Abstract
Background. Tamoxifen (TAM) is a nonsteroidal antiestrogen that has been widely used in the treatment of breast cancer through its anti-estrogen activity. Recent studies show that TAM is cytotoxic to both estrogen receptor (ER)-positive and ER-negative cells via the induction of apoptosis. However, the molecular mechanisms of this effect are not well understood. In the present study, we investigated the roles of c-Src, ERK, AKT and c-Cbl ubiquitin ligases during TAM-induced apoptosis of MCF-7 cells. Material and methods. MCF-7 cell proliferation and apoptosis were measured by 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, and flow cytometry, respectively. c-Cbl expression, and the activity of c-Src, ERK, AKT were assayed by Western blotting. Overexpression of the wild and the dominant-negative type of c-Cbl (70Z/Cbl) were achieved by transient transfection of plasmids encoding c-Cbl and 70Z/Cbl, respectively, and were confirmed by Western blotting. Statistical analysis was performed using the t-test, and a p-value <0.05 was considered to be statistically significant. Results. A high concentration of TAM (25 μM) induced a time-dependent apoptosis of MCF-7 cells. ERK1/2 and AKT were activated during TAM-induced apoptosis. The ERK1/2 inhibitor PD98059, the PI3K/Akt inhibitor LY294002, and the c-Src inhibitor PP2 all enhanced TAM action. Moreover, the ubiquitin ligase c-Cbl was up-regulated during this process. Over-expression of c-Cbl significantly enhanced the apoptosis-inducing effects of TAM, while 70Z/Cbl suppressed the apoptosis-inducing effects of TAM. Further investigation revealed that, overexpression of c-Cbl significantly downregulated the c-Src protein levels and TAM-induced AKT activity. But 70Z/Cbl significantly upregulated TAM-induced ERK and AKT activity. Conclusions. This study demonstrates that c-Src, ERK, and AKT played a protective role during TAM-induced apoptosis, and that c-Cbl sensitized MCF-7 cells to TAM by modulating the expression of c-Src, and TAM-induced ERK and AKT activity.
Breast cancer is one of the most common causes of cancer-related deaths in women. Since 1990, death rates from breast cancer have decreased by over 25%, and this is, at least in part, due to endocrine therapy [Citation1]. Despite recent advances in the treatment of breast cancer, Tamoxifen (TAM), a nonsteroidal antiestrogen is still a first-line drug in endocrine therapies. TAM acts primarily through the estrogen receptor (ER), modulates gene expression, and leads to growth arrest of ER-positive cells at lower concentrations, in the range 0.1–1 μM [Citation2]. High concentrations (above 5 μM) of TAM are cytotoxic to both ER-positive and ER-negative cells in vitro [Citation3] and shrink some tumors in vivo [Citation4]. The cytotoxic effect is thought to be mediated by the induction of apoptosis, mainly in an ER-independent manner. Several mechanisms have been proposed to explain the ER-independent apoptosis, including the generation of oxidative stress, triggering an increase of intracellular Ca2+, and secretion of active TGF-β [Citation5,Citation6]. Previous studies, however, focused mainly on death signals, and little research has been carried out to investigate the survival pathways.
c-Src is a member of membrane-associated non-receptor tyrosine kinases. The c-Src protein levels and its kinase activity are elevated frequently in breast cancer relative to normal tissue [Citation7]. In breast cancer, overexpression of c-Src has been implicated in the suppression of apoptosis induced by a number of anticancer drugs, including TAM, and results in TAM resistance [Citation7,Citation8]. Pharmacological inhibition of c-Src in MCF-7 cells enhances the growth-inhibitory and apoptosis-inducing effects of TAM [Citation9]. MEKERK1/2 and PI3K/Akt, which have been shown to mediate cell survival, are two main pathways leading to tamoxifen resistance [Citation10,Citation11]. Consequently, there is optimism that factors that downregulate the expression of c-Src, or the kinase activity of ERK and AKT, might have profound effects on TAM sensitivity. The Casitas B-lineage lymphoma (Cbl) family of ubiquitin ligases is a negative regulator of nonreceptor tyrosine kinase and some activated signaling pathways [Citation12,Citation13]. The TKB domain of the Cbl family proteins can interact with c-Src and the p85 subunit of phosphoinositide 3-kinase (PI3K), resulting in their ubiquitination and degradation [Citation12,Citation14]. Makishima et al. reported that patients with loss-of-function mutations of c-Cbl showed poor prognosis in myeloid malignancies [Citation15]. Our study demonstrated recently that the Cbl family can reverse multidrug resistance in solid cancers, and sensitize gastric cancer cells to anthracyclines through negative regulation the activity of ERK and Akt survival signals [Citation16,Citation17]. It is reported that c-Cbl-deficient mice show hyperplastic changes in the mammary glands [Citation18]. The role of c-Cbl in breast cancer, however, is still not clear. Furthermore, there are no data on whether c-Cbl could regulate TAM sensitivity.
In the present study, we evaluated primarily the role of c-Src, ERK, AKT and c-Cbl ubiquitin ligases during TAM-induced apoptosis. The results showed that c-Src, ERK, and AKT provided survival signals under this stress. For the first time, we described that c-Cbl sensitized MCF-7 cells to TAM by targeting the c-Src protein and TAM-induced ERK and AKT activity.
Material and methods
Reagents and antibodies
Rabbit anti-ERK, anti-β-actin, anti-phospho-ERK anti-AKT and mouse anti-Src antibodies were purchased from Santa Cruz Biotechnology (SantaCruz, CA, USA). Rabbit anti-p-Src (Y416) and anti-phospho-AKT (Ser-473) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti-c-Cbl antibody was purchased from Transduction Laboratories (Lexington, KY, USA). Tamoxifen (4-hydroxytamoxifen, TAM), PP2, LY294002 and PD98059 were purchased from Sigma–Aldrich (St Louis, Missouri, USA).
Cell culture
The human breast cancer MCF-7 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C under an atmosphere of 95% air and 5% CO2. Cells were routinely sub-cultured every two to three days and cell samples used were all in the logarithmic growth phase.
Cell viability assay
The effect of TAM on MCF-7 cell proliferation was measured using the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cells were seeded at a density of 5 × 103 cells/well in 96-well plates and incubated overnight, and then different concentrations of TAM were added and further incubated for the indicated times. Thereafter, 20 μl of MTT solution (5 mg/ml) was added to each well and the cells were incubated for another 4 h at 37°C. After the removal of the culture medium, the cells were lysed in 200 μl of dimethylsulfoxide (DMSO) and then the optical density (OD) was measured at 570 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). The following formula was used: cell viability = (OD of the experimental sample/OD of the untreated group) 3100%.
Flow cytometry analysis
Phase distributions of the cell cycle and hypodiploid DNA were determined by flow cytometry. Cells were seeded at a density of 1.5 × 105 cells/well in six-well plates and incubated overnight. After exposed to TAM for the indicated times, the cells were collected and washed twice with phosphate-buffered saline (PBS). After fixing in ice-cold 70% ethanol for 12 h, the samples were washed twice with PBS and then incubated with 20 μg/ml RNase A and 10 μg/ml propidium iodide (PI) for 30 min in the dark. Finally, the samples were evaluated by flow cytometry and data were analyzed using CellQuest software (Becton Dickinson, San Jose, CA, USA). The experiment was repeated three times.
Transfections of plasmid constructs
The cDNA of HA-c-Cbl and HA-70Z/Cbl were kindly provided by Dr Wallace Y. Langdon (University of Western Australia, Australia). HA-c-Cbl, HA-70Z/Cbl was subcloned into the pSVL expression vector (Amersham Biosciences, Piscataway, NJ). The dominant negative form of c-Cbl (70Z/Cbl), which is a deletion mutant form lacking 17 internal amino acids (amino acids 366 to 382), and has no ubiquitin-protein ligase function, was described previously [Citation19]. The cells were seeded at a density of 4 × 105 cells/well in six-well plates and incubated overnight, and then they were transiently transfected with the c-Cbl/PSVL and 70Z/Cbl/PSVL plasmids using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA), according to the protocol suggested by the manufacturer. Empty vector was used as the transfection control.
Western blot analysis
Western blotting was performed using standard techniques as previously described. Briefly, cells were washed twice with ice-cold PBS and solubilized in 1% Triton lysis buffer (1% Triton X-100, 50 mM Tris–Cl pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 2 μg/ml aprotinin) on ice. Protein concentration was determined by Lowry method. Total proteins (30–50 μg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to nitrocellulose membranes (Immoblin-P, Millipore, Bedford, MA, USA). Membranes were blocked with 5% skim milk in TBST [10 mM Tris (pH 7.4), 150 mM NaCl, 0.1% Tween 20] for 2 h at room temperature. Membranes were then incubated overnight at 4°C in 5% skim milk in TBST containing either p-Src, c-Src, β-actin, p-ERK, ERK, p-Akt, Akt, or c-Cbl antibodies. After washing with TBST, membranes were reacted with the appropriate horseradish peroxidase-conjugated secondary antibodies for 30 min at room temperature. After extensive washing with TBST, proteins were visualized by the enhanced chemiluminescence reagent (SuperSignal WesternPico Chemiluminescent Substrate; Pierce, Rockford, IL, USA).
Statistical analysis
All the presented data were confirmed in at least three independent experiments and are expressed as the mean ± SD. Statistical comparisons were made by Student's t-test. P<0.05 was considered statistically significant. IC50 values were calculated by non-linear regression analysis using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Effects of TAM on the cell viability and apoptosis of MCF-7 cells
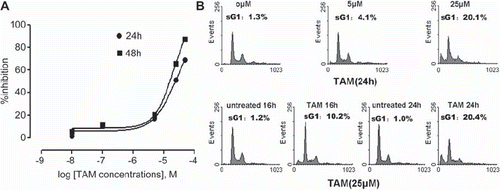
To evaluate the effect of TAM on the proliferation of MCF-7 cells, the cells were treated with indicated concentrations of TAM for 24 h and 48 h. As shown by MTT assay, TAM triggered a time- and dose-dependent inhibition of proliferation (). The 50% inhibitory concentrations (IC50) at 24 h and 48 h were 23.6 ± 1.2 μM and 14.4 ± 1.1 μM respectively. Flow cytometry analysis showed that TAM concentration up to 25 μM induced an obvious apoptosis (). After treatment with 25 μM TAM, the percentage of apoptotic cells was about 10% at 16 h, and 20% at 24 h, while it was only 1.2% at 16h, and 1.0% at 24h in the untreated cells (p < 0.05). These results indicated that a high concentration of TAM induced apoptosis in a time-dependent manner.
Figure 1. Effects of TAM on MCF-7 cell viability and apoptosis. (A) MCF-7 cells were treated with different concentrations of TAM for 24 h and 48 h. Cell growth inhibition was assessed by the MTT assay. Points represent means ± SD. Sigmoidal dose response curves were derived from fitting the data to a non-linear regression program (Graph Pad Prism). (B) MCF-7 cells were treated with different concentrations of TAM for the indicated times. The changes in cell cycle phase distribution were assessed using flow cytometry with propidium iodide (PI) staining.
Role of c-Src, ERK1/2 and AKT during TAM-induced apoptosis
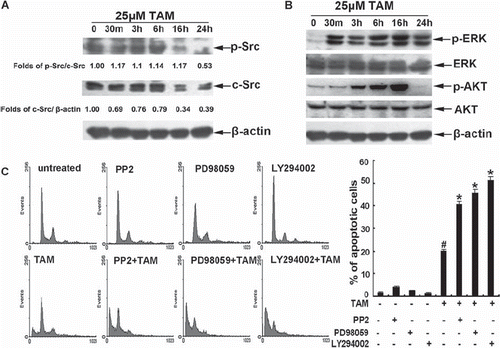
To assess the effect of TAM on the activity of survival signals, the cells were treated with 25 μM TAM for up to 24 h. Western blots showed that the cells exhibited endogenous c-Src activity, and both the activity and expression of c-Src was reduced significantly at 16 h and 24 h (). ERK was activated rapidly at 30 min, and lasted up to 24 h. The activity of AKT was upregulated at three hours and decreased below baseline at 24 h (). To further assess the role of c-Src, ERK, and AKT activation during TAM-induced apoptosis, the cells were pre-exposed to 10 μM of the c-Src inhibitor PP2, 20 μM of the ERK1/2 inhibitor PD98059 or 10 μM of PI3K/Akt inhibitor LY294002, respectively. And then the cells were treated with TAM for 24 h. Flow cytometry analysis showed that PP2, PD98059, and LY294002 significantly elevated the percentages of the sub-G1 population in TAM-treated cells, from approximately 20.1% up to 40.67%, 45.88%, and 51.4%, respectively (). These results showed clearly that the endogenous c-Src activity and TAM-induced ERK and AKT activity provided survival signals against TAM-induced apoptosis.
Figure 2. Role of c-Src, ERK1/2 , and AKT during TAM-induced apoptosis. (A) The cells were treated with 25μM TAM for the indicated times. The total protein was isolated for measuring phospho-Src, c-Src and β-tubulin levels by Western blotting. (B) The cells were treated with 25μM TAM for the indicated times. The total proteins were isolated for measuring phospho-ERK, ERK, phospho-AKT, AKT and β-tubulin levels by Western blotting. (C) The cells were pre-treated with 10 μM of PP2, 20 μM of PD98059, or 10 μM LY294002 for 1 h, and then treated with 25 μM TAM for 24 h. The changes in cell cycle phase distribution were assessed by flow cytometry analysis. Data are means ± SD of three independent experiments (#p < 0.05 compared to the untreated group cells; *p < 0.05 compared to cells treated with only TAM).
Effects of c-Cbl on the TAM sensitivity of MCF-7 cells
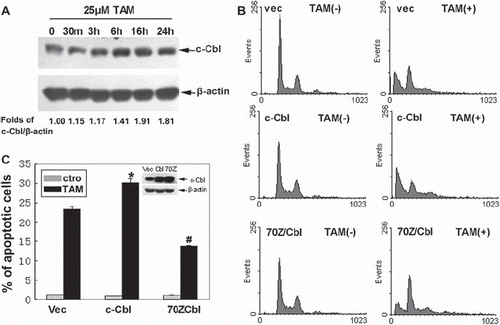
To explore whether c-Cbl was involved in TAM-induced apoptosis, we examined the expression level of c-Cbl. Western blots showed that c-Cbl protein was expressed in MCF-7 cells. After treatment with TAM for 24 h, the expression of c-Cbl was approximately 1.8-fold that of the untreated control group (p < 0.05) (). Furthermore, the cells were transiently transfected with empty vector, a plasmid encoding wild type c-Cbl or dominant negative type of c-Cbl (70Z/Cbl), losing the ability to promote the ubiquitination. Flow cytometric results showed that wild type c-Cbl enhanced apoptosis from 23.3 to 30.2% at 24 h of treatment with 25 μM TAM, while 70Z/Cbl reduced aopotosis from 23.3 to 13.75% (). These data implied that c-Cbl could promote TAM sensitivity, and that the promoting effects were dependent on its ubiquitin ligase activity.
Figure 3. Effects of c-Cbl on sensitivity of the cells to TAM. (A) Cells were untreated, or treated with 25 μM TAM for 30m, 3 h, 6 h, 16 h or 24 h. The expression of c-Cbl proteins was analyzed by Western blotting. (B) Cells were transiently transfected with plasmids encoding Vec, c-Cbl or 70Z/Cbl for 48 h. The cells were then untreated or treated with 25 μM TAM for 24 h, then the changes in cell cycle phase distribution were assessed by flow cytometric analysis. The final results are summarized in the bar graphs. (C) Data are means ± SD of three independent experiments (*p < 0.05 compared to the cells in the Vec group; #p < 0.05 compared to the cells in the Vec group).
Effects of c-Cbl on the expression of c-Src and TAM-induced ERK and AKT activation
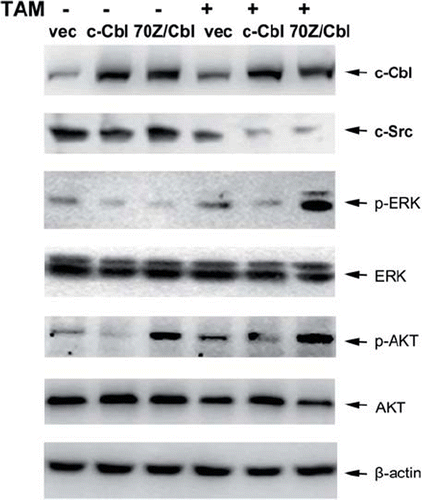
To elucidate the mechanism by which c-Cbl sensitize MCF-7 cells to TAM, the expression of c-Src was assessed. Western blot analysis showed that the levels of c-Src were decreased in the untreated c-Cbl-transfected cells, while 70Z/Cbl have no effect on the expression of c-Src. However, c-Src was downregulated in both c-Cbl and 70Z/Cbl transfected cells after TAM treatment (). On the other hand, compared with the untreated vector control, ERK activity was partly suppressed in c-Cbl-transfected cells, but strongly enhanced in 70Z/ Cbl-transfected cells after TAM treatment. The endogenous and TAM-induced AKT activity was suppressed in c-Cbl-transfected cells, and strongly upregulated in 70Z/Cbl-transfected cells. These results implied that c-Cbl might enhanced TAM-induced apoptosis by suppressing c-Src protein expression and restraining TAM-induced ERK and AKT activity.
Figure 4. Effect of c-Cbl on the survival signals. Cells were transiently transfected with plasmids encoding Vec, c-Cbl or 70Z/Cbl, and then the cells were untreated, or treated with 25 μM TAM for 6 h. Then, the changes of c-Src, and the activities of ERK and AKT were analyzed by Western blotting.
Discussion
TAM has been used in the treatment of breast cancer for more than three decades. Earlier studies have shown that TAM inhibits mainly the proliferation of ER-positive cells by competing with E2 and other estrogens for binding to the ligand binding site of ER [Citation20]. Recent studies have revealed that high concentrations (above 5 μM) of TAM induce apoptosis [Citation3], which mainly depends on the death signals. Little is known, however, about the role of survival signals. c-Src, ERK and AKT are main survival signals in breast cancer cells. Inhibition of these signals enhanced the cytostatic effects of TAM in breast cancer cells [Citation21,Citation22]. Tian et al. [Citation23] also reported that ERK was activated, and plays a protective role against high concentration of TAM-induced apoptosis in the human glioma cell line C6. Whether the activation of these pathways affect the apoptosis induced by high concentration of TAM in breast cancer cells still remains unclear. In the present study, MCF-7 cells had endogenous c-Src activity. TAM induced cells apoptosis, accompanied by downregulation of c-Src expression, the transient activation of AKT, and the continuous activation of ERK. Inhibition of these kinases with PP2, Ly294002 or PD98059, respectively, enhanced TAM-induced MCF-7 cells apoptosis. These results suggest that endogenous c-Src activity and TAM-stimulated ERK and AKT activation also antagonized TAM-induced apoptosis.
Data have emerged recently implicating the Cbl family of proteins, c-Cbl and Cbl-b, function as negative regulators of several signal transduction pathways through their E3 ubiquitin ligase activity [Citation24]. They can influence the balance between proliferation and apoptosis by mediating related protein degradation [Citation15]. Recent studies have suggested that Cbl family proteins enhanced apoptosis. Miyake et al. [Citation25] report that overexpression of c-Cbl sensitized NIH3T3 cells to undergo apoptotic cell death through negative regulation of PDGF-dependent protection. Our previous study also reported that Cbl-b, a homologue of c-Cbl, was upregulated during anthracycline-induced apoptosis, and overexpression of Cbl-b sensitized gastric cancer cells to chemotherapy [Citation17]. On the other hand, ubiquitin ligases inactivating mutations of c-Cbl and Cbl-b might confer a chemoresistant phenotype. The loss-of-function mutations of c-Cbl results in chemotherapy resistance and sequential poor prognosis in acute myeloid leukemia [Citation15,Citation26]. Our previous data showed that a loss-of-function mutation of Cbl-b leads to chemoresistance [Citation17], but the role of c-Cbl mutation in breast cancer endocrine therapy is not clear. The present results demonstrated that the expression of c-Cbl increased during TAM-induced apoptosis. Overexpression of c-Cbl enhanced TAM-induced MCF-7 cells apoptosis, while 70Z/Cbl, which was a deletion mutation of c-Cbl without E3 ubiquitin-ligase activity, significantly suppressed the apoptosis-inducing effects of TAM. Additionally, TAM-induced up-regulation of c-Cbl was accompanied by the downregulation of c-Src protein, which is one of the target proteins of c-Cbl in other cell lines [Citation12,Citation19]. The downregulation of c-Src was suppressed by 70Z/Cbl in untreated cells. However, this downregulation was not suppressed by 70Z/Cbl after TAM treatment. These observations suggest that a complicated mechanism other than the ubiquitin ligase activity of c-Cbl might be involved in the downregulation of c-Src after TAM treatment. It is reported that ERK and AKT are also modulated by Cbl proteins [Citation14,Citation16,Citation17]. In the present study, overexpression of c-Cbl also suppressed ERK and AKT activation, and 70Z/Cbl strongly enhanced TAM-stimulated ERK and AKT activity. Taken together, these results indicate that c-Cbl might enhance TAM sensitivity by downregulating c-Src expression, and limiting the activation of ERK and AKT, and this action was partially dependent on its ubiquitin ligase activity.
In conclusion, this study demonstrates that c-Src, ERK, and AKT played a protective role against TAM-induced apoptosis. c-Cbl acted as a brake to cut off the protective signaling pathway under this stress by downregulating the level of endogenous c-Src and restraining TAM-induced ERK and AKT activity. The results suggest that detecting the expression or the mutation of c-Cbl may provide the means to predict the therapeutic effect of TAM. Further studies are needed to obtain a better understanding of the role of the c-Cbl in endocrine therapy.
Acknowledgements
This work was supported by grants from the National Science Foundation of China (No. 30770993, No. 30700807), from Science and Technology Foundation of Liaoning Province (2004225004-12).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ 2001;322:1528–32.
- Otto A, Paddenberg R, Schubert S, Mannhertz H. Cell-cycle arrest, micronucleus formation, and cell death in growth inhibition of MCF-7 breast cancer cells by tamoxifen and cisplatin. J Cancer Res Clin Oncol 1996;22:603–12.
- Perry R, Kang Y, Greaves B. Effects of tamoxifen on growth and apoptosis of estrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol 1995;2: 238–45.
- Hui AM, Zhang W, Chen W, Xi D, Purow B, Friedman GC, . Agents with selective estrogen receptor (ER) modulator activity induce apoptosis in vitro and in vivo in ER-negative glioma cells. Cancer Res 2004;64:9115–23.
- Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001;6:469–77.
- Ferlini C, Scambia G, Marone M, Distefano M, Gaggini C, Ferrandina G, . Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor-negative human cancer cell lines. Br J Cancer 1999;79:257–63.
- Reissig D, Clement J, Sanger J, Berndt A, Kosmehl H, Bohmer FD. Elevated activity and expression of Src-family kinases in human breast carcinoma tissue versus matched non-tumor tissue. J Cancer Res Clin Oncol 2001;127: 226–30.
- Hiscox S, Morgan L, Green T, Nicholson RI. Src as a therapeutic target in anti-hormone/anti-growth factor-resistant breast cancer. Endocrine-Related Cancer 2006;13(Suppl 1): S53–S59.
- Planas-Silva MD, Hamilton KN. Targeting c-Src kinase enhances tamoxifen's inhibitory effect on cell growth by modulating expression of cell cycle and survival proteins. Cancer Chemother Pharmacol 2007;60:535–43.
- Ghayad SE, Vendrell JA, Larbi SB, Dumontet C, Bieche I, Cohen PA. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer 2010; 126:545–62.
- Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA 2002;99:14783–8.
- Yokouchi M, Kondo T, Sanjay A, Houghton A, Yoshimura A, Komiya S, . Src-catalyzed phosphorylation of c-Cbl leads to the interdependent ubiquitination of both proteins. J Bio Chem 2001;276:35185–93.
- Rao N, Dodge I, Band H. The Cbl family of ubiquitin ligases: Critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol 2002;71:753–63.
- Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem 2001; 276:4872–8.
- Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, . Mutations of E3 ubiquitin ligase Cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 2009;27:6109–16.
- Zhang Y, Qu X, Hu X, Yang X, Hou K, Teng Y, . Reversal of P-glycoprotein-mediated multi-drug resistance by the E3 ubiquitin ligase Cbl-b in human gastric adenocarcinoma cells. J Pathol 2009;218:248–55.
- Qu X, Zhang Y, Li Y, Hu X, Xu Y, Xu L, . Ubiquitin ligase Cbl-b sensitizes leukemia and gastric cancer cells to anthracyclines by activating the mitochondrial pathway and modulating Akt and ERK survival signals. FEBS Letters 2009;583:2255–62.
- Murphy, MA, Schnall, RG, Venter, DJ, Barnett L, Bertoncello I, Thien, CB, . Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol 1998;18:4872–82.
- Kyo S, Sada K, Qu X, Maeno K, Miah SM, Kawauchi-Kamata K, . Negative regulation of Lyn protein-tyrosine kinase by c-Cbl ubiquitin-protein ligase in FcεRI-mediated mast cell activation. Genes Cells 2003;8:825–36.
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002;295:2465–8.
- Herynk MH, Beyer AR, Cui Y, Weiss H, Anderson E, Green TP, . Cooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptorpositive human breast cancer cells. Mol Cancer Ther 2006;5: 3023–31.
- Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther 2002;1:707–17.
- Tian F, Wu H, Li Z, Wang N, Huang J, Li C, . Activated PKCα/ERK1/2 signaling inhibits tamoxifen-induced apoptosis in C6 cells. Cancer Invest 2009;27:802–8.
- Sanjay A, Horne WC, Baron R. The Cbl family: Ubiquitin ligases regulating signaling by tyrosine kinases. Sci STKE 2001;2001:pe40.
- Miyake S, Mullane-Robinson KP, Lill NL, Douillard P, Band H. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A critical role for Cbl tyrosine kinase-binding domain. J Biol Chem 1999;274:16619–28.
- Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, . Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 2007;110: 1004–12.