates MICA expression on tumour cells and further suggest that HSF1 plays a critical role in the induction of MICA on Colo205 cells.
Mutation of the HSF1 binding site results in the loss of thermally enhanced activity of the MICA promoter
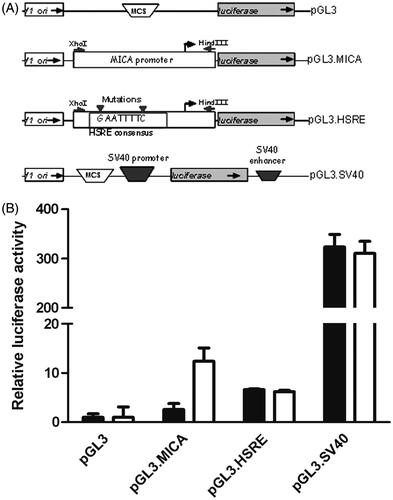
To further test the role of HSF1 in thermal induction of MICA, the minimal MICA promoter described in [Citation32] was cloned into the pGL3 luciferase reporter construct. HSF1 binding sites in the promoter were mutated by site-directed mutagenesis (). Promoter constructs were transfected into Colo205 cells; the cells were then subjected to thermal stress (). Mild thermal stress induced a significant increase in MICA promoter activity as measured by increased luciferase activity, and this was abrogated when the HSF1 binding site was mutated. Thermal stress had no effect on luciferase activity in cells transfected with either a promoter-less construct (pGL3) or when luciferase expression was driven by SV40 (pGL3.SV40).
Figure 3. Mutation of HSF1 binding site results in the loss of thermally enhanced activity of MICA promoter. (A) Schematic representation of plasmid constructs containing luciferase reporter gene driven by MICA promoter. Basic pGL3 construct (pGL3), pGL3 construct carrying the MICA promoter (pGL3.MICA), point mutations in HSRE in MICA promoter (pGL3.HSRE), and positive control construct carrying an SV40 promoter and enhancer (pGL3.SV40). (B) Colo205 cells were transfected with the reporter constructs described in (A) and treated at normothermic (▪) or hyperthermic (□) temperatures for 6 h. Relative luciferase activity was detected by dual luciferase reporter assay and normalised to Renilla luciferase activity as transfection control. *p ≤ 0.05 compared to the corresponding normothermic group.
MICA stability on tumour cell surface after thermal stress depends on thermally enhanced MICA synthesis
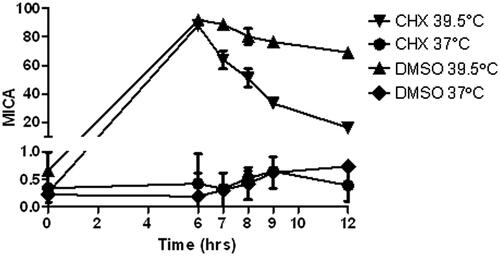
Previously we observed an increase in MICA message within 3 h after thermal stress in vitro, followed by an observed increase in MICA surface expression at 6 h [Citation19]. Cycloheximide, as an inhibitor of eukaryotic protein signalling, can block translational elongation; therefore, we used it to determine the extent of de novo translation-independent increase in surface MICA up-regulation after mild thermal stress. This experiment tests whether the thermal stress-induced increase in cell surface MICA is due to elevated transcription/translation, or is perhaps due to other events, such as cell membrane changes as we previously observed [Citation19]. Since treatment with cycloheximide following thermal stress leads to a faster loss of MICA surface expression than seen using DMSO alone following thermal stress (), the prolonged increase in MICA surface expression following thermal stress likely requires continual protein synthesis.
Figure 4. Cycloheximide inhibits induction of MICA by thermal stress in Colo205 cells. Colo205 cells were treated with mild thermal stress at 39.5 °C (▴, ▾) or at 37 °C (♦, •) with DMSO for 6 h in vitro, followed by cycloheximide (5 μg/mL in the culture medium) (▾, •) or DMSO, the solvent used for cycloheximide (▴, ♦) at normothermic temperatures. Cell surface MICA expression was detected with flow cytometry.
Whole body hyperthermia results in increased MICA in tumour cells in vivo
Our previous results demonstrated that in vitro thermal stress results in an increase in MICA levels on tumour cells, which correlates with enhanced NK cell cytotoxicity [Citation19]. To ask whether similar results would occur in vivo, i.e., in tumour cells in mice treated with WBH, SCID mice bearing Colo205 (), HT29 () and four different patient-derived colon tumour xenografts () were treated with WBH. Colo205-derived tumours were found to have up-regulated message levels for MICA, HSF1 and Hsp70 after WBH (), while HT29-derived tumours only showed an increase in Hsp70 (). Of the four patient xenografts, MICA was increased in two of the earlier passage tumours, 16115 (second passage) and 16460 (third passage), but not in two late passage tumours 13391 (17th passage) and 15053 (sixth passage).
Figure 5. Response of colon cell lines and patient tumour xenografts to mild thermal stress. Colo205 (A), or HT29 (B) tumour bearing SCID mice were treated with whole body hyperthermia (WBH) at 39.5 °C for 6 h in vivo. Fold change of MICA. HSF1 and HSP70 mRNA in the tumours compared to 18 S RNA or levels in tumours from control mice was measured by quantitative RT-PCR and calculated against 18 S RNA, using triplicate wells. The data shown represent the mean ± SD (n ≥ 3) of three experiments. (C) Colon tumours from four patients were implanted into SCID mice and treated with WBH. MICA mRNA expression was detected by qPCR and fold change determined compared to untreated controls (D, E) Kinetics of MICA mRNA levels in Colo205 and HT29 xenografts respectively at 3, 6, 12 and 24 h after WBH. (F) Kinetics of the murine MICA orthologue, Rae1 mRNA expression in CT26 tumours in BALB/c mice □ hyperthermic, ▪ normothermic.
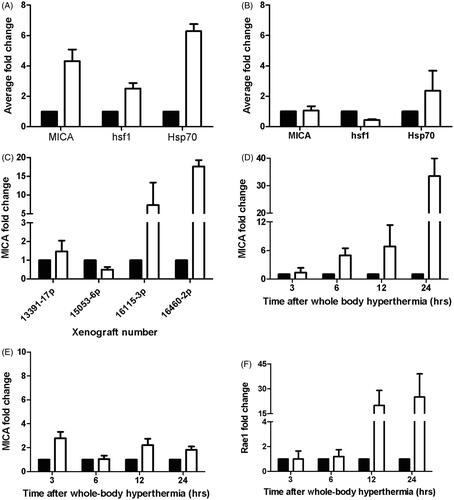
Previous kinetic studies of MICA expression with thermal stress showed that transcriptional regulation of MICA message starts as early as 3 h in vitro [Citation19]. Therefore we investigated the kinetics of MICA message in Colo205 and HT29 tumours within 24 h after WBH. (mRNA) MICA levels steadily increased in the Colo205 tumours up to 24 h after WBH (). On the other hand, MICA message levels did not increase over the course of 24 h in HT29 tumours (), in agreement with the lack of increase in MICA message expression at 6 h in (). Mouse MICA orthologue, Rae1, mRNA expression in CT26 tumours occurred at 12 h post-WBH () which is slightly slower than the analogous response in Colo205.
Normal human colonocytes do not up-regulate MICA after thermal stress
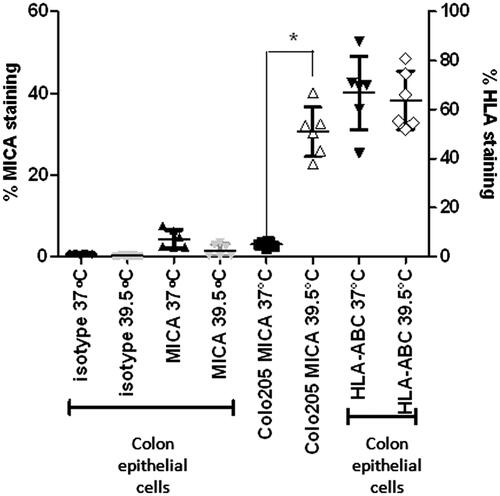
We examined whether mild thermal stress also enhanced surface expression of MICA by normal human colonocytes. Colonocytes isolated from colon samples were treated with mild thermal stress at 39.5 °C for 6 h in vitro, labelled for surface MICA protein expression and pan-HLA (since thermally mediated NK cell cytotoxicity against these cells could potentially change if surface MHC Class I expression is altered) and then compared to Colo205 cells treated under the same conditions. Although MICA and HLA are expressed in normal colonocytes, we observed no change in the expression of either following mild thermal stress (). This complements our previous studies which showed that there was no change in HLA expression on tumour cells following thermal stress [Citation19].
Figure 6. Expression levels of MICA and MHC Class I are not altered with thermal stress in non-malignant, normal human colon epithelial cells. Normal colon epithelial cells isolated from donors and Colo205 cells were treated with mild thermal stress at 39.5 °C for 6 h in vitro and cell surface levels of MICA and HLA-A, B, C were quantified by flow cytometry. The results are plotted showing means and standard deviations of six samples. *p = 0.0002 compared to the corresponding normothermic group using unpaired t-test with Welch’s correction.
Discussion
The initial data presented here () provides some additional descriptive evidence that following systemic heating in vivo, tumour cell apoptosis (in a patient tumour-derived xenograft model) increases () and, in a different tumour model, that there is an increase in the presence of NK cells, as detected by the marker NKp46+ cells within the tumour microenvironment (). NKp46 has recently been validated as a human NK cell marker in gut-associated tissues, including its association with innate lymphoid cells (ILC) in mice [Citation43]. These observations, combined with our previous studies demonstrating that NK cell cytotoxicity is enhanced by mild heating, provided an impetus to learn more about how mild heating affects tumour cell sensitivity to NK cells. Thus, the major goal of this study was to study effects of mild heating on tumour cell expression of the non-classical MHC-like molecule, MICA, which is a major ligand for NKG2D on NK cells, and more specifically, to determine whether any increased expression of MICA on the surface of tumour cells following mild heat exposure might be regulated by HSF1.
We demonstrated that down-regulation of (mRNA) HSF1 by use of two different siRNAs resulted in decreased HSF1 message, accompanied by down-regulation of thermally enhanced, but not endogenous, MICA levels. This confirms that the effect of thermal stress on MICA expression is at the transcriptional level through the activity of HSF1 (). Consistent results using two different siRNA constructs support that HSF1 silencing-mediated (mRNA) MICA down-regulation is not a non-specific, off-target effect (,B). Importantly, MICA message down-regulation resulted in decreased NK activity against Colo205 tumour cells in vitro (). The observed decrease in NK-mediated cytotoxicity with (mRNA) HSF1 siRNA at normothermic conditions suggests a role for HSF1 in NK cell function under normothermic conditions. While a role for other activating ligands that might be present on Colo205 cell surface was not suspected, since NKG2D/MICA blocking antibodies were previously shown to block endogenous and thermally enhanced cytotoxicity in this model [Citation19], our data does not exclude a possible role for other surface molecules that might be altered with thermal stress.
Promoter reporter assays revealed an up-regulation of MICA promoter activity with thermal stress which is dependent on the presence of intact HSF1 binding site in the promoter (). These results are in agreement with previous studies demonstrating the role of HSF1 binding site in MICA promoter in quiescent HCT116 colon tumour cells with heat shock [Citation32]. However, a correlation between HSF1-mediated transcriptional regulation of the MICA gene at mild thermal stress temperatures has not been previously described. The observed increase in luciferase activity at control temperatures when the HSF1 binding site is mutated suggests that MICA expression might be suppressed by HSF1 under normothermic conditions. However, these arguments would have been greatly supported by the use of a chromatin immunoprecipitation (ChIP) assay showing direct HSF1 binding to the MICA promoter.
We have previously observed a transient increase in mild thermal stress-enhanced NK cell cytotoxicity of Colo205 cells, which regresses to non-significant levels of cytotoxicity compared to control temperatures within 24 h in vitro (unpublished results). Here we show that thermally enhanced surface MICA expression on cell surface is dependent on sustained MICA synthesis, where disruption leads to faster levels of decay from the cell surface (). Our results indicate that MICA cell surface kinetics follow ‘one-phase exponential decay’, with significantly enhanced MICA levels on the cell surface without cycloheximide. While thermal stress clearly helps to regulate the transcription of MICA, the precise mechanisms by which MICA trafficking to the cell surface might be affected requires further research. In addition, it is important to remember that shedding of MICA molecules from the tumour cell surface occurs through metalloproteinase activity [Citation44,Citation45]; however, we have not yet addressed this issue under thermal stress conditions.
We also tested whether systemic heating in vivo could up regulate (mRNA) MICA expression, i.e., in Colo205 and HT29 tumour cells growing in mice (). We consistently observed an up-regulation of MICA message in Colo205 tumours following WBH, averaging a 4.2-fold change compared to tumours in mice maintained at normothermia. While Colo205 tumours in SCID mice show a consistent increase of MICA message levels up to 24 h following WBH, HT29 cells do not [Citation19]. However, HSP70 up-regulation with thermal stress is still intact in HT29 cells, albeit less than Colo205 cells. The role of HSF1 in regulation of HSP70 and MICA message may vary between different tumour cells, and the basis for these differences should be investigated.
Several studies have shown that classical MHC class I molecule expression on cell surface is the major signal for ‘self’ recognition by the immune system, and is also a major deterrent for NK cell-mediated cytotoxicity [Citation46,Citation47]. Previous reports for endogenous expression of MICA on human colonocytes [Citation11,Citation48], combined with the importance of HLA expression for limiting NK cell-mediated killing, led us to investigate the expression of HLA and MICA levels on colonocytes isolated from human donors and then treated in vitro with hyperthermia. Although we have observed relatively low expression of MICA message in normal colonocytes, our study suggests that mild heat stress does not lead to significant changes in MICA or HLA (A, B, C) levels (). This evidence for some specificity of heat stress on MICA expression on tumour cells versus normal cells requires further analysis to test whether there are changes in the transcriptional control of MICA expression during colon cancer progression.
This report has highlighted HSF1-mediated MICA expression on tumour cells as a potential molecular mechanism to explain the enhancement of NK cell cytotoxicity against tumours following mild thermal stress. However, there are other studies suggesting alternative regulation pathways for MICA regulation in the literature, mainly through other transcription factors. For example, a recent study shows that NF-κB may also be a transcription factor that controls MICA expression. Lin et al. [Citation49] demonstrated that the TNF-α-mediated MICA up-regulation on human umbilical vein endothelial cells is through NF-κB. In this model NF-κB acts on the −130 bp of MICA transcription start site, overlapping with heat shock response elements (HSRE). We also investigated this region in our study. Lin et al. [Citation49] found that TNF-α-mediated MICA up-regulation is inhibited with an inactive form of HSF1 (lacking a transactivation domain). These results further support our observations. Lin et al. further suggested a competition between NF-κB and HSF1 for the MICA promoter binding site, which is found to be responsive to heat shock (42 °C, 1 h). However Andresen et al. [Citation50] reported that histone deactylase inhibitors cause an increase in surface MICA/B expression in Jurkat T cells, which is completely abrogated by intracellular calcium depletion, independent of NF-κB but dependent on transcription factor Sp1. They identified −115 to −84 of MICA promoter as the strong regulatory region in Jurkat T cells. In our colon tumour models, we have not determined whether the effect of HSF1 on MICA expression is independent of NF-κB or TNF-α. Luo et al. [Citation51] stated that hypoxia inducible factor (HIF-1) regulates MICA expression in human renal proximal tubular epithelial cells HK-2. This observation of MICA up-regulation under hypoxic stress provides further support to our earlier observation [Citation19] of its up-regulation under thermal stress. Another transcription factor identified in MICA transcriptional regulation is STAT3 [Citation52]. Studies suggest that STAT3 negatively regulates MICA after it was observed that there was increased expression of MICA following STAT3 inhibition and direct binding of STAT3 on MICA promoter. MICA induction in mesenchymal stem cells after heat shock (45 °C, 1 h) is also prevented with constitutive activation of STAT3, suggesting that STAT3 might also play a role in thermal regulation of MICA up-regulation. Clearly, the role of other transcription factors in thermal modulation of NK cell activity requires further investigation.
Overall, the data shown here provides additional support for the notion that mild thermal stress can enhance the activity of immune cells such as NK cells. However, at the present time there are no clinical studies which have correlated MICA expression on tumour cells with patient survival following hyperthermia treatment and this is clearly an important gap in the field. The studies reported here predict that some patients may benefit from mild hyperthermia treatment through thermally enhanced tumour cell expression of MICA and increased anti-tumour NK cell cytolytic function. However, our data also reveals significant variation among tumour cells in terms of MICA expression following heating, suggesting that not all patients may benefit. Clearly new biomarkers or correlative studies which can link heat-induced changes in NK cell activity, and/or tumour cell sensitivity, could be very helpful in predicting which patients may benefit most by receiving clinical hyperthermia.
Declaration of interest
This study was supported by Komen Foundation DISS0402487 and Fulbright Scholarships to BED, National Institutes of Health grants P01 CA94045, R01 CA71599, R21 CA098852, and also Roswell Park Cancer Institute Core grant CA16056. The authors alone are responsible for the content and writing of the paper.
Acknowledgements
We are grateful to Julie Ostberg for her considerable advice at the onset of this project, Bonnie Hylander for expert advice on immunohistochemical staining, Hima Bansal for her help on reporter assays, Rose Pitoniak for help and advice on animal studies, Earl Timm for advice on flow cytometry analyses and Katie Kokolus and Bonnie Hylander for their help with manuscript preparation.
References
- Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer 1975;16:230–9
- Kiessling R, Klein E, Wigzell H. ‘Natural’ killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol 1975;5:112–17
- Glimcher L, Shen FW, Cantor H. Identification of a cell-surface antigen selectively expressed on the natural killer cell. J Exp Med 1977;145:1–9
- Gryllis C, Wainberg MA, Bentwich Z, Gornitsky M, Brenner BG. Increased LAK activity against HIV-infected cell lines in HIV-1+ individuals. Clin Exp Immunol 1992;89:356–61
- Smyth MJ, Cretney E, Takeda K, Wiltrout RH, Sedger LM, Kayagaki N, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med 2001;193:661–70
- Garner WL, Minton JP, James AG, Hoffmann C. Human breast cancer and impaired NK cell function. J Surg Oncol 1983;24:64–6
- Kobayashi H, Sigrid Dubois, Noriko Sato, Helen Sabzevari, Yoshio Sakai, Waldman TA, et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood 2005;105:721–7
- Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, et al. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J Exp Med 1993;178:597–604
- Braud VM, Allan DS, O’Callaghan CA, Söderström K, D'Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998;391:795–9
- Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 2001;19:197–223
- Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999;285:727–9
- Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol 1998;161:571–7
- Garni-Wagner BA, Purohit A, Mathew PA, Bennett M, Kumar V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol 1993;151:60–70
- Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996;4:573–81
- Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med 2001;194:235–46
- Lopez-Botet M, Carretero M, Bellón T, Pérez-Villar JJ, Llano M, Navarro F. The CD94/NKG2C-type lectin receptor complex in recognition of HLA class I molecules. Res Immunol 1997;148:155–9
- Long EO, Burshtyn DN, Clark WP, Peruzzi M, Rajagopalan S, Rojo S, et al. Killer cell inhibitory receptors: Diversity, specificity, and function. Immunol Rev 1997;155:135–44
- Dayanc BE, Beachy SH, Ostberg JR, Repasky EA. Dissecting the role of hyperthermia in natural killer cell mediated anti-tumor responses. Int J Hyperthermia 2008;24:41–56
- Ostberg JR, Dayanc BE, Yuan M, Oflazoglu E, Repasky EA. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J Leukoc Biol 2007;82:1322–31
- Bahram S, Inoko H, Shiina T, Radosavljevic M. MIC and other NKG2D ligands: From none to too many. Curr Opin Immunol 2005;17:505–9
- Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA 1996;93:12445–50
- Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2006;291:L222–31
- Gannage M, Buzyn A, Bogiatzi SI, Lambert M, Soumelis V, Dal Cortivo L, et al. Induction of NKG2D ligands by gamma radiation and tumor necrosis factor-alpha may participate in the tissue damage during acute graft-versus-host disease. Transplantation 2008;85:911–15
- Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res 2005;65:6321–9
- Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol 2006;176:5108–16
- Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood 2007;110:606–15
- Eagle RA, Traherne JA, Ashiru O, Wills MR, Trowsdale J. Regulation of NKG2D ligand gene expression. Hum Immunol 2006;67:159–69
- Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 2007;21:2103–8
- Molinero LL, Fuertes MB, Girart MV, Fainboim L, Rabinovich GA, Costas MA, et al. NF-kappa B regulates expression of the MHC class I-related chain A gene in activated T lymphocytes. J Immunol 2004;173:5583–90
- Rodriguez-Rodero S, González S, Rodrigo L, Fernández-Morera JL, Martínez-Borra J, López-Vázquez A, et al. Transcriptional regulation of MICA and MICB: A novel polymorphism in MICB promoter alters transcriptional regulation by Sp1. Eur J Immunol 2007;37:1938–53
- Skov S, Pedersen, MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res 2005;65:11136–45
- Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol 2007;178:961–9
- Schreiner B, Voss J, Wischhusen J, Dombrowski Y, Steinle A, Lochmuller H, et al. Expression of toll-like receptors by human muscle cells in vitro and in vivo: TLR3 is highly expressed in inflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. Faseb J 2006;20:118–20
- Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer 2003;104:354–61
- Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J Immunol 2003;171:5423–9
- Pritchard MT, Ostberg JR, Evans SS, Burd R, Kraybill W, Bull JM, et al. Protocols for simulating the thermal component of fever: Preclinical and clinical experience. Methods 2004;32:54–62
- Sistonen L, Sarge KD, Morimoto R. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol Cell Biol 1994;14:2087–99
- Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J Immunol 2009;183:4385–94
- Murapa P, Gandhapudi S, Skaggs HS, Sarge KD, Woodward JG. Physiological fever temperature induces a protective stress response in T lymphocytes mediated by heat shock factor-1 (HSF1). J Immunol 2007;179:8305–12
- Mohammadpour HA. Isolation and culture of human colon epithelial cells using a modified explant technique employing a noninjurious approach. Methods Mol Med 2005;107:237–47
- Burd R, Dziedzic ST, Xu Y, Caligiuri MA, Subjeck JR, Repasky EA. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J Cell Physiol 1998;177:137–47
- Loo DT, Rillema JR. Measurement of cell death. Methods Cell Biol 1998;57:251–64
- Tomasello E, Yessaad N, Gregoire E, Hudspeth K, Luci C, Mavilio D, et al. Mapping of NKp46(+) cells in healthy human lymphoid and non-lymphoid tissues. Front Immunol 2012;3:344
- Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007;447:482–6
- Salih HR, Rammensee HG, Steinle A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J Immunol 2002;169:4098–102
- Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 1990;11:237–44
- Gorelik E, Gunji Y, Herberman RB. H-2 antigen expression and sensitivity of BL6 melanoma cells to natural killer cell cytotoxicity. J Immunol 1988;140:2096–102
- Perera L, Shao L, Patel A, Evans K, Meresse B, Blumberg R, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis 2007;13:298–307
- Lin D, Lavender H, Soilleux EJ, O'Callaghan CA. NF-kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem 2012;287:4299–310
- Andresen L, Jensen H, Pedersen MT, Hansen KA, Skov S. Molecular regulation of MHC class I chain-related protein A expression after HDAC-inhibitor treatment of Jurkat T cells. J Immunol 2007;179:8235–42
- Luo L, Lu J, Wei L, Long D, Guo JY, Shan J, et al. The role of HIF-1 in up-regulating MICA expression on human renal proximal tubular epithelial cells during hypoxia/reoxygenation. BMC Cell Biol 2010;11:91
- Bedel R, Thiery-Vuillemin A, Grandclement C, Balland J, Remy-Martin JP, Kantelip B, et al. Novel role for STAT3 in transcriptional regulation of NK immune cell targeting receptor MICA on cancer cells. Cancer Res 2011;71:1615–26