
Abstract
Purpose: Hyperthermia is used in combination with conventional anticancer agents to potentiate their cytotoxicity. One of its key events is the synthesis of heat shock proteins (HSPs), which are able to associate with components from DNA repair mechanisms. However, little is known about their relationship with the mismatch repair system (MMR). Our aim was to study the effects of hyperthermia on cisplatin (cPt) sensitivity and to determine whether MLH1 and MSH2 associate with Hsp27 and Hsp72 in MMR-deficient(−)/-proficient(+) cells.
Materials and methods: HCT116+ch2 (MMR−) and HCT116+ch3 (MMR+) cell lines were exposed to cPt with or without previous hyperthermia (42 °C, 1 h). Clonogenic survival assays, MTT, confocal immunofluorescence, immunoprecipitation, immunoblotting and flow cytometry were performed.
Results: Hyperthermia increased the cPt resistance in MMR− cells 1.42-fold. Immunofluorescence revealed that after cPt, Hsp27 and Hsp72 translocated to the nucleus and colocalisation coefficients between these proteins with MLH1 and MSH2 increased in MMR+ cells. Immunoprecipitation confirmed the interactions between HSPs and MMR proteins in control and treated cells. Hyperthermia pretreatment induced cell cycle arrest, increased p73 expression and potentiated cPt sensitivity in MMR+ cells.
Conclusions: This is the first report showing in a MMR−/+ cellular model that MLH1 and MSH2 are client proteins of Hsp27 and Hsp72. Our study suggests that p73 might participate in the cellular response to hyperthermia and cPt in a MMR-dependent manner. Further functional studies will confirm whether HSPs cooperate with the MMR system in cPt-induced DNA damage response or whether these protein interactions are only the result of their chaperone functions.
Introduction
Hyperthermia induces complex cellular responses ranging from protein aggregation, misfolding or unfolding, to organelle fragmentation, changes in protein associations, and reactive oxygen species generation [Citation1,Citation2]. As a consequence of these events, at the genomic level hyperthermia may cause stalling of DNA replication fork and double strand breaks (DSBs), chromosome aberrations [Citation3], micronucleus formation [Citation4], gene amplification [Citation5], and inhibition of DNA repair processes [Citation6].
One of the most extensively described features of the heat shock response is the expression of the highly conserved heat shock proteins (HSPs). HSPs belong to a superfamily sharing a protection function against cellular stress [Citation7]. They are traditionally classified according to their molecular weights as Hsp110 (HSPH), Hsp90 (HSPC), Hsp70 (HSPA), Hsp60 (HSPD), Hsp40 (DNAJ) and small Hsps (HSPB). An updated nomenclature is shown in parentheses [Citation8]. Usually, there are basal levels of HSPs (constitutive or cognate HSPs) in cells, which often perform housekeeping chaperoning functions. They may also participate in cell development, differentiation, survival, ageing and death. HSPs can be induced by several pathological or stressful situations (inducible HSPs) to protect tissues from injury [Citation7]. Hsp27 (HSPB1) and Hsp72 (HSPA1A, inducible form of HSPA) are overexpressed in many tumour cells and are known to have anti-apoptotic properties. In addition, they have been implicated in resistance to antitumour drugs [Citation9,Citation10]. Hsp27 is up-regulated in response to cellular stress, accomplishing multifunctional roles such as increasing intracellular glutathione, antagonising mitochondria-dependent or independent apoptotic pathways or regulating actin cytoskeletal dynamics [Citation11,Citation12]. Hsp70, through its chaperone activity, exerts different cellular functions, such as inhibiting intrinsic and extrinsic apoptotic pathways, inhibiting senescence, promoting immune response, stabilising lysosome function and allowing autophagy [Citation13–15]. There is evidence of Hsp70 also participating in genome stability and DNA repair [Citation16,Citation17].
Over recent years, many studies have reported a link between HSPs and DNA repair pathways, which has become a special topic of interest in oncology research [Citation17]. The participation of Hsp70 in the base excision repair (BER) system has been well established; furthermore, it has been related to nucleotide excision repair (NER) [Citation18–20]. Little is known about the implications of Hsp27 in DNA repair. It has recently been reported that Hsp27 down-regulation impaired DNA DSB repair in irradiated tumour cell lines [Citation21]. In addition, a possible functional relationship between Hsp90 and MLH1, a component of the MMR system, has been proposed as a consequence of cisplatin-induced DNA damage [Citation22]. However, the relationship between Hsp27, Hsp72 and the MMR pathway still remains poorly explored.
Different cellular events occur after hyperthermia that involve multiple targets, among them DNA repair proteins [Citation6]. Although the effect of hyperthermia is pleotropic, certain DNA repair pathways may be directly inhibited by hyperthermia, such as homologous recombination (HR) repair [Citation23,Citation24]. These facts are of special interest, as in some cases the combination of hyperthermia with conventional strategies for cancer treatment has shown the ability to kill tumour cells synergistically [Citation25]. Cisplatin (cis-diamminedichloroplatinum(II), cPt) is one of the most widely used chemotherapeutic agents for the treatment of solid tumours. This drug forms a variety of DNA adducts (monoadducts, intra-strand adducts and inter-strand cross-links) that inhibit and block DNA replication and transcription, leading to cell death [Citation26]. The efficacy of cPt depends not only on its ability to induce DNA damage, but also on an efficient activation of the DNA damage response [Citation27]. Hyperthermia has been reported to enhance intracellular platinum accumulation and inhibit the repair of adducts, particularly in tumour cells with acquired platinum resistance [Citation28]. Hyperthermia effectiveness is, however, still controversial. The cPt-DNA adducts are recognised by MMR [Citation29]. The MMR system corrects mismatches and insertion/deletion loops generated during DNA replication. The MutSα complex (heterodimer of human MutS homologues, MSH2 and MSH6) recognises and binds to mismatches generated by cPt-DNA adducts. MuLα (heterodimer of MLH1 and PMS2) is then recruited to help organise other proteins, coordinating the repair of the lesion [Citation30]. Binding of the MMR complex to cPt–DNA adducts increases cytotoxicity by activating downstream signalling pathways, leading to apoptosis. However, cPt-DNA damage is poorly repaired in MMR-deficient (MMR−) tumour cells; cells proliferate and become resistant to the drug [Citation31].
It has been demonstrated that mild hyperthermia (41–42.5 °C) sensitises HR-proficient tumour cells to PARP1 inhibitors, given that heat shock leads to degradation of BRCA2, a client protein of Hsp90, inhibiting the repair of DSBs [Citation23]. The increasing knowledge of hyperthermia targets and HSPs client proteins will allow hyperthermia to become a powerful tool for cancer treatment. The role of hyperthermia in enhancing cPt delivery and cytotoxicity has been investigated in cPt resistant/sensitive tumour cells [Citation32–34]. Nevertheless, the molecular interactions underlying this enhancement and the implications of hyperthermia on cPt response due to differential MMR phenotype in cancer cells, are not yet fully understood.
Understanding the biological behaviour of HSPs and their target DNA repair proteins is key to optimising the therapeutic benefits of using hyperthermia in combination with cPt in cancer therapy. The interactions of Hsp27 and Hsp72 with their clients may be essential during stress, and even under normal physiological conditions. We have previously reported the nuclear colocalisation between Hsp27 and Hsp72 with MLH1 and MSH2 in human peripheral blood lymphocytes (PBL) exposed in vitro to hyperthermia and cPt [Citation35]. To further explore and establish associations between HSPs and MMR proteins in another biological model, we used MMR−/MMR-proficient (MMR+) human colon cancer cell lines exposed to cPt (alone or in combination with hyperthermia). In brief, the aim of our work was to investigate for the first time the effects of hyperthermia on cPt sensitivity in MMR−/+ cells and to determine whether MLH1 and MSH2 are client proteins of Hsp27 and Hsp72.
Materials and methods
Cell culture
Human colon adenocarcinoma cell lines HCT116+ch2 (MMR−) and HCT116+ch3 (MMR+) were used [Citation18]. They were kindly donated by C.R. Boland and M. Koi (Baylor University Medical Center, Dallas, TX, USA). Cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% foetal bovine serum (FBS) (Gibco, Invitrogen Life Technologies, Grand Island, NY, USA). Aliquots of 400 µg/mL of geneticin G418 (Gibco) were added to HCT116+ch2 and HCT116+ch3 cells to maintain chromosome additions.
Experimental treatments
Cisplatin was purchased from Sigma (Saint Louis, MO, USA). Cell lines were exposed to: 1) cisplatin 10 µM for 1 h (cPt) or 2) cisplatin 10 µM (1 h) 24 h after heat shock (42 °C, 1 h) in a water bath (H+cPt). Cells were allowed to recover at 37 °C and were collected 24 h after cPt incubation. The experiments were performed in triplicate.
Clonogenic survival assay
Untreated and heat shocked cells (300 per well) were seeded in a 6-well plate. Twenty-four hours after plating, cell lines were treated with 5, 10, 15, 20 or 25 µM of cPt for 1 h. The cells were then washed, and new drug-free medium was added. After 8–10 days, cells were fixed in methanol, stained with crystal violet and the number of colonies (colonies with > 50 cells) was counted. The experiment was performed a minimum of three times.
Immunofluorescence
The cells (1.8 × 104) were grown on coverslips in 12-well plates and exposed to experimental treatments. Briefly, the cells were fixed in 4% p-formaldehyde, washed with 1 × phosphate-buffered saline (PBS) and treated with 50 mM ammonium chloride in PBS. Cells were then permeabilised with 0.1% Triton X-100 in PBS followed by blocking with 1% bovine serum albumin (BSA) for 30 min. The coverslips were incubated overnight with mouse monoclonal antibody (mAb) anti-MLH1 (clone M1, Sigma, 1:100) or mouse mAb anti-MSH2 (clone FE11, Calbiochem, EMD Millipore, Billerica, MA, USA, 1:25) in a humidity chamber at 4 °C. The samples were rinsed with wash solution (0.05% saponin and 0.2% BSA in PBS) and probed with Alexa 488-labelled goat anti-mouse Ab (Molecular Probes, Invitrogen, 1:400). For colocalisation assay, the coverslips were incubated 2 h at room temperature with rabbit polyclonal antibody against Hsp72 (SPA-812 Stressgen, Enzo Life Sciences, Farmingdale, NY, USA, 1:100) or rabbit polyclonal antibody against Hsp27 (kindly donated by Dr Matthias Gaestel, Hannover Medical School, Germany, 1:1000). After washes, cells were probed with Cy3-labelled goat anti-rabbit Ab (Molecular Probes, 1:400). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). The coverslips were mounted on slides with Vectashield mounting medium (H1000, Vector Laboratories, Burlingame, CA, USA) for examination with confocal microscope (FV 1000, Olympus, Tokyo, Japan) using a Paplon 60× lens.
Quantitative co-localisation analysis
The co-localisation analysis was performed using the JACoP plugin for Image J software (National Institutes of Health, Bethesda, MD) according to Bolte and Cordelieres [Citation36]. Z-Stack images of cells stained for MLH1 or MSH2 and Hsp72 or Hsp27 were deconvolved. The channel-specific point spread functions were generated and the signal-to-noise ratio was adjusted until deconvolved images were free of pixel noise. The co-localisation analysis was made using two correlation coefficients, Pearson (PCC) and Manders (MCC). For PCC calculation, the dependency of pixels in dual-channel images (green and red channels for MMR and HSPs, respectively) was measured by plotting the pixel grey values of two images against each other. These values were displayed in a pixel-distribution diagram (scatter plot), and a linear equation of the relation between the intensities of the two images was calculated by linear regression. The value of the PCC can range from 1 to −1, with 1 standing for complete positive correlation, −1 for negative correlation, and 0 for no correlation.
The MCC is based on the PCC, with average intensity values being taken out of a mathematical expression [Citation37]. Two coefficients were obtained, MCC-M1 and MCC-M2, for the fraction of MMR protein overlapping with HSP protein and for the fraction of HSP overlapping with MMR protein, respectively. These values vary from 0 to 1, corresponding to non-overlapping images or complete co-localisation, respectively.
Western blotting and immunoprecipitation
Floating and adherent cells were collected, centrifuged and washed with 1 × PBS. RIPA buffer containing protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) was used for cell lysis. The lysates were quantified using the micro-BCA kit (Pierce, Thermo Fisher Scientific, Rockford, IL, USA). Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The blots were incubated overnight with the following primary antibodies and dilutions: anti-MSH2 mouse mAb (Calbiochem, 1:2000), anti-MLH1 mouse mAb (Sigma, 1:2000), anti-Hsp72 mouse mAb (SPA810, Stressgen, 1:8000), anti-Hsp27 mouse mAb (SPA800, Stressgen, 1:12000), anti-p53 mouse mAb (ab28, Abcam, Cambridge, MA, 1:2000), anti-p73 rabbit mAb (ab40658, Abcam, 1:1000), anti-Bax rabbit mAb (ab32503, Abcam, 1:2000), anti-Bcl-2 mouse mAb (M0887, Dako North America, Carpinteria, CA, USA, 1:2000), and anti-β-actin mouse mAb (A5441, Sigma, 1:8000). The membranes were washed with PBS-T (PBS Tween-20) and incubated with anti-mouse or anti-rabbit secondary antibodies (Dako) for 1 h. After washing, the blots were developed with chemiluminescence reagents (Sigma). The images were captured using ChemiDoc XRS+ System with Image Lab software (Bio-Rad, Hercules, CA).
Immunoprecipitation (IP) was performed as described by Fanelli et al. [Citation38]. Mouse mAbs anti-MLH1 (1.5 µg) and anti-MSH2 (1 µg) were covalently coupled to M280 Tosylactivated Dynabeads (Invitrogen). MLH1 and MSH2-coated beads were then incubated with 500 µg of proteins for each cell line experimental group. For western blotting, proteins were separated in SDS-PAGE. Blots were then probed with anti-MLH1 or anti-MSH2 (to verify the success of the IP), anti-Hsp27 and anti-Hsp72 as previously described.
Cell viability
The viability of cells to hyperthermia, cPt or combined treatment was evaluated using the 3-[4,5-dimethylthiazol-2-yl]-diphenyltetrazolium bromide (MTT) assay (Sigma). Cells (5 × 103) were seeded in 96-well microplates in growth medium (100 µL) and incubated at 37 °C in a humidified 5% CO2 atmosphere. A heat shock (42 °C, 1 h) was performed to the corresponding experimental group 24 h later. After an additional 24 h the medium was removed and replaced with serum-free medium containing cPt 10 µM. The drug was removed 1 h later, and serum-supplemented medium was added to the cells. Twenty-four hours after concluding the cPt treatment, a working solution of MTT (0.5 mg/mL) was added to each well and incubated at 37 °C in the dark for 3 h. Finally, the formazan rings were dissolved with 100 µL dimethyl sulphoxide (DMSO). The colour intensity was assessed with a microplate reader (Multiskan EX, Thermo Scientific, Lafayette, CO) at 570 nm wavelength. Viability was expressed as a percentage of mean absorbance for treated wells compared to the mean absorbance for the control wells.
Cell cycle analysis
Treated cells were harvested by trypsinisation, washed twice with cold 1 × PBS + 1%FBS and fixed in 70% ethanol (4 °C, 1 h). After that, cells were centrifuged (1500 rpm, 5 min), washed with cold 1 × PBS + 1%FBS, treated with RNase (0.2 mg/mL, Qiagen Sciences Inc, Germantown, MD, USA) at 37 °C for 30 min and stained with propidium iodide (40 µg/mL, Sigma). The cells were analysed on a flow cytometer FACS Aria III (BD, Franklin Lakes, NJ) and cell cycle analysis was performed using FCS Express version 4 (De Novo Software, Los Angeles, CA) from a total of 2 × 104 events.
Annexin V-FITC/propidium iodide analysis
Twenty-four hours after cPt or H+cPt treatment, attached and detached cells were collected, washed with cold 1 × PBS and centrifuged (1200 rpm, 5 min). Cells (1 × 105) were re-suspended in 1 × binding buffer and stained with FITC Annexin V (556547, BD Biosciences, San Jose, CA, USA) for 20 min in the dark. Propidium iodide was then added to the cells and the samples were analysed within 1 h by flow cytometry using a BD FACS Aria III. The proportion of stained cells was calculated with FlowJo software (Tree Star, San Carlos, CA) from a total of 1 × 104 events.
Statistics
All the experiments were performed at least three times and error bars indicate standard error of the means. The Student’s t-test or the non-parametric Kruskal-Wallis test was applied to calculate the significance of differences using the GraphPad Prism software version 6.01 (San Diego, CA). p < 0.05 was considered statistically significant.
Results
Effect of hyperthermia on the colony formation of cisplatin-treated colon cancer cell lines
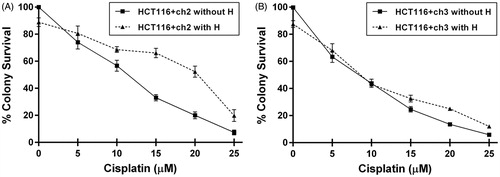
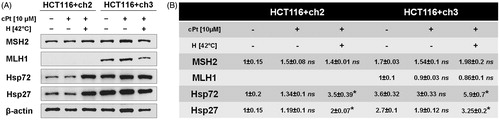
HCT116+ch3 (MMR+) and HCT116+ch2 (MMR−) cells were exposed to increasing concentrations of cPt (0, 5, 10, 15, 20 and 25 µM). One group of cells was exposed to hyperthermia (42 °C) 24 h before cPt treatment (). MMR+ cells were more sensitive to cPt (IC50 = 10.27 ± 1.40 µM) compared to MMR− cells (IC50 = 12.10 ± 0.89 µM). Hyperthermic treatment before cPt reduced the colony-forming ability at increasing drug concentrations in both MMR+/− cell lines. However, MMR− cells exposed to a mild heat were 1.42-fold more resistant to cPt (IC50 = 17.60 ± 2.10, ). Conversely, in MMR+ cells hyperthermia did not affect the number of colonies at concentrations lower than 10 µM, but it increased resistance at higher drug concentrations (IC50 = 11.50 ± 1.80, ). Therefore, for subsequent experiments we have chosen cPt IC50 value from MMR+ cells.
Figure 1. Clonogenic survival curves for MMR-deficient (HCT116 + ch2) and MMR-proficient (HCT116 + ch3) colon cancer cells exposed to cisplatin (cPt) and hyperthermia + cisplatin (H+cPt). (A) MMR- cells and (B) MMR+ cells treated with cPt for 1 h (closed squares) or treated with cPt 24 h after 42 °C hyperthermia (closed triangles). Each point represents the mean ± SEM of three experiments performed in duplicate.
Cellular distribution of Hsp27, Hsp72 and MMR proteins after cisplatin (cPt) or hyperthermia + cisplatin (H+cPt) treatment
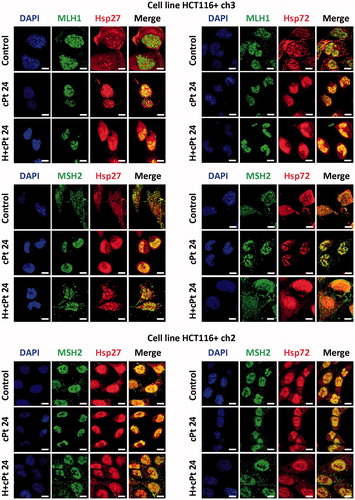
Hsp27, Hsp72, MLH1 and MSH2 proteins were studied by immunofluorescence. At basal conditions, MLH1 was exclusively expressed in the nucleus of HCT116+ch3 cells (MMR+), in agreement with DAPI staining (). HCT116+ch2 (MMR−) cells expressed a truncated non-functional MLH1 protein that was not recognised by the monoclonal antibody used (this observation was corroborated by immunoblotting, see below). MSH2 appeared in the nucleus of untreated MMR+/− cells, but weak expression of the protein was also noted in the cytoplasm. Observation of the cells 24 h post-treatment (cPt or H+cPt) revealed that MLH1 remains in the nucleus of MMR+ tumour cells. As expected, MSH2 was predominantly located in the nucleus after cPt treatment in both MMR+/− cells. Interestingly, the application of a mild hyperthermia before cPt treatment led to a small proportion of MSH2 being relocalised to the cytoplasm (more extensively than in untreated control cells). In the case of the HSPs, MMR+/− cell lines constitutively expressed Hsp27 and Hsp72, which increased in response to H+cPt treatment. Hsp27 and Hsp72 were localised within cytoplasmic and nuclear compartments of untreated and H+cPt treated cells. However, after cPt exposure, Hsp27 and Hsp72 moved to the nucleus; this behaviour was similar to that observed for MSH2.
Figure 2. MLH1 and MSH2 colocalise with Hsp27 and Hsp72 in HCT116+ch3 (MMR+) and HCT116+ch2 (MMR−) cells. Tumour cells were double-stained with MLH1 or MSH2 (green) and Hsp27 or Hsp72 (red) antibodies. Colocalisation was revealed by the signals overlap (yellow) in the merged images. A scale bar was included in each microphotograph (20 μm). Control, untreated cells; cPt 24, cells collected 24 h after 10 μM cPt exposure. H+cPt 24, cells treated with cPt 24 h after H, and collected 24 h later.
Hsp27 and Hsp72 associate and interact with MMR proteins
The association between Hsp27 and Hsp72 with MLH1 and MSH2 proteins was determined using two colocalization coefficients: Pearson and Manders (PCC and MCC, respectively). Immunofluorescence images from MMR+ cells revealed that Hsp27 and Hsp72 colocalised with MLH1 and MSH2, especially after cPt treatment. These observations were supported by PCC values, which significantly increased in cPt-treated cells (p < 0.05, ). In cPt-treated MMR+ cells, MCC coefficient measures showed a high degree of overlap between MLH1 and Hsp72, and also between MSH2 and Hsp27. In both cases we observed that MCC-M1 significantly increased 24 h after cPt exposure.
Figure 3. Changes of Pearson’s correlation coefficient (PCC) and Manders’s correlation coefficients (MCC-M1 and MCC-M2) following cPt and H+cPt administration. (A) HCT116+ch3 cells (MMR+). Coefficients show that MLH1 colocalises with Hsp27 and Hsp72. MSH2 colocalises with Hsp27 and Hsp72. (B) HCT116+ch2 cells (MMR−). Graphs show colocalisation coefficients for MSH2 protein with Hsp27 and Hsp72. Bars represent mean ± SEM. Significant differences were calculated with respect to the control group. *p < 0.05; **p < 0.01; ***p < 0.001.
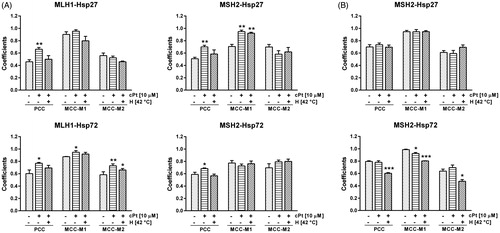
On the other hand, untreated MMR− cells had higher PCC values for Hsp27-MSH2 and Hsp72-MSH2 than control MMR+ cells. The PCC values of these proteins were, however, not modified by cPt treatment in MMR− cells ().
Hyperthermia reduced PCC values of MLH1-Hsp27, MLH1-Hsp72, MSH2-Hsp27 and MSH2-Hsp72 in cPt-treated MMR+ cells almost to control levels. MCC-M1 and MCC-M2 coefficients also diminished in cells previously exposed to hyperthermia for MLH1-Hsp27 and MLH1-Hsp72. There were no significant differences between the MCC coefficients of H+cPt-treated and cPt-treated MMR+ cells in comparisons of MSH2-Hsp27 and MSH2-Hsp72.
In MMR− cells hyperthermia did not change PCC values for MSH2-Hsp27. However, PCC and MCC values for MSH2-Hsp72 were lower than those observed in cells exposed to cPt only ().
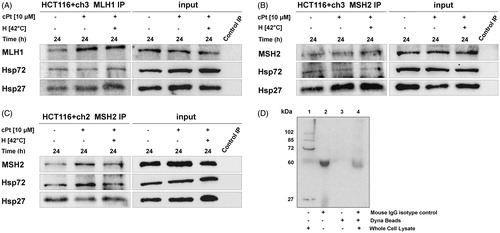
Our immunostaining findings were corroborated using IP, which confirmed interactions between Hsp27 and Hsp72 with MLH1 and MSH2 proteins in untreated and treated HCT116+ch3 cells (MMR+). In addition, the interactions between Hsp27 and Hsp72 with MSH2 were also demonstrated in MMR− cells (). Negative controls were included in order to test the specificity of the IP bands ().
Figure 4. MLH1 and MSH2 interact with Hsp27 and Hsp72 in HCT116 + ch3 and HCT116 + ch2 cells. MLH1 (A) and MSH2 (B) were immunoprecipitated from MMR+ cell extracts treated with cPt or H+cPt and analysed by immunoblotting for co-precipitation with Hsp27 and Hsp72. (C) MSH2 immunoprecipitates from MMR- cells exposed to cPt and H+cPt. (D) Negative controls included whole cell lysate 20 µg (lane 1), mouse monoclonal IgG isotype control (lane 2), unconjugated Dynabeads (lane 3), incubation of whole cell extract with isotype IgG/Dynabeads complexes (lane 4). Input, total protein from cell lysates; Control IP, control mouse IgG.
Total expression of HSPs and MMR proteins
After studying the subcellular localisation of HSPs and MMR proteins in response to cPt or H+cPt, we further investigated changes in protein expression using immunoblotting (). HCT116+ch2 and HCT116+ch3 cells constitutively expressed elevated Hsp27 and Hsp72 levels, especially in MMR+ cells. The expression of HSPs did not significantly change in MMR−/+ cells following 24 h of recovery after cPt incubation. As expected, mild hyperthermia 24 h before cPt exposure significantly increased the expression of Hsp27 and Hsp72 in both cell lines. As mentioned above (see Materials and methods section), the HCT116+ch2 cell line is MLH1 deficient. The expression of MLH1 in MMR+ cell line did not differ significantly between the experimental groups at 24 h following cPt treatment. In the case of MSH2, hyperthermia followed by cisplatin (H+cPt) increased its expression in both cell lines, but protein expression was higher in MMR+ cells. MMR+ cells exposed to cPt without hyperthermia treatment, however, displayed a modest decrease in MSH2 protein levels.
Figure 5. Immunoblot analysis of Hsp27, Hsp72 and DNA repair proteins MSH2 and MLH1 after cPt or H+cPt treatment in colon cancer cells. (A) Immunoblots using the total protein extracts from HCT116+ch2 (MMR−) and HCT116+ch3 (MMR+) cells; 20 µg of proteins were loaded and resolved by SDS-PAGE using 7.5% polyacrylamide gels. (B) Densitometric quantification of immunoblots (Mean ± SEM). Protein bands were normalized to β-actin (optical density ratio). Statistical comparisons were calculated with respect to untreated control cells. *p < 0.05.
Hyperthermia enhances the reduction in cellular viability induced by cisplatin in MMR-competent cells
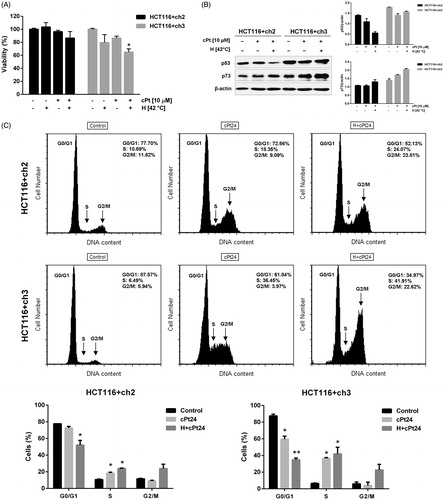
Given that intact MMR machinery is required for cPt sensitivity, we used MMT to explore the influence of mild hyperthermia (42 °C) on the cytotoxicity of cPt in MMR−/+ cells (). No statistically significant changes were observed in the cell viability of MMR−/+ cells exposed to hyperthermia or cPt alone. Hyperthermia potentiated the cPt sensitivity in MMR+ cells (p < 0.05).
Figure 6. Hyperthermia followed by cPt treatment decreases MMR+ cells (HCT116 + ch3) viability, and induces p73 expression and cell cycle arrest. (A) Cell viability studied by MTT assay in MMR− and MMR+ cells (black and grey bars, respectively). (B) Immunoblotting of wild type p53 and p73 after cPt or H+cPt treatments. Proteins (30 µg) were loaded and resolved by SDS-PAGE using 7% polyacrylamide gels. β-actin was used as loading control. Quantification graphs were included. (C) Representative histograms and proportion of HCT116+ch2 (MMR−) and HCT116+ch3 (MMR+) cells in G0/G1, S and G2/M phases. Bars represent mean ± SEM. Significant differences were calculated with respect to the control group. *p < 0.05.
Expression of p53 and p73 after cPt and H+cPt treatment
We explored the expression of p53 and p73 (proteins involved in cell cycle arrest and cell death signalling in response to genotoxic damage or cellular stress) in MMR+/− cell lines (). Twenty-four hours after cPt treatment, p53 expression showed a slight decrease in both MMR+/− cells. Interestingly, hyperthermia followed by cPt (H+cPt) caused a decrease in p53 levels in MMR− cells, but not in MMR+ cells. On the other hand, the treatment with cPt increased p73 protein in MMR+ cells, but levels of this protein remained stable in MMR− cells. The combined treatment (H+cPt) greatly induced expression of p73 in MMR+ cells, but protein levels only slightly increased in MMR− cells.
Hyperthermia contributes to cell cycle arrest in cisplatin-treated MMR−/+ cells
Flow cytometry was applied to study the cell cycle distribution of MMR−/+ cells after cPt and H+cPt exposure (). The proportion of G1 phase cells decreased in response to the treatments in both cell lines, but this decay was more accentuated in H+cPt treated cells. The percentage of S-phase cells significantly increased after treatments in MMR−/+ cells. Treatment with cPt and H+cPt caused an accumulation of MMR+ cells in S phase (p < 0.05), even to a level above that observed in MMR− cells. The exposure to cPt did not change the proportion of MMR−/+ cells in G2/M phase, but hyperthermia (H+cPt) increased G2/M phase arrest (p = 0.057 in MMR+ cells, ).
Effects of cPt and H+cPt on cell death
We performed Annexin V/IP analysis by flow cytometry 24 h after cPt and H+cPt exposure (). The dot plots (Annexin V vs. IP) from the gated cells showed the percentage of events corresponding to necrotic (Q1, Annexin V−/IP+), late apoptotic (Q2, Annexin V+/IP+), early apoptotic (Q3, Annexin V+/IP−) and viable cells (Q4, Annexin V−/IP−). The percentage of late apoptotic cells slightly increased in cPt treated MMR− cells, but not in the H+cPt group. Treatments with cPt and H+cPt failed to induce apoptotic changes in the MMR+ cell line.
Figure 7. Hyperthermia does not induce apoptotic changes or affect the Bax/Bcl-2 ratio in either MMR− or + cells. (A) Flow cytometry analysis of apoptosis by Annexin V and PI staining, 24 h after cPt and H+cPt. The dot plots showed the percentage of events corresponding to: necrotic (Q1, Annexin V−/IP+), late apoptotic (Q2, Annexin V+/IP+), early apoptotic (Q3, Annexin V+/IP−) and viable cells (Q4, Annexin V−/IP−). (B) Immunoblotting of Bax and Bcl-2 in HCT116+ch2 (MMR−) and HCT116+ch3 (MMR+) cells. Proteins (30 µg) were resolved by SDS-PAGE using 12% polyacrylamide gels. β-actin was used as loading control. The image shows Bax/Bcl-2 ratio (mean ± SEM).
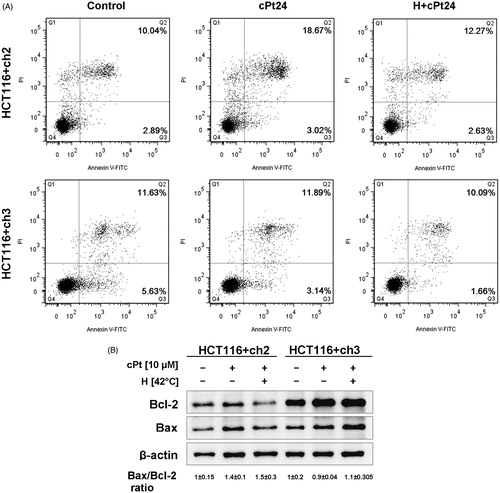
In addition, we studied the expression of the apoptosis-related proteins Bcl-2 and Bax. The behaviour of these proteins differed between the two cell lines. As a result of the combination of hyperthermia with cPt, MMR+ cells displayed increased Bcl-2 and Bax levels, while MMR− cells showed lower levels. However, comparison of Bax/Bcl-2 ratio levels between treatments (cPt or H+cPt) in MMR−/+ cells revealed no statistically significant changes ().
Discussion
Hyperthermia is used in combination with chemotherapy/radiotherapy to potentiate their cytotoxicity. It is well known that hyperthermia induces the synthesis of HSPs [Citation39,Citation40]. Several proteins have been related to drug resistance, among them HSPs and members of the DNA repair systems. Little is known, however, about the participation of HSPs in the repair of DNA damage induced by specific drugs such as cPt.
We have previously reported that Hsp27 (HSPB1) and Hsp72 (HSPA1A) colocalise with MLH1 and MSH2 in peripheral blood lymphocytes (PBL) from healthy subjects exposed in vitro to hyperthermia and cPt [Citation35]. The insufficient sample material obtained from PBL became a limiting factor to performing IP assays. For this reason and in order to investigate associations between HSPs and molecules from the MMR machinery, we have chosen human colon cancer MLH1−/+ cells as experimental model. We have recently shown that mild hyperthermia (42 °C) induces the nuclear accumulation of Hsp27 and Hsp72 and affects the subcellular localisation of MMR proteins MLH1 and MSH2 in MMR+ cells [Citation41].
Although several studies have shown the biological effects of hyperthermia on the clonogenic cell survival of cPt-sensitive/-resistant cells lines, none of them has characterised the behaviour of MMR−/+ cell lines exposed to cPt 24 h after hyperthermia [Citation32–34,Citation42–45]. There is a broad consensus that hyperthermia administered simultaneously or immediately before cPt enhances the sensitivity of tumour cells to the drug [Citation25]. However, when hyperthermia precedes cPt treatment, sensitisation gradually diminishes as the time interval between heat and drug exposure is extended, producing an additive effect or a similar cytotoxicity in non-heated and pre-heated cells. This behaviour has also been described for other anti-neoplastic drugs, such as carboplatin and mitomycin-C [Citation42,Citation43]. A comparative study of sensitivity to cPt in unheated and preheated HA-1 cells demonstrated that hyperthermia 12 h before cPt caused higher resistance to cPt [Citation44]. In contrast, Majima et al. [Citation45] reported that the thermal potentiation of cPt remained stable 12 h after hyperthermia in CHO cells. Furthermore, in these cells exposed to 43 °C 24 h before cPt, drug sensitivity was not modified when cPt was applied at 37 °C, while the cells showed more resistance to cPt when drug was incorporated at 43 °C as a consequence of development of thermotolerance [Citation45]. These results suggest that different cPt sensitivity and biological effects exist as a result of thermotolerance development among different cell lines, which may depend on the timing of application of hyperthermia (before or during drug exposure) or due to intrinsic characteristics of each cell line. In our study, after hyperthermia pretreatment (24 h prior to cPt), MMR− cells became more thermotolerant than MMR+ cells, and MLH1 deficiency might compromise thermally induced cPt cytotoxicity. We have previously reported that MMR− cells were more resistant to heat treatment (41 °C and 42 °C) and they better tolerated the effects of mild hyperthermia at the DNA damage level [Citation41].
In addition to time interval, the hyperthermic potentiation depends on cell growth phase; sensitivity may be higher in plateau phase cells, whereas changes in the number of cells during the experiment can affect cytotoxicity in exponentially growing cells [Citation43]. Glioma and ovarian carcinoma cell lines in plateau phase are more sensitive to cPt than exponentially growing cells, with hyperthermia (42 °C) being more effective in exponentially growing cPt sensitive cells than in resistant cells [Citation33]. In summary, the effect of hyperthermia on chemosensitisation may be affected by different factors: temperature and duration of heat, interval time between drug and heat treatments, temperature at which the drug is applied, cell growth phase, and drug sensitivity of the cell line. The participation of the MMR system in the development of thermotolerance remains, however, to be explored.
We have also characterised the localisation of Hsp27, Hsp72, MLH1 and MSH2 proteins in cellular compartments in untreated and cPt/H+cPt treated tumour cells. At basal conditions, MSH2 appeared in the nucleus of MMR−/+ cells, but weak expression was additionally seen in the cytoplasm, although cells exposed to hyperthermia showed higher cytoplasmic protein content. In accordance with our previous report, a cytoplasmic expression of MSH2 was also shown in PBL from cancer patients, although its clinical significance remains unclear [Citation35]. Immunoblot results showed no significant differences in total MSH2 protein levels among treatments, suggesting that hyperthermia before cPt treatment induced the translocation of a small portion of MSH2 out of the nucleus. These results are in agreement with our previous studies, in which hyperthermia effect in MMR+ cells led to the release of MLH1and MSH2 proteins from the nucleus into the cytoplasm [Citation41]. Other authors have also reported the nuclear delocalisation of DNA repair proteins as a consequence of hyperthermia. Xu et al. have shown that temperatures of 41 °C or higher will drive the repair protein MRE11 out of the nucleus [Citation46].
In the case of the HSPs, Hsp27 and Hsp72 were localised within cytoplasmic and nuclear compartments in control MMR−/+ cell lines, while they were almost exclusively present in cell nuclei after cPt exposure. Given that western blot results did not show an increased total HSP expression in this group, we assume that Hsp27 and Hsp72 moved to the nucleus after cPt exposure. There are several reports showing that Hsp27 and Hsp70 (HSPA) can translocate to the nucleus and accumulate there under stressful conditions, and are probably involved in either protection or recovery of DNA damage [Citation41,Citation47–50]. Hyperthermia prior to cPt significantly increases total expression (nuclear and cytoplasmic) of Hsp27 and Hsp72 in both MMR−/+ cell lines, although HSP fold change with respect to the control group was higher in thermotolerant MMR− cells than in MMR+ cells. Laszlo et al. demonstrated that Hsc70 (constitutive Hsp70, HSPA8) and Hsp72 are the major components of the heat-induced excess nuclear proteins, which associate with a faster recovery of cytoplasmatic and nuclear processes from the initial damage and thermotolerance development in heat-resistant cells [Citation51,Citation52]. Taking these results together, we can assume that hyperthermia may facilitate the recovery of the cPt-damaged cells in a MMR−/+ cellular model, thus the induction of HSPs can explain in part the MMR− cells’ thermotolerant state.
We have also determined the association of Hsp27 and Hsp72 with MLH1 and MSH2 proteins in MMR+ cells using PCC and MCC and IP assays. In MMR+ cells, associations between the pairs of proteins studied significantly increased especially after cPt treatment but decreased almost to basal levels in preheated cells. In contrast, this behaviour was not observed in MMR− cells. The decrease in the values of these coefficients may not be explained merely by relocation of Hsp27 and Hsp72 proteins from the nucleus to the cytoplasm. It is well known that hyperthermia not only induces HSPs, but also helps in recruiting HSPs attending misfolded or unfolded proteins, leaving their regular partners and establishing associations with other molecules to assist them in a wide variety of cellular processes [Citation6]. IP assays confirmed the interactions of Hsp27 and Hsp72 with MLH1 and MSH2 proteins in untreated and H+cPt-treated HCT116+ch3 cells (MMR+). In addition, the interactions of Hsp27 and Hsp72 with MSH2 were also demonstrated in MMR− cells.
This is the first report of interactions of Hsp27 and Hsp72 with MMR proteins in MMR−/+ colon cancer cell lines under hyperthermia and cisplatin effects. We have recently reported that Hsp27 and MSH2 interact in glioma cells exposed to temozolomide. In agreement with our present work, this colocalisation was found in Gli36 cell nuclei at basal levels, but it significantly increased after drug treatment [Citation53]. The participation of the molecular chaperones in DNA repair mechanisms has been reported by a number of authors. Several DNA repair proteins have demonstrated to be interacting partners of HSPs, although new client proteins of Hsp27 and Hsp72 remains to be identified [Citation11]. Kenny et al. found that Hsp70 binds to the human APE1 endonuclease and enhances its endonuclease activity 10–100-fold [Citation18]. The same group also demonstrated that the N-terminal domain of Hsp70 was responsible for stimulating Polβ enzymatic activity 5–10-fold, thus improving repair efficiency [Citation19]. Guttmann et al. found that Hsp27 colocalised with ATM in untreated and irradiated cancer cell lines and that Hsp27 knockdown attenuated DNA DSB repair, resulting in radiosensitisation. They postulated a role for Hsp27 in DBSs’ repair signalling, and proposed it as a potential clinical target [Citation21].
We have also explored whether hyperthermia pretreatment modulates cPt chemosensitivity and whether its effect is MMR-dependent. Although cPt induced an S-phase arrest in both cell lines, this ability was reduced in cells lacking a functional MMR system. In addition, hyperthermia pretreatment induced accumulation of the cells in S and G2/M phases 24 h after cPt in both cell lines. Furthermore, the effect of hyperthermia exposure was greater in MMR-competent cells. This means that hyperthermia delayed cell cycle progression and inhibited cell growth, probably to complete the repair of the damage. It is well known that p73 is involved in cPt DNA damage signalling [Citation26,Citation27], but its participation in the response to the combination of heat shock and cPt treatments is not fully understood. We demonstrated that exposure of MMR+ cells to hyperthermic stress decreased cell viability and increased p73 expression. According to our results, p73 may participate in the hyperthermia potentiation of cPt action in a MMR-dependent manner. However, more extensive studies are needed to better understand the biological effects of hyperthermia on the cisplatin-induced activation of p73-dependent pathways in MMR−/+ cells.
Nevertheless, the experimental treatments did not exert a significant effect on the expression of p53 in both cell lines. P53 is not only induced by DNA damage, but also by other cellular stress conditions, such as heat shock [Citation54]. Our findings might be partially explained by the fact that hyperthermia at temperatures above 41 °C denatures thermolabile cellular proteins (Lepock, 2004), such as p53. Moreover, heat induction of p53 is shorter than that induced by DNA damage [Citation55]. On the other hand, Matsumoto et al. described that heat-induced p53 activation was suppressed by cPt in A-172 cells but not in T98G cells [Citation56]. In agreement with our study, Van Bree et al. have shown that 41 °C hyperthermia (60 min) simultaneously with cPt enhanced the cytotoxicity of the drug irrespective of p53 function [Citation57]. In summary, a short induction of p53 by heat, inhibition of protein expression by cPt or a cell line-dependent behaviour may explain the absence of significant changes in p53 protein levels after treatments found in our cellular model. All these observations together support that hyperthermia potentiated cPt sensitivity in both MMR−/+ cells, but significantly in the MMR+ cell line.
Further, Annexin V/PI and Bax/Bcl-2 ratio assays revealed that cPt or H+cPt did not induce significant apoptotic changes in MMR−/+ cells. In order to initiate programmed cell death, long drug exposure would be necessary, or different cPt concentrations and several drug-free recovery periods. Cell viability measured by the MTT assay estimates the number of viable cells in a linear relationship with the mitochondrial activity. Our results showed a decreased cell viability of MMR+ cells exposed to hyperthermic stress before cPt treatment, thereby without activating the apoptotic programme, resulting in a cytostatic effect. In other words, H+cPt treatment induced S, and G2/M phase arrest in the absence of apoptotic changes, especially in MMR+ cells, may be to allow the repair of the DNA damage.
Finally, this is the first study that reports MLH1 and MSH2 as client proteins of Hsp27 and Hsp72 in a MMR+/− cellular model. Because MSH2 interacts with Hsp27 and Hsp72 in MMR+ as well as in MMR− cells, further studies are needed to elucidate whether HSPs are facilitating the repair of cPt-induced DNA damage by the MMR system or if these interactions are only a result of their chaperone functions. Our results provide novel insights into the complex interplay between MMR and the chaperone function of Hsp27 and Hsp72. The role of the heat shock response in the MMR system might be a valuable tool for facing chemotherapy resistance.
Acknowledgements
We are grateful to R.W. Caron who kindly provided the antibodies against p53 and Bax proteins, and to the Flow Cytometry Service of the Medical School, National University of Cuyo.
Declaration of interest
This study was supported by the National Research Council of Argentina (PIP112 201101 00836) and National University of Cuyo (06/J392 and J026). The authors alone are responsible for the content and writing of the paper.
References
- Roti Roti JL. Cellular responses to hyperthermia (40–46 °C): Cell killing and molecular events. Int J Hyperthermia 2008;24:3–15
- Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res 2002;30:1354–63
- Roti Roti JL. Heat-induced alterations of nuclear protein associations and their effects on DNA repair and replication. Int J Hyperthermia 2007;23:3–15
- Hintzsche H, Riese T, Stopper H. Hyperthermia-induced micronucleus formation in a human keratinocyte cell line. Mutat Res 2012;738–9:71–4
- Yan B, Ouyang R, Huang C, Liu F, Neill D, Li C, Dewhirst M. Heat induces gene amplification in cancer cells. Biochem Biophys Res Commun 2012;427:473–7
- Eppink B, Krawczyk PM, Stap J, Kanaar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int J Hyperthermia 2012;28:509–17
- Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst 2000;92:1564–72
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009;14:105–11
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006;5:2592–601
- Ciocca DR, Calderwood SK. Heat shock proteins in cancer: Diagnostic, prognostic, predictive and treatment implications. Cell Stress Chaperones 2005;10:86–103
- Arrigo AP, Gibert B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int J Hyperthermia 2013;29:409–22
- Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J Leukoc Biol 2007;81:15–27
- Murphy ME. The Hsp70 family and cancer. Carcinogenesis 2013;34:1181–8
- Guzhova IV, Shevtsov MA, Abkin SV, Pankratova KM, Margulis BA. Intracellular and extracellular Hsp70 chaperone as a target for cancer therapy. Int J Hyperthermia 2013;29:399–408
- Murshid A, Eguchi T, Calderwood SK. Stress proteins in aging and life span. Int J Hyperthermia 2013;29:442–7
- Pandita TK, Higashikubo R, Hunt CR. Hsp70 and genomic stability. Cell Cycle 2004;3:591–2
- Nadin SB, Ciocca DR. Participation of heat shock proteins in DNA repair mechanisms in cancer. In: Columbus F, editor. DNA Repair: Damage, Repair Mechanisms and Aging. New York: Nova Science, 2010, pp. 165–86
- Kenny MK, Mendez F, Sandigursky M, Kureekattil RP, Goldman JD, Franklin WA, Bases R. Heat shock protein 70 binds to human apurinic/apyrimidinic endonuclease and stimulates endonuclease activity at abasic sites. J Biol Chem 2001;276:9532–6
- Mendez F, Kozin E, Bases R. Heat shock protein 70 stimulation of the deoxyribonucleic acid base excision repair enzyme polymerase beta. Cell Stress Chaperones 2003;8:153–61
- Duan Y, Huang S, Yang J, Niu P, Gong Z, Liu X, et al. HspA1A facilitates DNA repair in human bronchial epithelial cells exposed to Benzo[a]pyrene and interacts with casein kinase 2. Cell Stress Chaperones 2014;19:271–9
- Guttmann DM, Hart L, Du K, Seletsky A, Koumenis C. Inhibition of Hsp27 radiosensitizes head-and-neck cancer by modulating deoxyribonucleic acid repair. Int J Radiat Oncol Biol Phys 2013;87:168–75
- Fedier A, Stuedli A, Fink D. Presence of MLH1 protein aggravates the potential of the Hsp90 inhibitor radicicol to sensitize tumor cells to cisplatin. Int J Oncol 2005;27:1697–705
- Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 2011;108:9851–6
- Genet SC, Fujii Y, Maeda J, Kaneko M, Genet MD, Miyagawa K, Kato TA. Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol 2013;228:1473–81
- Kampinga HH. Cell biological effects of hyperthermia alone or combined with radiation or drugs: A short introduction to newcomers in the field. Int J Hyperthermia 2006;22:191–6
- Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci 2000;57:1229–35
- Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005;4:307–20
- Hettinga JV, Lemstra W, Meijer C, Dam WA, Uges DR, Konings AW, et al. Mechanism of hyperthermic potentiation of cisplatin action in cisplatin-sensitive and -resistant tumour cells. Br J Cancer 1997;75:1735–43
- Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehmé A, et al. The role of DNA mismatch repair in platinum drug resistance. Cancer Res 1996;56:4881–6
- Jiricny J. The multifaceted mismatch repair system. Nat Rev Mol Cell Biol 2006;7:335–46
- Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, et al. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res 1998;58:3579–85
- Eichholtz-Wirth H, Hietel B. Heat sensitization to cisplatin in two cell lines with different drug sensitivities. Int J Hyperthermia 1990;6:47–55
- Raaphorst GP, Chabot P, Doja S, Wilkins D, Stewart D, Ng CE. Effect of hyperthermia on cisplatin sensitivity in human glioma and ovarian carcinoma cell lines resistant and sensitive to cisplatin treatment. Int J Hyperthermia 1996;12:211–22
- Hettinga JV, Konings AW, Kampinga HH. Reduction of cellular cisplatin resistance by hyperthermia – A review. Int J Hyperthermia 1997;13:439–57
- Nadin SB, Vargas-Roig LM, Drago G, Ibarra J, Ciocca DR. Hsp27, Hsp70 and mismatch repair proteins hMLH1 and hMSH2 expression in peripheral blood lymphocytes from healthy subjects and cancer patients. Cancer Lett 2007;252:131–46
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006;224:213–32
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 1992;103:857–62
- Fanelli MA, Montt-Guevara M, Diblasi AM, Gago FE, Tello O, Cuello-Carrión FD, et al. P-cadherin and beta-catenin are useful prognostic markers in breast cancer patients: Beta-catenin interacts with heat shock protein Hsp27. Cell Stress Chaperones 2008;13:207–20
- Lindquist S, Craig EA. The heat shock proteins. Annu Rev Genet 1988;22:631–77
- Singh IS, Hasday JD. Fever, hyperthermia and the heat shock response. Int J Hyperthermia 2013;29:423–35
- Nadin SB, Cuello-Carrión FD, Sottile ML, Ciocca DR, Vargas-Roig LM. Effects of hyperthermia on Hsp27 (HSPB1), Hsp72 (HSPA1A) and DNA repair proteins hMLH1 and hMSH2 in human colorectal cancer hMLH1-deficient and hMLH1-proficient cell lines. Int J Hyperthermia 2012;28:191–201
- Cohen JD, Robins HI. Hyperthermic enhancement of cis-diammine-1,1-cyclobutane dicarboxylate platinum(II) cytotoxicity in human leukemia cells in vitro. Cancer Res 1987;47:4335–7
- Wallner KE, Li GC. Effect of drug exposure duration and sequencing on hyperthermic potentiation of mitomycin-C and cisplatin. Cancer Res 1987;47:493–5
- Hahn GM. Thermotolerance and drugs. In: Henle KJ, editor. Thermotolerance, Vol 1. Thermotolerance and Thermophily. Boca Raton (FL): CRC Press, 1987, pp. 97–126
- Majima H, Kashiwado K, Egawa S, Suzuki N, Urano M. Interaction between the kinetics of thermotolerance and effect of cis-diamminedichloroplatinum(II) or bleomycin given at 37 or 43 °C. Int J Hyperthermia 1992;8:431–42
- Xu M, Myerson RJ, Xia Y, Whitehead T, Moros EG, Straube WL, Roti JL. The effects of 41 °C hyperthermia on the DNA repair protein, MRE11, correlate with radiosensitization in four human tumor cell lines. Int J Hyperthermia 2007;23:343–51
- Arrigo AP, Suhan JP, Welch WJ. Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol 1988;8:5059–71
- McClaren M, Isseroff RR. Dynamic changes in intracellular localization and isoforms of the 27-kD stress protein in human keratinocytes. J Invest Dermatol 1994;102:375–81
- Ohtsuka K, Laszlo A. The relationship between hsp70 localization and heat resistance. Exp Cell Res 1992;202:507–18
- Lepock JR, Frey HE, Heynen ML, Senisterra GA, Warters RL. The nuclear matrix is a thermolabile cellular structure. Cell Stress Chaperones 2001;6:136–47
- Laszlo A, Wright W, Roti Roti JL. Initial characterization of heat-induced excess nuclear proteins in HeLa cells. J Cell Physiol 1992;151:519–32
- Laszlo A. The thermoresistant state: Protection from initial damage or better repair? Exp Cell Res 1992;202:519–31
- Castro GN, Cayado-Gutiérrez N, Zoppino FC, Fanelli MA, Cuello-Carrión FD, Sottile M, et al. Effects of temozolomide (TMZ) on the expression and interaction of heat shock proteins (HSPs) and DNA repair proteins in human malignant glioma cells. Cell Stress Chaperones 2015;20:253–65
- Ohnishi K, Ohnishi T. Heat-induced p53-dependent signal transduction and its role in hyperthermic cancer therapy. Int J Hyperthermia 2001;17:415–27
- Lepock JR. Role of nuclear protein denaturation and aggregation in thermal radiosensitization. Int J Hyperthermia 2004;20:115–30
- Guan J, Stavridi E, Leeper DB, Iliakis G. Effects of hyperthermia on p53 protein expression and activity. J Cell Physiol 2002;190:365–74
- Matsumoto H, Hayashi S, Shioura H, Ohtsubo T, Nishida T, Kitai R, et al. Suppression of heat-induced p53 accumulation and activation by CDDP or X-rays in human glioblastoma cells. Int J Oncol 1998;13:741–7
- van Bree C, Franken NA, Snel FA, Haveman J, Bakker PJ. Wild-type p53-function is not required for hyperthermia-enhanced cytotoxicity of cisplatin. Int J Hyperthermia 2001;17:337–46