
Abstract
Purpose: Despite clinical progress, mechanisms involved in cellular responses to low and high doses of hyperthermia are not entirely clear. This study investigates the role of Bcl-2 family proteins in control of the mitochondrial pathway of apoptosis during hyperthermia at 42–43 °C and the protective effect of a low dose adaptive survival response, mild thermotolerance induced at 40 °C.
Materials and methods: Levels of Bcl-2 family proteins were detected in HeLa cells by western blotting, caspase activation by spectrofluorimetry and apoptosis by chromatin condensation.
Results: Hyperthermia (42–43 °C) decreased total and mitochondrial expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, while expression of pro-apoptotic proteins Bax, Bak, Puma and Noxa increased. Hyperthermia perturbed the equilibrium between these anti- and pro-apoptotic Bcl-2 family proteins in favour of pro-apoptotic conditions. Hyperthermia also caused activation of caspases-9 and -3, and chromatin condensation. Disruption of the balance between Bcl-2 family proteins was reversed in thermotolerant (40 °C) cells, thus favouring cell survival. Bcl-2/Bcl-xL inhibitor ABT-737 sensitised cells to apoptosis, which indicates that Bcl-2 family proteins play a role in hyperthermia-induced apoptosis. The adaptive response of mild thermotolerance (40 °C) was still able to protect cells against hyperthermia (42–43 °C) when Bcl-2/Bcl-xL were inhibited.
Conclusions: These results improve knowledge about the role of Bcl-2 family proteins in cellular apoptotic responses to hyperthermia (42–43 °C), as well as the adaptive survival response induced by exposure to mild stresses, such as a fever temperature (40 °C). This study could provide rationale to explore the manipulation of Bcl-2 family proteins for increasing tumour sensitivity to hyperthermia.
Introduction
Over the past three decades, there have been major advances in the use of hyperthermia as a tumour-targeted approach in cancer treatment, mainly in combination with radiotherapy, and/or chemotherapy [Citation1–4]. Hyperthermia (41–45 °C) is one of the most effective radiation sensitisers known and can eliminate radio-resistant tumour cells [Citation1]. Hyperthermia displays synergistic interactions with different anticancer drugs including bleomycin, alkylating agents and platinum compounds, and can increase the effectiveness of chemotherapy [Citation2].
Despite important clinical progress with hyperthermia, the molecular mechanisms involved in cellular responses to heat stress remain unclear [Citation5]. Hyperthermic temperatures, which are only a few degrees above normal, can cause protein denaturation and aggregation which results in the inactivation of protein synthesis, cell cycle progression and DNA repair processes [Citation6,Citation7]. Consequently, cells either die by apoptosis and/or necrosis or become sensitised to other cytotoxic modalities such as radiation. Hyperthermia (41 to 45 °C) is known to activate apoptosis through the death receptor, mitochondrial [Citation5,Citation8,Citation9] and endoplasmic reticulum (ER) pathways [Citation10].
The preconditioning of cells at elevated temperatures induces thermotolerance, which is associated with accumulation of heat shock proteins (HSPs) [Citation8,Citation11–14]. Thermotolerance can render cells resistant to subsequent toxic doses of insults such as heat shock, chemotherapeutic agents, radiation and environmental stress [Citation15,Citation16]. Thermotolerance is transient and usually declines within several days. Therefore, there is no interference with clinical use of hyperthermia that is applied at intervals beyond the time frame of thermotolerance. Thermotolerance can be induced by short exposures (e.g. 30 min) to higher temperatures (42–45 °C), or by continuous heating (e.g. 3–24 h) at mild, non-lethal temperatures (39.5–41.5 °C) [Citation11–14]. Thermotolerance induced at higher temperatures (>42.5 °C) has been widely studied, whereas thermotolerance induced by lower, fever-range temperatures has received little attention. Mild thermotolerance developed at 40 °C protected cells against hyperthermia-induced activation of mitochondrial and death receptor-mediated apoptosis [Citation8,Citation9]. The phenomenon whereby cellular defence mechanisms are triggered by low doses of stress such as heat can be of great interest in terms of different pathologies such as ischaemia-reperfusion and responses to the toxic effects of drugs and environmental toxins.
The B-cell lymphoma 2 (Bcl-2) family consists of a large number of proteins that play a pivotal role in apoptosis by determining cell fate upstream of mitochondrial outer membrane permeabilisation (MOMP) [Citation17,Citation18]. Bcl-2 family proteins are classified into two groups: anti-apoptotic molecules such as Bcl-2, Bcl-2-like 1 (Bcl-xL), Bcl-2-like 2 (Bcl-w), myeloid cell leukemia-1 (Mcl-1), and pro-apoptotic molecules such as Bcl-2-associated X protein (Bax), Bcl-2 homologous antagonist killer (Bak), BH3-interacting-domain death agonist (Bid), p53-upregulated modulator of apoptosis (Puma) and Noxa. The ratio between levels of pro-apoptotic and anti-apoptotic proteins at the membranes of organelles such as mitochondria is an important determinant in cellular sensitivity to apoptosis [Citation19]. Anti-apoptotic proteins such as Bcl-2 and Bcl-xL are integral membrane proteins found at subcellular compartments such as mitochondria. They can exert their anti-apoptotic actions by heterodimerisation with pro-apoptotic proteins, such as Bax and Bak. Most pro-apoptotic Bcl-2 family proteins are found in the cytosol and can translocate to organelles such as mitochondria under stress stimuli. Pro-apoptotic proteins such as truncated Bid (tBid) and Puma interact with and enhance the activity of other pro-apoptotic proteins such as Bax and Bak, leading to their activation and MOMP. Other pro-apoptotic proteins such as Bad and Noxa neutralise anti-apoptotic Bcl-2 proteins by displacing sequestered Bax and Bak, facilitating their activation and MOMP.
Apoptosis plays a key role in the maintenance of cellular homeostasis by removing damaged or compromised cells, such as tumour cells. The overexpression of pro-survival members of the Bcl-2 family of proteins (e.g. Bcl-2, Bcl-xL, Mcl-1) occurs in several types of tumours, including haematopoietic and lymphoid cancers, and is often correlated with poor survival [Citation20]. The overexpression and anti-apoptosis action of Bcl-2 appears to co-operate to facilitate proto-oncogene MYC-driven cell transformation or tumorigenesis [Citation21].
Although the role of Bcl-2 family proteins in the regulation of apoptosis is well established [Citation22], their role during cellular responses to hyperthermia is not entirely clear. This study investigates the effect of hyperthermia at 42° and 43 °C on the expression of several pro-apoptotic and anti-apoptotic Bcl-2 family proteins that are involved in the mitochondrial pathway of apoptosis in HeLa cells. The ability of a low dose adaptive survival response, mild thermotolerance induced at 40 °C, to reverse hyperthermia-induced changes in Bcl-2 family proteins is also evaluated. The Bcl-2/Bcl-xL inhibitor ABT-737 will be used to assess the role of these anti-apoptotic proteins in modulating hyperthermia-induced apoptosis. Moreover, the role of reactive oxygen species (ROS) and p53 in mediating changes in the expression of Bcl-2 family proteins will be clarified using the antioxidant PEG-catalase and the p53 inhibitor pifithrin-α, respectively. This study could provide the rationale to explore the manipulation of Bcl-2 family proteins for increasing tumour sensitivity to hyperthermia.
Materials and methods
Cell culture
Human cervical adenocarcinoma HeLa cells (ATCC no.CCL-2) were cultured as monolayers in Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% fetal bovine serum (PAA Laboratories, Toronto, ON), L-glutamine (2 mM) and sodium pyruvate (1 mM) [Citation8]. Cells were maintained in tissue culture flasks (Sarstedt, Saint-Laurent, QC) at 37 °C in a humidified atmosphere of 5% CO2. Culture medium was replaced with fresh medium 24 h before experiments. To induce thermotolerance, confluent cells were transferred to a CO2 incubator for 3 h at 40 °C (±0.1 °C) following a period of 20 min to allow culture medium to reach 40 °C. Cells were harvested using 0.5 mg/mL trypsin/0.2 mg/mL EDTA in phosphate-buffered saline (PBS) and washed by centrifugation (1,000 × g, 3 min). There was no loss of cell viability in cells heated at 40 °C for 3 h (trypan blue exclusion).
Heat treatment
Freshly harvested thermotolerant (3 h at 40 °C) and non-thermotolerant cells (3 h at 37 °C) were heated for 3 h at 42° or 43 °C, relative to controls (37 °C), in temperature-controlled precision waterbaths (±0.02 °C) (Haake D8, Fisher Scientific, Montreal, QC) [Citation8]. One mL of cell suspension reached a temperature within 0.1 °C of the waterbath temperature within 3 min.
Treatment with inhibitors
The Bcl-2 inhibitor ABT-737 (1 µM) (Selleck Chemicals, Houston, TX) was added to cells 24 h before experiments. The p53 inhibitor pifithrin-α (10 µM) and antioxidant polyethylene glycol (PEG)-catalase (300 µM) were added to cells 1 h and 3 h, respectively, prior to experiments. In Hela cells, PEG-catalase increased intracellular catalase activity by 47%, from 3.71 ± 0.67 to 6.20 ± 0.30 μmol/min/106 cells (n = 3).
Preparation of whole cell lysates
Cells were washed by centrifugation (1000 × g, 3 min) in buffer A (100 mM sucrose, 1 mM EGTA, 20 mM MOPS, pH 7.4) [Citation8,Citation23]. The supernatant was discarded, pelleted cells were resuspended in lysis buffer B (buffer A plus 5% Percoll, 0.01% digitonin, 1 mM phenylmethylsulfonyl fluoride (PMSF) and a cocktail of protease inhibitors: 10 µM aprotinin, 10 µM pepstatin A, 10 µM leupeptin, 25 µM calpain inhibitor I, pH 7.4), and then incubated on ice for 1 h. Whole cell lysates were isolated in the supernatant by a 10 min centrifugation step at 2500 × g to remove nuclei and unbroken cells.
Subcellular fractionation: isolation of mitochondrial and cytosolic fractions
Subcellular fractionation was performed as described previously [Citation8,Citation23], with modifications. Cells were washed in buffer A, and then resuspended in buffer B containing 0.1 mM dithiothreitol (DTT). Membranes were broken using a dounce homogeniser (200 strokes/sample). After 30 min incubation on ice, debris and unbroken cells were removed by centrifugation (500 × g, 10 min) and then supernatants were centrifuged (2500 × g, 5 min) to separate nuclei (pellet). Supernatants were then centrifuged (15,000 × g, 15 min) to separate mitochondria. The pellet containing the mitochondrial fraction was then resuspended in buffer C (300 mM sucrose, 1 mM EGTA, 20 mM MOPS, cocktail of protease inhibitors, pH 7.4) containing 0.1 mM DTT. Supernatants were further centrifuged (100,000 × g, 1 h) to separate the cytosolic fraction (supernatant). The purity of cytosolic and mitochondrial fractions was confirmed by western blotting using glutathione S-transferase (GST-π1) (Calbiochem, La Jolla, CA, USA) and cytochrome oxidase (Molecular Probes, Eugene, OR), respectively.
Western blot analysis
Proteins (30 µg) [Citation24] were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (8–15%) [Citation25] and immunodetected using primary antibodies (1:1000) recognising Bcl-2, Bcl-xL, Bax, Bak, Puma and Noxa (Santa Cruz Biotechnology, Santa Cruz, CA) [Citation8]. Horseradish peroxidase (HRP)-conjugated polyclonal secondary antibodies (1:10000) were from Biosource (Camarillo, CA) and Santa Cruz Biotechnology. Protein expression was analysed relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) loading controls, using a laser scanning densitometer (Alpha Innotech, San Leandro, CA) and Fluorchem and QuantityOne software.
Caspase activity
Caspase activity was determined in cell lysates using the substrates (200 µM) Ac-Asp-Glu-Val-Asp-amino-4-methyl-coumarin (Ac-DEVD-AMC) for caspase-3 and Ac-Leu-Glu-His-Asp-amido-4-trifluoromethylcoumarin (Ac-Leu-Glu-His-Asp-AFC) for caspase-9 (Calbiochem) [Citation8]. The kinetic reaction for caspase activity was followed at respective excitation and emission wavelengths of 380 and 460 nm for caspase-3, and 400 and 505 nm for caspase-9, using a spectrofluorimeter (Spectra Max Gemini, Molecular Devices, Sunnyvale, CA).
Chromatin condensation
Apoptotic and necrotic cells were visualised by fluorescence microscopy (model IM, Carl Zeiss Canada, St Laurent, QC) using Hoechst 33 258 (5 µg/mL) (Sigma-Aldrich), which binds to condensed chromatin in the nucleus of apoptotic cells, and propidium iodide (PI) (50 µg/mL), respectively [Citation8]. For each condition, at least 300 cells were counted.
Statistics
Data represent means ± SEM from at least three independent experiments. Comparisons of mean values with the control were analysed by the Student’s bilateral t-test. The Bonferroni-Holmes stepwise adjustment was used to control for the Family-wise error rate at a desired level (α = 5%). Comparisons among multiple groups were made by one-way ANOVA, which measures the linear contrast of means, with either Bonferroni or Dunnett adjustments. Software used was JMP Statistical Discovery 4.0 (SAS Institute, Cary, NC) and GraphPad Prism5 (San Diego, CA). For significant differences, p < 0.05.
Results
Hyperthermia (42–43 °C) causes pro-apoptotic changes in total cellular levels of Bcl-2 family proteins
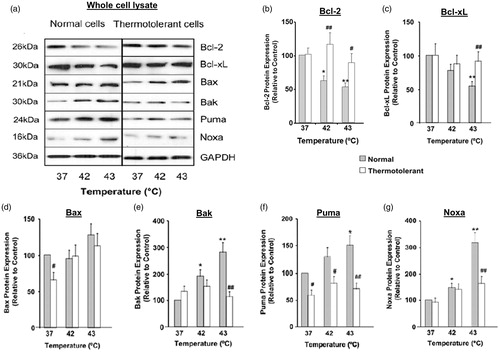
The ability of hyperthermia (42–43 °C) to alter the total protein expression of several different Bcl-2 family proteins was evaluated in whole cell lysates of HeLa cells (). Indeed, hyperthermia caused significant decreases in total expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL, compared to untreated controls (37 °C) (). In contrast, total cellular expression of the pro-apoptotic proteins Bax, Bak, Puma and Noxa increased during high heat stress at 42–43 °C (). Hyperthermia (42–43 °C) therefore changes the overall cellular ratio between these anti-apoptotic and pro-apoptotic Bcl-2 family proteins, which would sensitise cells to apoptosis.
Figure 1. Heat shock (42–43 °C) alters the cellular balance between pro-apoptosis and anti-apoptosis Bcl-2 family proteins: protective role of thermotolerance (40 °C). Non-thermotolerant (3 h at 37 °C) and thermotolerant (3 h at 40 °C) cells were exposed to heat shock (42–43 °C) for 3 h. (a) Western blots for protein expression in whole cell lysates are representative of at least three independent experiments. Means and SEM are shown for densitometric analysis of proteins (b) Bcl-2, (c) Bcl-xL, (d) Bax, (e) Bak, (f) Puma, (g) Noxa. Protein expression in thermotolerant and non-thermotolerant cells was normalised to GAPDH loading controls and is relative to non-thermotolerant controls at 37 °C (100%). For significant differences between heated (42–43 °C) cells and the control (37 °C), p < 0.05 (*), p < 0.001 (**). For significant differences between thermotolerant and non-thermotolerant cells at each specific temperature, p < 0.05 (#), p < 0.001 (##).
Mild thermotolerance developed at 40 °C reverses hyperthermia-induced pro-apoptotic changes in total cellular expression of Bcl-2 family proteins
Next we examined whether mild thermotolerance (40 °C) could protect cells against hyperthermia (42–43 °C)-induced pro-apoptotic changes in total cellular expression of the Bcl-2 family proteins. Indeed, mild thermotolerance reversed the hyperthermia-induced decreases in total cellular levels of anti-apoptotic proteins Bcl-2 (at 42 and 43 °C) and Bcl-xL (at 43 °C) (). Furthermore, the hyperthermia-induced increases in expression of pro-apoptotic proteins Bak, Puma and Noxa were reversed by preconditioning of cells at 40 °C (). The increase in Bax expression was not reversed at 40 °C (). Therefore, mild thermotolerance (40 °C), an adaptive survival response, protected cells against pro-apoptotic changes in total levels of several Bcl-2 family proteins triggered by hyperthermia (42–43 °C), thus maintaining cellular homeostasis.
The development of thermotolerance at mild, fever-range temperatures such as 39.5 °C to 40 °C leads to the accumulation of different heat shock proteins (HSP) such as Hsp27, Hsp32, Hsp60, Hsp70, Hsp90 and Hsp110 [Citation9,Citation26]. To our knowledge, it is not known whether mild thermotolerance can alter the expression of Bcl-2 family proteins. Thermotolerance (40 °C) itself caused only minor changes in the expression of Bcl-2 family proteins in non-heated cells (). The basal levels of the pro-apoptotic proteins Bax and Puma decreased in thermotolerant (40 °C, 3 h) cells compared to normal (37 °C, 3 h) cells (). Cellular levels of Bcl-2, Bcl-xL, Bak or Noxa did not change in thermotolerant (40 °C, 3 h) cells ().
Hyperthermia (42–43 °C) causes subcellular relocalisation of Bcl-2 family proteins between mitochondrial and cytosolic compartments
The level of protein expression and subcellular localisation of different Bcl-2 family proteins is critical to their function during stress-induced apoptosis [Citation17]. Hyperthermia (42–43 °C) altered the subcellular localisation of several Bcl-2 family proteins in HeLa cells (, ). Levels of anti-apoptotic proteins Bcl-2 and Bcl-xL decreased in the mitochondrial fraction, relative to controls (37 °C) (). Hyperthermia (42–43 °C) also decreased Bcl-xL levels in the cytosol ().
Figure 2. Heat shock (42–43 °C) causes relocalisation of Bax, Bak, Bim, Puma and Noxa to mitochondria while expression of Bcl-2 and Bcl-xL decreases: reversal by thermotolerance (40 °C). Non-thermotolerant (3 h at 37 °C) or thermotolerant (3 h at 40 °C) cells were heated (42–43 °C) for 3 h. (a) Western blots for protein expression in mitochondrial fractions are representative of at least three independent experiments. (B) The purity of cytosolic and mitochondrial fractions was confirmed using GST-π1 and cytochrome c oxidase (Cox2) antibodies, respectively. The average purity of the cytosolic fraction from at least four separate experiments was 89.72 ± 1.49% and that of the mitochondrial fraction was 92.33 ± 2.57%. Means and SEM are shown for densitometric analysis of (c) Bcl-2, (d) Bcl-xL, (e) Bax, (f) Bak, (g) Puma, (h) Noxa. Protein expression is relative to non-thermotolerant controls at 37 °C (100%). For significant differences between heated (42–43 °C) cells and control (37 °C), p < 0.05 (*), p < 0.001 (**). For significant differences between thermotolerant and non-thermotolerant cells at each specific temperature, p < 0.05 (#), p < 0.001 (##).
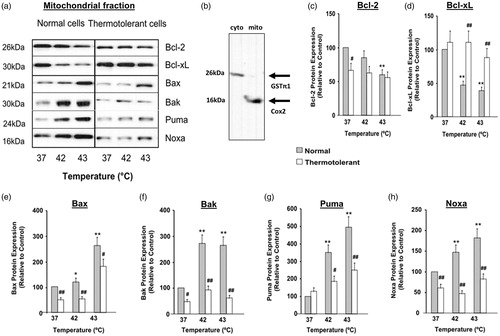
Figure 3. Decreased expression of Bcl-2 family proteins in cytosolic fractions during heat shock (42–43 °C): reversal by 40 °C thermotolerance. Thermotolerant (3 h at 40 °C) and non-thermotolerant (3 h at 37 °C) cells were heated (42–43 °C) for 3 h. (a) Western blots for protein expression in cytosolic fractions are representative of at least three independent experiments. + is a positive control for Bcl-2 expression in whole cell lysates. Means and SEM are shown for densitometric analysis of (b) Bcl-xL, (c) Bax, (d) Puma, (E) Noxa. Protein expression is relative to non-thermotolerant controls at 37 °C (100%). For significant differences between hyperthermia-treated (42–43 °C) cells and the control (37 °C), p < 0.05 (*), p < 0.001 (**). For significant differences between thermotolerant and non-thermotolerant cells at each specific temperature, p < 0.05 (#), p < 0.001 (##).
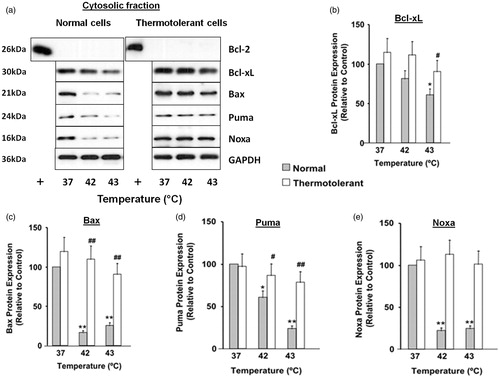
For the pro-apoptotic proteins, hyperthermia (42–43 °C) caused significant increases in levels of Bax, Bak, Puma and Noxa in the mitochondrial fraction (). There were corresponding decreases in cytosolic levels of Bax, Puma and Noxa, relative to controls (37 °C) (). Together, these findings show that hyperthermia (42–43 °C) perturbs the equilibrium between anti-apoptotic and pro-apoptotic Bcl-2 family proteins in the mitochondrial compartment, in favour of pro-apoptotic conditions.
Thermotolerance developed at 40 °C reverses hyperthermia (42–43 °C)-induced changes in subcellular localisation of Bcl-2 family proteins
Subsequently, the ability of mild thermotolerance (40 °C) to reverse the hyperthermia (42–43 °C)-induced alterations in subcellular distribution of Bcl-2 family proteins was examined (, ). In thermotolerant cells the hyperthermia (42–43 °C)-induced decreases in levels of anti-apoptotic protein Bcl-xL were reversed in the mitochondrial () and cytosolic () fractions. However at 40 °C, mitochondrial levels of Bcl-2 were not restored to control levels (). For pro-apoptotic proteins there was pronounced reversal of hyperthermia (42–43 °C)-induced changes in mitochondrial () and cytosolic () levels of Bax, Puma and Noxa following mild heat preconditioning at 40 °C. Mitochondrial levels of Bak were also reversed at 40 °C (). Therefore, mild thermotolerance (40 °C) protected cells against hyperthermia (42–43 °C)-induced alterations in the distribution of diverse Bcl-2 family proteins between the cytosolic and mitochondrial compartments, thus favouring anti-apoptotic conditions and cell survival.
Implication of ROS and p53 in the expression of Bcl-2 family proteins during hyperthermia
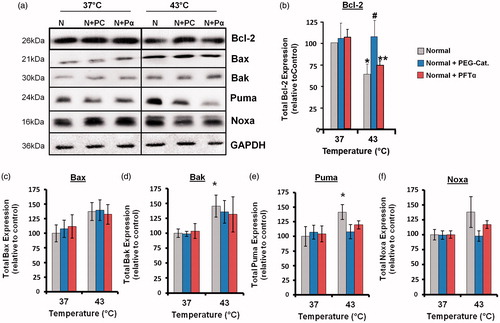
ROS and p53 are known to have a role in the activation of apoptosis [Citation26,Citation27]. To investigate whether these molecules could affect levels of Bcl-2 family proteins, cells were incubated with the antioxidant PEG-catalase (PC) or the p53 inhibitor pifithrin-α (Pα). In whole cell lysates, the hyperthermia (43 °C)-induced decrease in Bcl-2 expression was inhibited by PEG-catalase (). The hyperthermia (43 °C)-induced increases in expression of the pro-apoptotic proteins Puma and Noxa were inhibited by both PEG-catalase and pifithrin-α (). The cellular levels of Bcl-xL (data not shown), Bax and Bak () were not affected by these inhibitors.
Figure 4. Hyperthermia-induced down-regulation of Bcl-2 involves ROS, and up-regulation of Puma and Noxa involves p53 and ROS. Cells were pretreated with PEG-catalase (PC) or pifithrin-α (Pα) and then heated (43 °C) for 3 h. (a) Western blots for protein expression in whole cell lysates are representative of at least three independent experiments. Means and SEM are shown for densitometric analysis of proteins: (b) Bcl-2, (c) Bax, (d) Bak, (e) Puma, (f) Noxa. Protein expression was normalised to GAPDH loading controls and is relative to non-treated controls at 37 °C. For significant differences between heated (43 °C) cells and the control (37 °C), p < 0.05 (*). For significant differences between treated and non-treated cells at each specific temperature, p < 0.05 (#).
Activation of the mitochondrial pathway of apoptosis by hyperthermia (42–43 °C)
Hyperthermia (42–43 °C)-induced pro-apoptotic changes in Bcl-2 family proteins at the level of mitochondria were reflected by activation of caspase-9, the initiator caspase in mitochondrial apoptosis (). Hyperthermia also activated caspase-3 () and caused apoptosis (chromatin condensation) (). Caspase activation and apoptosis were significantly diminished in thermotolerant (40 °C) cells (). Levels of necrotic cells were very low ().
Figure 5. Bcl-2 inhibitor ATB-737 enhances hyperthermia-induced apoptosis: protective role of mild thermotolerance (40 °C). Relative activities of caspase-9 (a) and caspase-3 (b), and levels of apoptosis (c) and (d) necrosis in thermotolerant (3 h at 40 °C) and non-thermotolerant (3 h at 37 °C) cells, with or without pretreatment with ABT-737 (1 µM). Means ± SEM are from at least three independent experiments. For significant differences between heated (42–43 °C) cells and the non-thermotolerant control (37 °C), p < 0.05 (*), p < 0.001 (**). For significant differences between thermotolerant and non-thermotolerant cells at each temperature, p < 0.05 (#), p < 0.001 (##). For cells with or without ABT-737, p < 0.05 (+), p < 0.01 (++), p < 0.001 (+++).
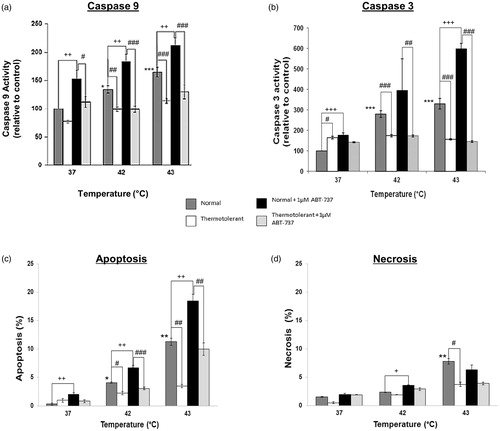
Sensitisation to hyperthermia-induced apoptosis by Bcl-2/Bcl-xL inhibitor ABT-737: protective effect of mild thermotolerance (40 °C)
To clarify the role of anti-apoptotic Bcl-2 family proteins in hyperthermia-induced apoptosis, cells were treated with ABT-737 [Citation28]. The activation of caspase-9 () and caspase-3 () by hyperthermia (42–43 °C), as well as the induction of apoptosis () were significantly increased by ABT-737. In addition, ABT-737 increased caspase activation and apoptosis in non-heated cells at 37 °C (). The inhibitor had little effect on the level of necrotic cells, which were low (). Mild thermotolerance (40 °C) proved once more to be effective by decreasing caspase-9 and -3 activation and the induction of apoptosis by hyperthermia (42–43 °C), even in the presence of the inhibitor ().
Discussion
This study shows that hyperthermia (42–43 °C) perturbed the equilibrium between several anti-apoptotic and pro-apoptotic Bcl-2 family proteins in HeLa cells, thus favouring pro-apoptotic conditions. The equilibrium was perturbed with respect to total Bcl-2 protein expression, as well as their levels in mitochondria. Hyperthermia (42–43 °C) triggered overall pro-apoptotic conditions by decreasing total cellular expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, while the expression of pro-apoptotic proteins Bax, Bak, Puma and Noxa was increased (). The imbalance in the expression of Bcl-2 family proteins in favour of the proapoptotic members resulted in activation of caspase-9, the initiator caspase downstream from mitochondria, and the activation of execution phase events including caspase-3 activation and nuclear chromatin condensation ().
Scheme 1. Heat shock (42–43 °C) alters the pro-apoptosis/anti-apoptosis balance in Bcl-2 family proteins during apoptosis in HeLa cells. (1) Heat shock induced translocation of Bax, Puma and Noxa to mitochondria. (2) Heat shock decreased levels of Bcl-2 and Bcl-xL in mitochondria. (3) Heat shock increased the pro-apoptosis/anti-apoptosis balance in Bcl-2 family proteins in mitochondria, which led to cytochrome c release, activation of caspase-9 and caspase-3, and execution of apoptosis. (4) Thermotolerance (TT) at 40 °C decreased the translocation of Bax, Noxa and Puma to mitochondria, and inhibited cytochrome c release, caspase activation and apoptosis. (5) Inhibition of the anti-apoptotic proteins Bcl-2 and Bcl-xL by ABT-737, preventing them from interfering with heat shock-induced apoptosis.
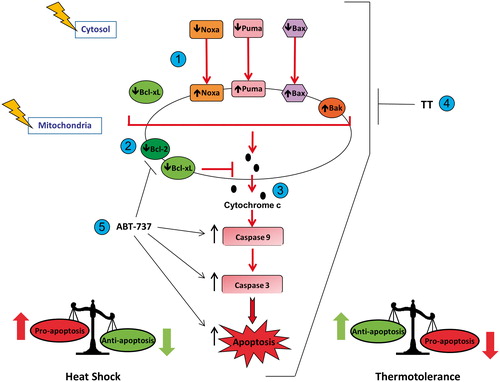
The hyperthermia (42–43 °C)-induced increases in the expression of Puma and Noxa in HeLa cells were inhibited by pifithrin-α, which indicates that they were dependent on p53 transactivation in the nucleus. Noxa is a p53-inducible gene that antagonises Mcl-1 and A1, whereas Puma can bind to all of the anti-apoptotic Bcl-2 family proteins [Citation29]. The induction of Puma expression releases cytosolic p53 from binding to Bcl-xL, which keeps it inactive in the cytosol [Citation27]. Once released, cytosolic p53 can then promote Bax oligomerisation and translocation to mitochondria. Earlier, we reported that hyperthermia (42–45 °C) causes Bax translocation to mitochondria, MOMP and cytochrome c release from mitochondria in HeLa and CHO cells [Citation8].
Pifithrin-α is known to block p53-dependent transactivation of apoptotic genes [Citation30]. The transcription factors p63 and p73 are homologues of p53. They can activate most of p53’s target genes, including those responsible for apoptosis [Citation31]. p73 can directly transactivate PUMA to induce apoptosis [Citation32]. p63 and p73 can also promote apoptosis through transcription-independent mechanisms. They can bind to different members of the Bcl-2 family: p63 and p73 can bind to the anti-apoptotic proteins Bcl-xL and Mcl-1 and p63 to the pro-apoptotic protein Bak [Citation33]. Due to their high degree of similarity, it is possible that the p53 inhibitor pifithrin-α could have an effect on these homologues. Pifithrin-α was shown to act on p73 in zebra fish embryo [Citation34]; however, its effect on p63 and p73 in mammalian cells is not known. We cannot rule out the possibility that pifithrin-α is inhibiting not only p53, but also p63 and p73. This would not change the outcome of the data and would mean that the effects are due to the inhibition of several p53 members and not only p53 alone.
The increases in the expression of Puma and Noxa in HeLa cells were inhibited by PEG-catalase, which implies that ROS are involved in their activation by hyperthermia. Hyperthermia can increase the generation of ROS [Citation35], which are known to up-regulate the expression of Noxa and Puma [Citation36,Citation37]. In addition, the hyperthermia (42–43 °C)-induced decrease in Bcl-2 expression was reversed by PEG-catalase in HeLa cells. ROS can play a pro-apoptotic role by causing down-regulation and degradation of the Bcl-2 protein through the ubiquitin-proteasome pathway [Citation38].
Cancer cells have deregulated many of the physiological mechanisms that tightly regulate the different apoptotic signalling pathways in order to evade death by apoptosis [Citation39]. For example, mechanisms have evolved to inactivate pro-apoptotic molecules and many anti-apoptotic factors are expressed at high levels in cancer cells to confer resistance to apoptosis. Therefore, anti-apoptosis proteins such as Bcl-2 can promote tumour cell survival, which suggests that impaired apoptosis could be one of the critical steps in tumour formation and progression [Citation20]. Furthermore, tumour cells are often inherently insensitive to cancer treatments such as chemotherapy. Multiple mechanisms appear to be involved, which include the increased expression of drug efflux transporters and cellular detoxification systems, altered drug targets, activation of pro-survival pathways, and enhanced repair of DNA damage [Citation40]. Drug resistance could also be linked to defects in the apoptosis machinery, thus favouring tumour cell survival [Citation39]. The results from this study showed that hyperthermia (>42 °C) caused an imbalance between several members of the Bcl-2 family in favour of pro-apoptotic conditions. The establishment of pro-apoptotic conditions could be beneficial for the sensitisation of cancer cells to the cytotoxic effects of radiation and chemotherapy treatments by hyperthermia [Citation1–4].
It is not known whether thermotolerant cells induced at mild temperatures such as 40 °C are resistant to chemotherapy and radiation. Thermotolerance induced at higher temperatures (e.g. 43–45 °C) may be associated with different forms of drug resistance including multidrug resistance (MDR) [Citation41]. This did not appear to be the case for resistance to radiation.
It is well established that hyperthermia causes protein degradation but can also cause conformational changes to proteins that could expose previously concealed regions [Citation6]. Conformational changes to proteins during hyperthermia could alter binding interactions between different Bcl-2 family proteins and facilitate the activation of BH3-only proteins such as Bax and Bak. Indeed, heat (43 °C, 1 h) directly activated recombinant Bax and Bak in isolated mitochondria, resulting in a loss of mitochondrial membrane potential and cytochrome c release [Citation42].
Several studies have examined the role of Bcl-2 family proteins in hyperthermia-induced cell death, but there is no clear consensus. It was reported that hyperthermia causes apoptosis through a caspase-2/Bid pathway with activation of Bax/Bak-dependent mitochondrial apoptosis in mouse embryonic fibroblasts (MEFs) [Citation43]. Another study reported that hyperthermia-induced apoptosis was dependent on Bim through a Bax/Bak-dependent pathway in MEFs [Citation44]. These different pathways appear to act independently and in parallel and/or their respective roles could depend on cell type and severity of heat stress. In Jurkat cells, however, caspase-9 activation was essential for hyperthermia-induced apoptosis, but not caspase-8 or caspase-2 [Citation45]. The anti-apoptotic protein Mcl-1 appears to be a critical heat-sensitive step leading to Bax activation in the human acute lymphoblastic T-cell line (PEER) [Citation46].
The overexpression of several anti-apoptotic proteins protected cells against hyperthermia-induced apoptosis in several cell types. Bcl-2 overexpression inhibited apoptosis induced by hyperthermia (44 °C, 40 min) in mouse haematopoietic cell lines [Citation47]. Different leukaemic haematopoietic cells were less sensitive to hyperthermia-induced apoptosis, due to an imbalance in expression of the Bcl-2 family proteins in favour of pro-apoptotic Bcl-2 proteins [Citation48]. Overexpression of Bcl-xL in murine IL-3-dependent prolymphoid progenitor cells (FL5.12) prevented the induction of apoptosis by acute heat stress (42 °C, 1 h) [Citation49]. Jurkat cells overexpressing Bcl-2/Bcl-xL were resistant to apoptosis induced by hyperthermia (44 °C, 1 h) [Citation45].
While the exposure to higher doses of hyperthermia (>42 °C) is cytotoxic to cancer cells [Citation3], the preconditioning of cells by lower doses of heat induces thermotolerance [Citation8,Citation11–14]. This study shows that mild thermotolerance induced at 40 °C in HeLa cells reversed the hyperthermia (42–43 °C)-induced disruption in the anti-apoptosis/pro-apoptosis balance between Bcl-2 family proteins. This protective, pro-survival effect occurred at both the cellular and mitochondrial levels. Low dose exposure to different stresses, including hyperthermia, can lead to adaptive survival responses that enable cells and organisms to continue normal function in the face of a toxic insult [Citation50]. Adaptive responses often involve the induction of defence systems (e.g. HSPs, antioxidants, anti-apoptotic proteins) that allow cells to protect themselves against different toxic and environmental stresses [Citation51]. However, if the adaptive response cannot protect the cell against an adverse stress exposure, then the damaged cell will be removed, for example, by apoptosis.
In mild thermotolerant (40 °C) HeLa cells, the protective reversal of changes in the balance between pro-apoptotic and anti-apoptotic Bcl-2 family proteins may be related to HSPs. The HSPs 27, 32, 60, 72, 90 and 110 were induced at 40 °C in HeLa cells [Citation8]. HSPs have a complex role in the regulation of apoptosis [Citation52]. Hsp70 is able to modulate Bcl-2-dependent apoptosis [Citation53]. Hsp70 and Hsp27 can inhibit apoptosis by interfering with events upstream of MOMP that ultimately suppress the activation of Bax, thereby inhibiting the release of pro-apoptotic factors such as cytochrome c from mitochondria [Citation54,Citation55]. This appears to involve Hsp70-mediated inhibition of c-Jun N-terminal kinase (JNK), which can activate pro-apoptotic Bcl-2 family proteins such as tBid that are able to promote Bax and Bak activation and induce MOMP [Citation5,Citation56,Citation57]. More recently, the overexpression of Hsp70 was shown to stabilise Mcl-1 protein levels and prevented Bax activation by hyperthermia [Citation46]. This resulted from reduced Mcl-1 ubiquitination and degradation, as well as enhanced Mcl-1 expression. In addition, Hsp70 overexpression allowed for new synthesis to replace degraded Mcl-1 [Citation46]. Hyperthermia (40–44 °C)-induced apoptosis was inhibited by pre-inducing Hsp70 in H9c2 cells [Citation58]. Findings from these different studies suggest that several HSPs could prevent the activation and mitochondrial translocation of pro-apoptotic proteins such as Bax and tBid, and maintain levels of anti-apoptotic proteins such as Bcl-2 and Mcl-1, and hence mitochondrial integrity, during hyperthermia.
The use of inhibitors of anti-apoptotic Bcl-2 family proteins has long been considered for bypassing the immortality of cancer cells. The small molecule inhibitor ABT-737 is a Bcl-2 homology domain 3 (BH3)-mimetic that binds strongly to Bcl-2, Bcl-xL and Bcl-w through an interaction mediated by the BH3 domain [Citation28,Citation59,Citation60]. This interaction releases pro-apoptotic proteins such as Bax/Bak from the complexes they form with their anti-apoptotic counterparts, allowing them to trigger apoptosis. This study shows that ABT-737 alone activated caspase-9 and caspase-3 and triggered apoptosis in HeLa cells. ABT-737 enhanced caspase-9 and caspase-3 activation at 42 and 43 °C and sensitised cells to hyperthermia-induced apoptosis. ABT-737 also sensitised MEFs to hyperthermia (44 °C, 1.5 h) [Citation44]. Since the pro-apoptotic protein Bim is critical for hyperthermia-induced killing in MEFs, it was suggested that ABT-737 could liberate Bim from Bcl-2 or Bcl-xL, which in turn could activate Bax and/or Bak. These studies emphasise the importance of the anti-apoptotic role of Bcl-2 during hyperthermia-induced apoptosis. ABT-737 is effective as a single agent against cancer cell lines including leukaemias, lymphomas, neuroblastoma and small cell lung carcinoma, primary tumour cells and animal models, but it is also useful in combination with other anticancer treatments such as radiation and chemotherapy [Citation28,Citation61,Citation62]. The oral analogue of ABT-737 (ABT-263, Navitoclax) has now entered phase I/II clinical trials [Citation63,Citation64]. The combined use of hyperthermia with an inhibitor such as ABT-737 could be a promising strategy to trigger apoptosis for the elimination of tumour cells. However, our results show that ABT-737 was unable to block the protective effects of thermotolerance (40 °C) against hyperthermia (42–43 °C)-induced apoptosis in HeLa cells. This suggests that ABT-737 cannot sensitise cells that overexpress HSPs to hyperthermia. In addition, ABT-737 does not inhibit the anti-apoptotic proteins Mcl-1 or A1 [Citation59]. Given that many tumours overexpress HSPs [Citation65], strategies that combine hyperthermia and ABT-737 with approaches that target HSPs and Mcl-1 could be envisaged.
Conclusion
This study establishes that (1) hyperthermia (42–43 °C) created an imbalance between members of the Bcl-2 family of proteins in favour of pro-apoptotic conditions at the cellular and mitochondrial levels, (2) thermotolerance, induced by mild hyperthermia (40 °C), reversed the imbalance between pro-apoptotic and anti-apoptotic Bcl-2 proteins that was triggered by hyperthermia (42–43 °C), (3) inhibition of anti-apoptotic Bcl-2 family proteins by ABT-737 sensitised HeLa cells to hyperthermia-induced apoptosis. These findings improve our understanding about hyperthermia-induced apoptosis, as well as the protective ability of adaptive survival responses such as thermotolerance against toxic stresses.
Declaration of interest
The University Mission of Tunisia in North America provided PhD scholarship support (to A.B.), the Society for Thermal Medicine (STM) awarded a student travel award to attend the 30th Annual conference of STM in April 2013 (to A.G.). Contract grant sponsor was NSERC Canada; Contract grant number: 36725-11 (to D.A.A.B.). The authors alone are responsible for the content and writing of the paper.
Acknowledgements
The authors thank Bertrand Fournier (Service de consultation en analyse de données, Université du Québec à Montréal) for statistical analyses.
References
- Horsman MR, Overgaard J. Hyperthermia: A potent enhancer of radiotherapy. Clin Oncol (R Coll Radiol) 2007;19:418–26
- Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer 2008;44:2546–54
- van der Zee J. Heating the patient: A promising approach? Ann Oncol 2002;13:1173–84
- van der Zee J, Vujaskovic Z, Kondo M, Sugahara T. The Kadota Fund International Forum 2004 – Clinical group consensus. Int J Hyperthermia. 2008;24:111–22
- Milleron RS, Bratton SB. ‘Heated’ debates in apoptosis. Cell Mol Life Sci 2007;64:232:9–33
- Lepock JR. How do cells respond to their thermal environment? Int J Hyperthermia 2005;21:681–7
- Richter K, Haslbeck M, Buchner J. The heat shock response: Life on the verge of death. Mol Cell 2010;40:253–66
- Bettaieb A, Averill-Bates DA. Thermotolerance induced at a mild temperature of 40 degrees C protects cells against heat shock-induced apoptosis. J Cell Physiol 2005;205:47–57
- Bettaieb A, Averill-Bates DA. Thermotolerance induced at a fever temperature of 40 degrees C protects cells against hyperthermia-induced apoptosis mediated by death receptor signalling. Biochem Cell Biol 2008;86:521–38
- Shellman YG, Howe WR, Miller LA, Goldstein NB, Pacheco TR, Mahajan RL, et al. Hyperthermia induces endoplasmic reticulum-mediated apoptosis in melanoma and non-melanoma skin cancer cells. J Invest Dermatol 2008;128:949–56
- Subjeck JR, Sciandra JJ, Johnson RJ. Heat shock proteins and thermotolerance: A comparison of induction kinetics. Br J Radiol 1982;55:579–84
- Landry J, Bernier D, Chretien P, Nicole LM, Tanguay RM, Marceau N. Synthesis and degradation of heat shock proteins during development and decay of thermotolerance. Cancer Res 1982;42:2457–61
- Przybytkowski E, Bates JH, Bates DA, Mackillop WJ. Thermal adaptation in CHO cells at 40 degrees C: The influence of growth conditions and the role of heat shock proteins. Radiat Res 1986;107:317–31
- Singh IS, Hasday JD. Fever, hyperthermia and the heat shock response. Int J Hyperthermia 2013;29:423–35
- Gill RR, Gbur CJ Jr, Fisher BJ, Hess ML, Fowler AA III, Kukreja RC, et al. Heat shock provides delayed protection against oxidative injury in cultured human umbilical vein endothelial cells. J Mol Cell Cardiol 1998;30:2739–49
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: Signaling for suicide and survival. J Cell Physiol 2002;192:1–15
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 2009;122:437–41
- Youle RJ, Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008;9:47–59
- Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol 2009;296:C941–53
- Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 2008;7:989–1000
- Shortt J, Johnstone RW. Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol 2012;4:a009829
- Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: A review. Biochim Biophys Acta 2011;1813:238–59
- Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999;18:2040–8
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–5
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000;5:415–18
- Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. The role of p53 in apoptosis. Discov Med 2010;9:145–52
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677–81
- Zhang LN, Li JY, Xu W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther 2013;20:1–7
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999;285:1733–7
- Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010;2:a004887
- Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, et al. p73 induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem 2004;279:8076–83
- Muller M, Schleithoff ES, Stremmel W, Melino G, Krammer PH, Schilling T. One, two, three – p53, p63, p73 and chemosensitivity. Drug Resist Updat 2006;9:288–306
- Davidson W, Ren Q, Kari G, Kashi O, Dicker AP, Rodeck U. Inhibition of p73 function by Pifithrin-alpha as revealed by studies in zebrafish embryos. Cell Cycle 2008;7:1224–30
- Bettaieb A WP, Averill-Bates DA. Hyperthermia: Cancer treatment and beyond. In: Rangel L, editor. Cancer Treatment – Conventional and Innovative Approaches. Book 2: InTech Open Access 2013, pp. 257–83. doi: 10.5772/45937
- Tonino SH, van Laar J, van Oers MH, Wang JY, Eldering E, Kater AP. ROS-mediated upregulation of Noxa overcomes chemoresistance in chronic lymphocytic leukemia. Oncogene 2011;30:701–13
- Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene 2008;27:S71–83
- Azad N, Iyer A, Vallyathan V, Wang L, Castranova V, Stehlik C, et al. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann N Y Acad Sci 2010;1203:1–6
- Fulda S. Targeting apoptosis for anticancer therapy. Semin Cancer Biol 2014. doi: 10.1016/j.semcancer.2014.05.002
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer. 2013;13:714–26
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, et al. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002;43:33–56
- Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, et al. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA 2005;102:17975–80
- Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR, Newmeyer DD. Caspase-2-induced apoptosis requires bid cleavage: A physiological role for bid in heat shock-induced death. Mol Biol Cell 2006;17:2150–7
- Mahajan IM, Chen MD, Muro I, Robertson JD, Wright CW, Bratton SB. BH3-only protein BIM mediates heat shock-induced apoptosis. PloS One 2014;9:e84388
- Shelton SN, Dillard CD, Robertson JD. Activation of caspase-9, but not caspase-2 or caspase-8, is essential for heat-induced apoptosis in Jurkat cells. J Biol Chem 2010;285:40525–33
- Stankiewicz AR, Livingstone AM, Mohseni N, Mosser DD. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ 2009;16:638–47
- Strasser A, Anderson RL. Bcl-2 and thermotolerance cooperate in cell survival. Cell Growth Differ 1995;6:799–805
- Setroikromo R, Wierenga PK, van Waarde MA, Brunsting JF, Vellenga E, Kampinga HH. Heat shock proteins and Bcl-2 expression and function in relation to the differential hyperthermic sensitivity between leukemic and normal hematopoietic cells. Cell Stress Chaperon 2007;12:320–30
- Robertson JD, Datta K, Kehrer JP. Bcl-xL overexpression restricts heat-induced apoptosis and influences Hsp70, Bcl-2, and Bax protein levels in FL5.12 cells. Biochem Biophys Res Commun 1997;241:164–8
- Holsapple MP, Wallace KB. Dose response considerations in risk assessment – An overview of recent ILSI activities. Toxicol Lett 2008;180:85–92
- Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000;50:279–89
- Beere HM. Death versus survival: Functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest 2005;115:2633–9
- Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene 2004;23:2907–18
- Steel R, Doherty JP, Buzzard K, Clemons N, Hawkins CJ, Anderson RL. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J Biol Chem 2004;279:51490–9
- Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 2005;280:38729–39
- Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol 2002;22:3415–24
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol 2002;22:816–34
- Hsu SF, Chao CM, Huang WT, Lin MT, Cheng BC. Attenuating heat-induced cellular autophagy, apoptosis and damage in H9c2 cardiomyocytes by pre-inducing Hsp70 with heat shock preconditioning. Int J Hyperthermia 2013;29:239–47
- Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis 2012;3:e366
- Andreu-Fernandez V, Genoves A, Messeguer A, Orzaez M, Sancho M, Perez-Paya E. BH3-mimetics- and cisplatin-induced cell death proceeds through different pathways depending on the availability of death-related cellular components. PloS One 2013;8:e56881
- Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 2009;9:321–6
- Fang H, Harned TM, Kalous O, Maldonado V, DeClerck YA, Reynolds CP. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin Cancer Res 2011;17:7093–104
- Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 2010;11:1149–59
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 2011;29:909–16
- Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem Sci 2006;31:164–72