Abstract
Background. Despite significant improvement in the outcome of patients with Ewing Sarcoma Family of Tumors (ESFT), second malignancies remain a problem that may compromise the outcome of some survivors. The Surveillance, Epidemiology and End-Results (SEER) database offers an opportunity to study second malignancies in a population-based cohort of patients. Methods. Cancer incidence rates were compared between the ESFT survivors and the general population using observed-to-expected ratios (O/E). Also, we studied the characteristics of patients with ESFT who developed second malignancies and compared them to those who did not. Results. We studied 1 166 patients with ESFT who were diagnosed from January 1973 to December 2005. Among them, 35 (3%) patients had records of second malignancy. Patients who received radiotherapy as part of their primary therapy had a higher chance of developing a second malignancy (odds ratio, 2.55; 95% CI, 1.09 to 6.00). Most solid tumors (78%) were diagnosed more than 5 years after diagnosis of ESFT while the majority (83%) of lymphatic/hematopoietic malignancies developed within five years of diagnosis. The 5-, 10-, and 20-year probability of developing a second malignancy were 2.1% ± 0.56%, 4.4% ± 0.95% and 8.0% ± 1.7%, respectively. The O/E ratio for developing a second malignancy was 4.10 (95%CI, 2.87 to 5.68) but was higher in children/adolescents (O/E, 9.94; 95%CI, 6.30 to 14.91). Conclusion. Having a second cancer following a diagnosis of ESFT is a known risk that may be increased by current therapies. This modest increase is justified by the benefit of these therapies in the majority of patients with ESFT.
The Ewing Sarcoma Family of Tumors (ESFT) encompasses a group of rare malignancies with varying degrees of neuroectodermal differentiation unified by similar chromosomal translocations and mainly affects children and young adults [Citation1]. Better characterization of the entity and improvements in diagnosis and treatment over the last three decades has contributed to steadily improved outcomes. Contemporary regimens are based on an intensive use of anthracyclines, alkylators, and topoisomerase inhibitors. With current multimodality therapy, more than two thirds of patients with localized disease are expected to be cured, while those with metastatic disease at presentation have significantly worse outcome [Citation2].
Patients surviving ESFT have an increased risk to develop second malignancies [Citation3]. Similar to other patients with cancer, genetic predisposition may play a role; however, it is believed that treatment modalities are the main contributors to this predicament [Citation3,Citation4]. In a previous population-based study, childhood cancer survivors observed for an average of 8.9 years were at six-fold increased risk of developing a second cancer relative to the general population [Citation5]. Patients with Ewing Sarcoma were at an increased risk in comparison to other patients and ranked third after survivors of retinoblastoma and primitive neuroectodermal tumors of the central nervous system.
In order to further define the risk of second malignancies in this group of patients, we performed a population-based study using the Surveillance, Epidemiology and End-Results (SEER) Program and estimated the incidence and type of second malignancies occurring in patients diagnosed with ESFT over a 33-year period.
Patients and methods
Data source and study population
The data was obtained from the Surveillance, Epidemiology, and End Results (SEER) 9 registries (http://seer.cancer.gov/data/) [Citation6]. The SEER Program of the National Cancer Institute (NCI) is an authoritative source of information on cancer incidence and survival in the United States [Citation7]. The SEER 9 registries database offers the longest follow-up for registered patients (since 1973) and thus was selected to conduct this study. This database covers 9.5% of the United States population [Citation8]. We used Case listing session of the SEER*Stat 6.4.4 program [Citation9] to generate a matrix of all individuals reported with a diagnosis of ESFT from January 1973 to December 2005. A selection query was designed to retrieve tumors based on the International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3). The following diseases were retrieved: (9260/3) Ewing Sarcoma, (9364/3) Peripheral Neuroectodermal Tumor, and (9365/3) Askin Tumor. In the SEER database, the use of radiotherapy during “primary therapy” of cancer is recorded and was used in this study with no reference to the site or dose of radiation which are not available. Patients were considered “metastatic” if they had a “distant” SEER stage, defined as a neoplasm that has spread to parts of the body remote from the primary tumor either by direct extension or by discontinuous metastasis to distant organs and tissues or via the lymphatic system to distant lymph nodes [Citation10]. The database provides a unique item entitled the “number of primaries” that indicates the number of records for each patient. To identify patients with second cancer we selected patients with number of primaries more than one. Exclusion criteria included 1) no microscopic confirmation, which is essential to confirm accurate diagnosis of ESFT, 2) a previous recorded malignancy – so that the occurrence of subsequent malignancy related to therapy of a previous tumor is excluded, and 3) the diagnosis of an “in situ” tumor. For some analyses, we compared three separate eras, considering 1985 and 1995 as cut-off points. The first cut-off (1985) marks the incorporation of etoposide and ifosfamide in most treatment regimens [Citation4], while the second (1995) was arbitrarily chosen to study the last decade of available data.
Data analysis
The resulting matrix from SEER*Stat was transferred to MedCalc for Windows, version 9.4.2.0 (MedCalc Software, Mariakerke, Belgium) to perform statistical calculations. Patients with more than one primary were identified and were compared with those who had only one primary. The Multiple primary - Standardized Incidence Ratios (MP-SIR) session of the SEER*Stat program was used to generate a list of second cancers along with associated demographic and clinical data. Only the first malignancy after the first cancer was used for analysis. These cancers were classified into “lymphatic and hematopoietic” vs. “solid tumors”. The χ2 test and the rank-sum (Mann-Whitney U) test were used to compare categorical and numeric variables, respectively. The Kaplan-Meier method and the log-rank test were used to construct and compare survival curves. Events were defined as all-cause mortality for calculating overall survival and the development of second cancer for event probability. Finally, the MPSIR session was used to calculate the observed to expected (O/E) ratio and excess risk in the SEER 9 registries database. The O/E is the ratio of the observed number of individuals with cancer in a selected population (O) to the expected number (E) based on the registry rate file in a similar standard population. An O/E ratio of 1 indicates similar occurrence of cancer in the studied population in comparison to the general population. An O/E ratio above or below one indicates higher or lower incidence of cancer in the studied population, respectively.
Results
Patient characteristics
We identified 1 208 patients with ESFT who were registered in the SEER9 open-access database with diagnosis dates between January 1973 and December 2005. Thirty seven patients were excluded for having no histologic confirmation (n = 10) or for having ESFT as a second malignancy (n = 27). Additionally, five more patients were excluded for having in-situ tumors in the breast (n = 2) or the cervix (n = 3).
Among 1 166 patients, there were 423 (36%) children/adolescents (birth to 20 years old). The average follow-up for all patients was 6.7 years. There were 1 131 patients (median age at first diagnosis, 17 years; 39% females) with no second cancers and 35 patients (median age at first diagnosis, 15 years; 54% females) with a second cancer recorded. There was no significant difference between the two groups in terms of sex, race, age at diagnosis, presence of metastasis or site of first primary tumor (). However, patients with second cancers were more likely to have received radiation as part of primary therapy for ESFT (odds ratio, 2.55; 95% confidence intervals, 1.09 to 6.0). More cases of second cancers were reported in patients whose ESFT was diagnosed in the first period (1973 through 1985; χ2 test, p = 0.0065) but that may simply reflect the longer period of follow-up for this group of patients.
Table I. Ewing Sarcoma Family of Tumors cases included in the analysis by sex, race, age, stage, therapy, site and period of diagnosis, stratified by having or not having a second cancer.
Patients with second cancer
Among 35 patients with second cancers, there were 23 patients with solid tumors and 12 patients with lymphatic and hematopoietic malignancies (). Two patients with second cancers had subsequent records of further cancers and these records – not patients – were not included in further analysis. These included a male patient (27 years old at diagnosis of ESFT) who developed testicular teratocarcinoma at the age of 32 and lung adenocarcinoma at the age of 42 and a female patient (14 years old at diagnosis of ESFT) who developed Hodgkin lymphoma at the age of 21 and cervical cancer at the age of 31.
Table II. Details of second cancer (n = 35) that were recorded in 1 166 patients with Ewing Sarcoma.
When patients who developed second solid tumors were compared to those who developed hematopoietic malignancies (), no significant differences were noticed in sex, race or age at diagnosis of the ESFT. Radiotherapy use was documented in 66% of the patients with second cancers with no significant difference between the two groups. Most second solid tumors (70%) were reported in patients who were treated for ESFT in the first period of incidence (1973 through 1985), while most lymphatic/hematopoietic malignancies (75%) were reported in patients diagnosed after 1986 (p = 0.043). The median interval to the development of second solid tumors was 98 months vs. 36 months for lymphatic/hematopoietic malignancies (p = 0.0007); subsequently, most solid tumors (78%) were diagnosed more than five years after diagnosis of the ESFT while most lymphatic/hematopoietic malignancies (83%) were diagnosed less than five years after ESFT diagnosis. The 5-year survival following the diagnosis of second cancer was better for patients who developed second solid tumors than those who developed lymphatic/hematopoietic malignancies (74% ± 10% vs. 42% ± 14%, respectively) but the difference did not reach statistical significance (p = 0.068). We searched the SEER database specifically for patients with ESFT with records of myelodysplastic syndrome (MDS) and therapy-related MDS but none were identified.
Table III. Patients with second tumor by age at diagnosis, therapy, period of diagnosis of primary tumor, time to develop second cancers, age at diagnosis of second cancer and its outcome, stratified by type of tumor.
Estimating the risk of developing a second cancer
The probability of developing a second malignancy by five, 10 and 20 years after a diagnosis of ESFT was 2.1% ± 0.56%, 4.4% ± 0.95% and 8.0% ± 1.7%, respectively ().
Figure 1. Probability of developing second solid tumors and lymphatic/hematopoietic (hematologic) malignancies following diagnosis of Ewing Sarcoma Family of Tumors in 1 166 patients, calculated using Kaplan-Meier method.
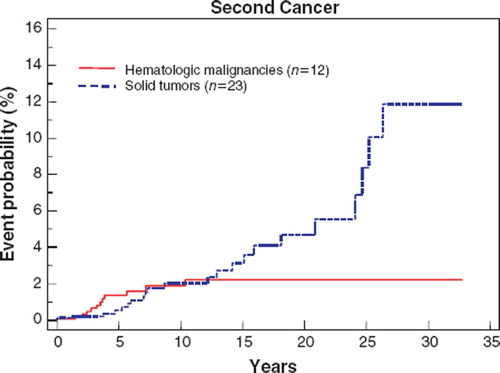
The expected number of cancers in the studied population based on cancer incidence in the general population was 8.77 as calculated by the MP-SIR session of the SEER*Stat program. With 35 events observed, the correlating O/E ratio for all second cancers was 4.01 (95% confidence intervals [CI], 2.80 to 5.58) with an excess of 33.18 per 10 000 persons. To assess the risk excluding cases diagnosed shortly after the diagnosis of ESFT, we calculated the O/E for developing a second cancer after a latency period of one year and the estimated risk was similar (O/E, 4.11; 95% CI, 2.81 to 5.81; excess risk, 35.41). . shows the O/E for different second malignancies in the studied population along with the associated excess risk. The two malignancies that were associated with the highest O/E were acute non-lymphocytic leukemia (ANLL, n = 6, O/E = 51.09; 95% CI = 18.66 to 111.21) and osteosarcoma (n = 4, O/E = 51.08; 95% CI = 13.74 to 130.77). There were six cases of soft tissue sarcomas (O/E, 35.56; 95% CI = 12.98 to 77.40). Eight of 10 patients who developed sarcoma (four with osteosarcoma and four with soft tissue sarcoma) had received radiotherapy as part of primary therapy for ESFT.
Table IV. Observed numbers and standardized incidence ratios of second malignancies.
Patients diagnosed with ESFT before the age of 20 years (children/adolescents) were analyzed separately. They showed higher O/E for developing a second malignancy (9.94; 95% CI = 6.30 to 14.91; excess rate, 36.73 per 10 000) than adults (O/E, 2.01; 95% CI = 1.07 to 3.44; excess rate, 28.58 per 10 000). The increased risk was particularly high for acute non-lymphocytic leukemia (ANLL) (O/E, 71.17; 95% CI, 19.15 to 182.21) and osteosarcoma (O/E, 66.44; 95% CI, 17.9 to 170.10). Children/adolescents were at significantly higher risk for developing breast cancer as second cancer (n = 2; O/E, 10.98; 95% CI = 1.23 to 39.65).
As mentioned above, the year 1985 marks the introduction of etoposide in the front-line therapy of ESFT, which was followed shortly by intensification of alkylating agents. Indeed, the percentage of patients who developed lymphatic/hematopoietic malignancies increased from 0.8% (O/E, 33.24) in patients diagnosed before this year to 2.5% (O/E, 69.68) in patients diagnosed afterwards, but the difference is not statistically significant (χ2 test, p = 0.89). On the other hand, the O/E ratio for developing second solid tumors decreased from 5.93 (95% CI, 3.32 to 9.78) to 1.34 (95% CI, 0.49 to 2.91). Interestingly, the use of beam radiotherapy decreased from 77% in patients diagnosed before 1985 to 58% in patients diagnosed after.
Discussion
In this population-based study, we reviewed the occurrence of second cancer among 1 166 patients with ESFT who were diagnosed from 1973 to 2005 and followed for an average of 6.7 years. There was a 4.4% (±0.95%) probability of having a second malignancy at ten years after the diagnosis of ESFT and 8.0% (±1.7%) at 10 years. In order to further define the magnitude of the problem, we used the observed-to-expected (O/E) ratio to estimate the increased risk. This was provided by the SEER*Stat software and was based on the incidence of cancer in patients with ESFT compared to the incidence in the SEER 9 registries general population. The O/E for developing a second malignancy in ESFT patients was 4.1 and was higher in those diagnosed first time before the age of 20 years (9.94). Previously published reports showed that the incidence of second cancers among ESFT survivors is variable with estimated risk at 5-years of 0.4–8.4% and at 10-years of 2.9 to 35% [Citation3,Citation11–20]. Clearly, our study shows similar results but with possibly more accurate risk estimation due to the large number of patients followed.
Twelve patients in our study were diagnosed with lymphatic/hematopoietic malignancies; among these patients, six had ANLL with more than 50 times increased incidence in comparison to the general population. The percentage of patients developing a second hematopoietic malignancy increased after 1986. This increase may be explained by the introduction of etoposide and intensification of alkylators in the frontline therapy of ESFT [Citation4]. In a study conducted by the Children's Oncology Group, [Citation21] the cumulative incidence of ANLL and therapy-related MDS in patients treated with an etoposide-free standard arm (regimen A, n = 262) was 0.4% at five years. In patients treated using the experimental arm that included a total of 5 grams of etoposide (regimen B, n = 256) the incidence was 0.9%. A markedly increased risk was seen in patients treated with regimen C for metastatic disease (cumulative incidence, 11% at five years). This regimen had higher doses of alkylating agents (cyclophosphamide, ifosfamide) and anthracyclines (doxorubicin) but the same dose of etoposide as regimen B. Five of six patients who developed ANLL/therapy related-MDS had deletion of chromosome 5 or 7 while one patient had 11q23 abnormality. Another factor that may play an important role in increasing the risk of ANLL is the use of granulocyte colony-stimulating factor (a cytokine) [Citation22]. Despite the fact that growth factors (cytokines) allow dose escalations and intensifications, they also permit the survival of genetically damaged stem cells and prevent apoptosis [Citation23]. Thus, it is possible that the interaction of many factors, including high cumulative doses of alkylators, anthracyclins and etoposide, as well as the use of growth factors may cause subclinical stem cell damage, as manifested early by subtle changes in the platelet count mainly in the form of thrombocytopenia and red cell macrocytosis [Citation24]. This damage may be the initiating event of a multi-step process that would eventually result in florid ANLL or therapy related MDS in some patients [Citation24]. Previous reports have demonstrated a direct relation between the degree and duration of the thrombocytopenia and the risk of MDS or ANLL [Citation25,Citation26]. Our study did not identify any patients with a specific diagnosis of MDS. The registration of MDS in the SEER program began in the year 2001, [Citation27] which may largely explain this finding.
Solid tumors developed in 23 patients, representing more than three times increase in the incidence when compared to the general population. The majority of these tumors developed more than five years after the diagnosis of ESFT. Among those, 10 were sarcomas (four osteosarcomas and six soft tissue sarcomas). Fifteen patients with second solid tumors (including eight with sarcoma) had received radiotherapy as part of the primary therapy for ESFT. There was a trend for better 5-year survival in patients who developed solid tumors when compared to lymphatic/hematopoietic malignancies (74% ± 10% vs. 42% ± 14%, respectively; log rank test, p = 0.068). Studies have shown that the probability of developing second solid tumors, particularly sarcomas, correlates with younger age at diagnosis of ESFT and increasing dose of radiotherapy used [Citation28,Citation29]. While we were able to show that second solid tumors were more common in patients diagnosed before the age of 20 years, we were unable to assess the impact of radiotherapy dose due to the lack of radiation-specific information in the SEER database. The O/E ratio for developing second solid tumors declined after the year 1985 and this coincided with a decrease in the use of radiotherapy. The decline in radiotherapy use occurred at a time of improved surgical techniques and systemic therapy intensification using etoposide and alkylators. A remaining factor in developing a second cancer in ESFT survivors is genetic susceptibility. We have shown that younger patients with ESFT were at increased risk of developing a second cancer, which may support a role of genetic predisposition.
This study has the largest number of patients with ESFT being analyzed for the development of second cancers. The strengths of a population-based study, like ours, include the large and unselected cohort of patients with long period of follow-up. The limitations of our study include the lack of complete information, including the chemotherapeutic regimens used, details of radiotherapy and the use of growth factors. In addition, second cancers might have been underreported because of migration of patients from the SEER Registry catchment areas. We excluded patients who had ESFT diagnosed following a previous cancer, those who had no documented histologic confirmation and those with in-situ tumors. The total number of the excluded patients (n = 42) represents a small fraction of the studied population (4%) and is unlikely to have affected our results.
In conclusion, this population-based study confirms that ESFT survivors have a modest, but significantly increased risk of developing second malignancies, and close surveillance should be implemented. This risk is higher in patients diagnosed before 20 years of age and in those patients treated with radiotherapy. Importantly, the risk of developing a second solid tumor continues to increase with time. With the current survival rates for patients with ESFT, it is unlikely that major de-escalation of therapy will take place. Furthermore, intensification of therapy with interval compression [Citation30] and aggressive use of growth factors may even result in a further increase in the risk of developing second malignancies. Nevertheless, this risk appears to be minimal compared with the gains obtained with treatment intensification.
Acknowledgements
The authors would like to acknowledge the King Hussein Cancer Foundation and the American Lebanese Syrian Associated Charities for their support to King Hussein Cancer Center and St. Jude Children's Research Hospital, respectively. The authors have no conflict of interest or financial interest to disclose.
References
- Dorfman HD, Czerniak B. Bone cancers. Cancer 1995; 75:203–10.
- Esiashvili N, Goodman M, Marcus RB, Jr. Changes in incidence and survival of Ewing Sarcoma patients over the past 3 decades: Surveillance epidemiology and end results data. J Pediatr Hematol Oncol 2008; 30:425–30.
- Navid F, Billups C, Liu T, Krasin MJ, Rodriguez-Galindo C. Second cancers in patients with the Ewing Sarcoma Family of Tumours. Eur J Cancer 2008; 44:983–91.
- Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, . Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's Sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003; 348:694–701.
- Inskip PD, Curtis RE. New malignancies following childhood cancer in the United States, 1973-2002. Int J Cancer 2007; 121:2233–40.
- Surveillance, Epidemiology and End Results (SEER) Program(www.seer.cancer.gov)SEER*Stat Database: Incidence - SEER 17 Regs Limited-Use + Hurricane Katrina Impacted Louisiana Cases, Nov 2007 Sub (1973–2005 varying) - Linked To County Attributes - Total U.S., 1969–2005 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, released April 2008, based on the November 2007 submission.
- Wingo PA, Jamison PM, Hiatt RA, Weir HK, Gargiullo PM, Hutton M, Lee NC, Hall HI: Building the infrastructure for nationwide cancer surveillance and control–a comparison between the national program of cancer registries (npcr) and the surveillance, epidemiology, and end results (seer) program (united states). Cancer Causes Control 2003;14:175–93.
- Yu JB, Gross CP, Wilson LD, Smith BD. NCI seer public-use data: Applications and limitations in oncology research. Oncology (Williston Park) 2009; 23:288–95.
- Surveillance research program, national cancer institute seer*stat software (www.Seer.Cancer.Gov/seerstat) version 6.4.4.
- Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International classification of childhood cancer, 3rd. Cancer 2005; 103:1457–67.
- Aparicio J, Segura A, Montalar J, Verdeguer A, Castel V, Sanchez-Heras AB. “Secondary cancers after Ewing Sarcoma and Ewing Sarcoma as second malignant neoplasm”. Med Pediatr Oncol 1998; 30:259–60.
- Bacci G, Longhi A, Barbieri E, Ferrari S, Mercuri M, Briccoli A, . Second malignancy in 597 patients with Ewing Sarcoma of bone treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. J Pediatr Hematol Oncol 2005; 27:517–20.
- Dunst J, Ahrens S, Paulussen M, Rube C, Winkelmann W, Zoubek A, . Second malignancies after treatment for Ewing's Sarcoma: A report of the cess-studies. Int J Radiat Oncol Biol Phys 1998; 42:379–84.
- Gasparini M, Lombardi F, Ballerini E, Gandola L, Gianni MC, Massimino M, . Long-term outcome of patients with monostotic Ewing's Sarcoma treated with combined modality. Med Pediatr Oncol 1994; 23:406–12.
- Kuttesch JF, Jr., Wexler LH, Marcus RB, Fairclough D, Weaver-McClure L, White M, . Second malignancies after Ewing's Sarcoma: Radiation dose-dependency of secondary sarcomas. J Clin Oncol 1996; 14:2818–25.
- McLean TW, Hertel C, Young ML, Marcus K, Schizer MA, Gebhardt M, . Late events in pediatric patients with Ewing Sarcoma /primitive neuroectodermal tumor of bone: The dana-farber cancer institute/children's hospital experience. J Pediatr Hematol Oncol 1999; 21:486–93.
- Paulussen M, Ahrens S, Lehnert M, Taeger D, Hense HW, Wagner A, . Second malignancies after Ewing tumor treatment in 690 patients from a cooperative German/Austrian/Dutch study. Ann Oncol 2001; 12:1619–30.
- Smith LM, Cox RS, Donaldson SS. Second cancers in long-term survivors of Ewing's Sarcoma. Clin Orthop Relat Res 1992; :275–81.
- Travis LB, Curtis RE, Hankey BF, Fraumeni JF, Jr. Second cancers in patients with Ewing's Sarcoma. Med Pediatr Oncol 1994; 22:296–7.
- Strong LC, Herson J, Osborne BM, Sutow WW. Risk of radiation-related subsequent malignant tumors in survivors of Ewing's Sarcoma. J Natl Cancer Inst 1979; 62:1401–6.
- Bhatia S, Krailo MD, Chen Z, Burden L, Askin FB, Dickman PS, . Therapy-related myelodysplasia and acute myeloid leukemia after Ewing Sarcoma and primitive neuroectodermal tumor of bone: A report from the children's oncology group. Blood 2007; 109:46–51.
- Relling MV, Boyett JM, Blanco JG, Raimondi S, Behm FG, Sandlund JT, . Granulocyte colony-stimulating factor and the risk of secondary myeloid malignancy after etoposide treatment. Blood 2003; 101:3862–7.
- Collins MK, Marvel J, Malde P, Lopez-Rivas A. Interleukin 3 protects murine bone marrow cells from apoptosis induced by DNA damaging agents. J Exp Med 1992; 176:1043–51.
- Rodriguez-Galindo C, Poquette CA, Marina NM, Head DR, Cain A, Meyer WH, . Hematologic abnormalities and acute myeloid leukemia in children and adolescents administered intensified chemotherapy for the Ewing Sarcoma Family of Tumors. J Pediatr Hematol Oncol 2000; 22:321–9.
- van Leeuwen FE, Chorus AM, van den Belt-Dusebout AW, Hagenbeek A, Noyon R, van Kerkhoff EH, . Leukemia risk following Hodgkin's disease: Relation to cumulative dose of alkylating agents, treatment with teniposide combinations, number of episodes of chemotherapy, and bone marrow damage. J Clin Oncol 1994; 12:1063–73.
- Morley A, Trainor K, Blake J. A primary stem cell lesion in experimental chronic hypoplastic marrow failure. Blood 1975; 45:681–8.
- Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: Incidence and survival in the United States. Cancer 2007; 109:1536–42.
- Henderson TO, Whitton J, Stovall M, Mertens AC, Mitby P, Friedman D, . Secondary sarcomas in childhood cancer survivors: A report from the childhood cancer survivor study. J Natl Cancer Inst 2007; 99:300–8.
- Tucker MA, D'Angio GJ, Boice JD Jr., Strong LC, Li FP, Stovall M, . Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med 1987; 317:588–93.
- Womer RB, Daller RT, Fenton JG, Miser JS. Granulocyte colony stimulating factor permits dose intensification by interval compression in the treatment of Ewing's Sarcomas and soft tissue sarcomas in children. Eur J Cancer 2000; 36:87–94.