Abstract
Background. One of many approaches being evaluated in experimental models and in the clinic for the treatment of cancer is the use of antibodies conjugated to various drugs or radionuclides. The aim of the present study was to compare the toxicity profiles of radioimmunoconjugates and drug-immunoconjugates based on the same monoclonal antibody, evaluated in the same experimental model, that much resembles human studies. The pattern of dose-limiting toxicity of a monomethylauristatin-conjugated monoclonal antibody (BR96) was compared to that of the same antibody conjugated with lutetium-177, and to the same non-conjugated antibody. Material and methods. Rats with established colon carcinoma were injected with monomethylauristatin-conjugated mAb-BR96, 177Lu-BR96, or non-conjugated BR96. Liver, kidney, and myelotoxicity were assessed for 100 days by analysis of blood parameters. Body weight and therapeutic effects was also monitored. Results. Myelotoxicity was found to be dose limiting for the radionuclide BR96 conjugate. The dose-limiting factor was prolonged suppression of leukocytes (>28 days) with increased risk of infections. For monomethylauristatin-conjugated BR96, liver toxicity was dose limiting, whereas no dose-limiting toxicity was observed with non-conjugated BR96. Both the drug-immunoconjugate and the radioimmunoconjugate resulted in decreased platelet counts, but the time to nadir and duration differed. Conclusion. The two conjugates resulted in different patterns of toxicity. By using the two conjugates of BR96 in a sequential therapeutic design it could be possible to increase the therapeutic window and hence probably the efficacy without significantly increasing the toxicity. This concept is regarded as valid regardless of conjugate or model chosen.
One of many approaches being evaluated in experimental models and in the clinic for the treatment of cancer is the use of antibody-drug conjugates [Citation1,Citation2]. The strategy is to target a tumor-associated marker with an antibody that can deliver a toxic payload to the cancer cell. The specific targeting of cancer cells means that the toxic payload has much greater potency than conventional chemotherapeutics, while reducing the risk of toxic side effects.
The auristatins are synthetic antimitotic agents that destabilize microtubules, and are structurally related to the potent marine cyclic pentapeptide dolastatin-10 [Citation3]. These drugs are much more potent than conventional chemotherapeutic drugs. A dipeptide (valine-citrulline) linker, cleavable by lysosomal proteases (e.g. cathepsin B), has recently been used to link auristatins to different monoclonal antibodies [Citation4]. Monomethylauristatin F (MMAF) is an auristatin that possesses a negatively charged C-terminal phenylalanine residue which limits cell permeability, in contrast to monomethylauristatin E (MMAE), which is uncharged and able to pass through the cell membrane. Intracellular linker proteolysis of MMAE conjugates may therefore expose surrounding normal tissues to free drug. MMAF does not exhibit this so-called “bystander effect”, i.e. the exposure of non-conjugate-binding tumor cells to extracellular free drug. MMAF is of great interest for targeted delivery, since the free drug has higher cell cytotoxicity, lower normal tissue toxicity, and much higher aqueous solubility than MMAE [Citation4]. An anti-CD30-MMAE conjugate has been evaluated in a phase I clinical trial on patients with relapsed or refractory Hodgkin's lymphoma [Citation5]. A phase I/II study using CR011-vcMMAE in patients with unresectable stage III or stage IV melanoma is ongoing [Citation6].
In our experimental model with colon carcinoma in syngeneic immunocompetent rats, administration of radionuclide-conjugated mAbs (e.g. 90Y- and 177Lu-conjugated BR96) has previously been shown to result in efficient tumor therapy of inoculated local tumors at the maximal tolerable dose (MTD) [Citation7]. However, treated rats with persistent local complete remission finally developed metastatic disease, indicating the need for new therapeutic strategies.
The aim of the present study was to compare the toxicity profiles of radioimmunoconjugates and drug-immunoconjugates based on the same monoclonal antibody, evaluated in the same experimental model, greatly resembling human studies. The dose levels chosen for each of the conjugates corresponded to the MTDs established in previous studies using the same model [Citation8,Citation9]. This syngeneic tumor model in immunocompetent rats is relevant to the clinical situation in humans, since the Lewis Y glycoprotein (Ley) recognized by BR96 is also expressed in sensitive normal tissues such as gastrointestinal epithelium. A fraction of Ley-positive tumors in locally cured rats developed distant metastases in the later course of the study, as is the case in humans. By using a subperitoneal tumor model, the tumor mimics the clinical situation by growing invasively in the surrounding tissue. (Furthermore, the size of the tumor can be easily assessed by palpation). Doxorubicin-conjugated BR96 has previously been evaluated in our rat colon carcinoma model [Citation10] and in combination with docetaxel in lung cancer patients [Citation11].
Material and methods
Monoclonal antibody
The chimeric (mouse/human) monoclonal IgG1 antibody BR96 (Seattle Genetics Inc., Seattle, WA, USA), binding the tumor-associated Ley, was used. Ley is expressed on the majority of human epithelial tumors, including breast, gastrointestinal tract, pancreas, non-small-cell lung, cervical, and ovarian cancers, as well as some melanomas. As with the majority of tumor-associated mAbs, BR96 also reacts with some normal tissues, primarily the epithelial cells of the gastrointestinal tract.
The drug-immunoconjugate
To 500 mg BR96 at a concentration of 10 mg/ml in phosphate-buffered saline (PBS), the following were added: 50 ml of a solution of 500 mM sodium borate, 500 mM NaCl, pH 8.0, and 110 μl 0.5 M diethylenetriaminepentaacetic acid (DTPA) (Aldrich). The mixture was heated to 37°C, and 85.1 μl 100 mM tris-carboxyethyl phosphine (TCEP) (Aldrich) (2.5 equivalents) was added. The mixture was maintained at 37°C for two hours. To the reduced antibody, 5.3 ml dimethyl sulfoxide (DMSO) and 897 μl of a DMSO solution of maleimidocaproylvaline-citrulline-p-aminobenzyl carbamoyl-monomethyl auristatin F (20.5 mM) was added. When a test using Ellman's reagent (5,5′-dithio-bis(2-nitrobenzoic acid)) showed that antibody thiols had been consumed, the maleimido reagent was quenched by the addition of 817 μl of a neutralized aqueous solution of N-acetylcysteine. The conjugate was diluted 1:10 using 25 mM NaOAc buffer, pH 5.0 (loading buffer), then applied to a sulfopropyl cation-exchange column, washed with loading buffer, and eluted with three times the standard concentration of PBS. Late-eluting fractions were analyzed separately, and then pooled. The collected product was diluted three times with water and concentrated. A total of 385 mg of antibody drug conjugates was obtained in a volume of 37 ml.
To the isolated conjugate 4 ml of 500 mM sodium borate, 500 mM NaCl solution, pH 8.0 were added, followed by 100 μl of a 100 mM DMSO solution of NHS-LC-biotin (Pierce; 3.8 equivalents). The reaction mixture was left at room temperature for three hours, and then purified using cation-exchange chromatography, in the same manner as the original conjugate. The eluate collected was diluted three times in water, concentrated, and filtered through a 0.2 μm membrane, yielding 21.5 ml of biotinylated conjugate at a concentration of 14.9 mg/ml in PBS.
The radioimmunoconjugate
BR96 was conjugated with the biotinylated DOTA reagent 1033 [Citation12], as previously described [Citation8,Citation13]. The following procedure was used for labeling with 177LuCl3 (MDS Nordion, Vancouver, Canada). Both the 1033-antibody conjugate in 0.25 M ammonium acetate buffer and the radionuclide solution were preheated at 45°C for 10 min. The 1033-antibody solution was then added to the radionuclide-containing vial and incubated at 45°C for 15 minutes. The reaction was quenched with an excess of DTPA for five minutes. The radiolabeled immunoconjugate was then diluted in 1% human serum albumin (HSA). The radiochemical purity of the labeled immunoconjugate was determined by instant thin-layer chromatography (ITLC) (1 × 9 cm silica-gel-impregnated fiberglass sheet, eluted in 0.1 M EDTA). Separation by size-exclusion chromatography together with high-performance liquid chromatography (HPLC) (7.8 3 300 mm molecular sieving column, Phenomenex SEC S3000; Phenomenex, Torrance, CA, USA, eluted in 0.05 M sodium phosphate at 1.0 ml/min) was used to control the radiochemical purity and to detect signs of aggregation or fragmentation.
Syngeneic rat tumor model
Immunocompetent rats of the Brown Norwegian (BN) strain (Harlan, Horst, the Netherlands) were used. As demonstrated by immunohistochemistry, BN rats express the BR96-binding epitope in sensitive normal tissues, such as the gastrointestinal organs (esophagus, stomach, intestines, pancreas), hence mimicking the human situation [Citation10]. BN7005-H1D2 is a single-cell clone of a rat colon carcinoma originally induced by 1,2-dimethylhydrazine in a BN rat. BN7005-H1D2 cells were cultured in RPMI 1640 medium (Euroclone, Devon, UK) supplemented with 10% fetal calf serum (FCS), 1 mM sodium pyruvate, 10 mM HEPES buffer solution and 29.3 μM gentamicin at 37°C in a humidified atmosphere containing 5% CO2. Cells were washed in saline, trypsinized and washed in medium + 10% FCS. Animals were inoculated subperitoneally with 3 × 105 cells (in 50 μl medium). Experiments were initiated 12–14 days after inoculation (tumor size 10 × 15 mm). The animals were kept under standard conditions and fed with standard pellets and fresh water. Studies were conducted in compliance with Swedish legislation on animal rights and protection, and were approved by the Animal Ethics Committee of Lund University.
Experimental design
The dose levels chosen for each of the conjugates corresponded to their respective MTD, established in previous studies using the same model [Citation8,Citation9]. One group of rats (group 1; N = 6) was injected intravenously with 15 mg/kg body weight (3.6 mg MMAF/m2) of b-BR96-MMAF. The other group (group 2; N = 6) was injected intravenously with 600 MBq/kg (6904 MBq/m2; 625 μg BR96/kg body weight) of biotinylated 177Lu-BR96. A control group (group 3; N = 6) was given 15 mg/kg body weight of non-conjugated BR96.
The body surface area of the rats was calculated as [9.1 × (body weight in grams)0.66] [Citation14] to make it possible to compare our results with those of other studies in which the administered dose is given in terms of mg/m2 body surface.
Measures of toxicity
To evaluate myelotoxicity, blood samples were collected from the tail artery twice a week during the first 28 days post-injection (p.i.) and then once weekly up to day 100 p.i. White blood cell counts (WBC), red blood cell counts (RBC), and platelet counts (PLT) were analyzed in a Medonic Cell Analyzer (Vet CA530, Boule Medical, Stockholm, Sweden).
In addition, plasma was collected, and the levels of aspartate aminotransferase (ASAT), alanine aminotransferase (ALAT), gamma-glutamyl transferase (G-GT), alkaline phosphatase (ALP), bilirubin, and creatinine were measured to determine liver and kidney toxicity.
The body weight, tumor size and general status of the animals were recorded at the time of blood sampling.
The toxicity was graded according to the National Cancer Institute Common Terminology for Adverse Events (CTCAE version 3.0) to compare the severity of the toxicity between the various regimens used (). The WBC, PLT, and RBC are not included in as the CTCAE definitions of grades for these parameters cannot be easily applied to rat data.
Table I. Toxicity.
Therapeutic efficacy and endpoints of the study
The size of the primary tumor (i.e. at the site of inoculation) was measured in two perpendicular directions using a caliper, twice a week up to day 28 p.i. and then once weekly. The tumor volume was calculated using the formula: 0.4 × (L × W2), where L = the length of the longest axis and W = the length of the axis perpendicular to L. Complete remission is used to describe a tumor that regressed completely and was not palpable for at least a week.
Body weight, general status, toxicity based on plasma analyses, effects on the primary tumor, and outcome were monitored during a total of 100 days p.i. The animals were sacrificed when the tumor burden became high (> 25 × 25 mm), when a marked decrease in general status was seen, or at the end of the study (100 days p.i.).
Results
Preparation of immunoconjugates
The drug-antibody conjugates were formed through controlled reduction of interchain disulfide bonds within the BR96 mAb using TCEP as a reducing agent [Citation15]. The drug linker derivative was then added, and the conjugate was purified before biotinylation. The final antibody-drug conjugate had an average of 4.4 MMAF molecules per mAb (21μg MMAF/mg BR96) and 3.2 biotin molecules per mAb. No aggregation or fragmentation was observed by size-exclusion HPLC, and no free drug was detected.
After conjugation, the average number of 1033 molecules per BR96 molecule was determined to be 2.6. After radiolabeling, the specific activity of the 177Lu-1033-BR96 conjugate was 53 MBq/nmole (820 MBq/mg). ITLC showed the radiochemical purity to be 99%. No aggregation or fragmentation was observed with HPLC.
Comparison of toxicity
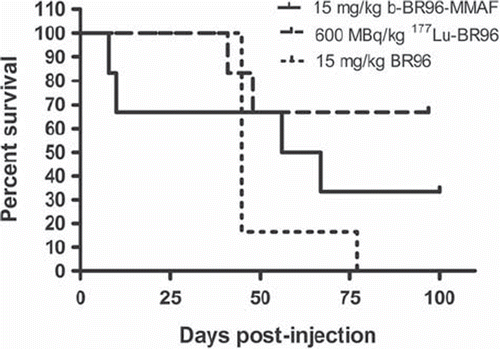
Survival. Deaths before day 21 were attributed to toxicity. All late deaths (after day 45) were attributed to metastatic disease. Only two tumor-bearing animals injected with b-BR96-MMAF (group 1) survived to the end of the study (day 100). Two of the rats that survived, for eight and ten days, died from side effects, probably due to liver toxicity, whereas the two other rats, which survived for 56 and 67 days, were sacrificed because of extensive growth of distant metastases (). All rats given 177Lu-BR96 (group 2) survived for at least 41 days; two rats were sacrificed on day 41 and 48 because of distant metastases (large tumor burden), whereas the other four rats survived the entire observation period (100 days). Five of the six rats given non-conjugated BR96 (group 3) were sacrificed 42 days p.i. because of high tumor burdens (four local recurrences and one with distant metastases), and the sixth rat in this group was sacrificed 77 days p.i. because of extensive growth of distant metastases.
Figure 1. Survival of animals given b-BR96-MMAFconjugates (group 1), 177Lu-labeled BR96 (group 2), and unconjugated BR96 (group 3) monitored for 100 days. Deaths before day 21 were attributed to toxicity. All the late deaths (after day 45) were attributed to metastatic disease. Four of the six animals treated with unconjugated BR96 (group 3) died due to local recurrences and the other two due to distant metastases. No animal treated with conjugated BR 96 (group 1&2) had a local recurrence. At termination of the study (day 100), two rats in group 1, and four rats in group 2 were still alive. In one rat in group 2, metastatic disease was detected.
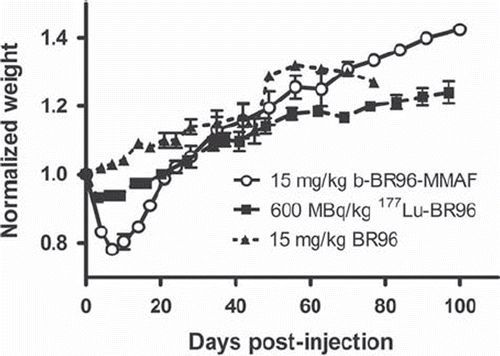
Body weight loss. All animals injected with conjugated BR96 lost weight (); the nadir being reached on days 4–7. In rats injected with 15 mg/kg b-BR96-MMAF (group 1), the mean peak weight loss was 22% (20–26%), followed by rapid individual recovery. The two rats surviving for ≤ ten days had body weight losses of 21% and 24%, and did not differ in this respect from the other rats in the group. The rats injected with 600 MBq/kg 177Lu-BR96 (group 2) had a mean peak weight loss of 7% (6–8%). These animals started to gain weight on day 7 p.i., and they regained their initial weight on day 21 p.i., but still had a lower body weight than the rats in group 3. The weight progression of animals injected with unconjugated BR96 (group 3) was unaffected by the administration.
Figure 2. Normalized body weights of animals given b-BR96-MMAFconjugates (group 1), 177Lu-labeled BR96 (group 2), and unconjugated BR96 (group 3).
Blood parameters. Myelotoxicity was monitored by quantification of blood cell counts. Both groups injected with conjugated BR96 showed a decrease in platelets to about the same level. The b-BR96-MMAF group (group 1) reached the nadir on day 10, and the values had recovered in all animals 21 days p.i. (). The two rats not surviving beyond day 10 did not show the greatest decrease in platelets. The group given 177Lu-BR96 (group 2) reached the nadir later, on day 16, and also recovered more slowly, reaching pre-treatment values on day 34.
Figure 3. Numbers of leukocytes (A) and platelets (B) in animals given b-BR96-MMAF conjugates (group 1), 177Lu-labeled BR96 (group 2), and unconjugated BR96 (group 3).
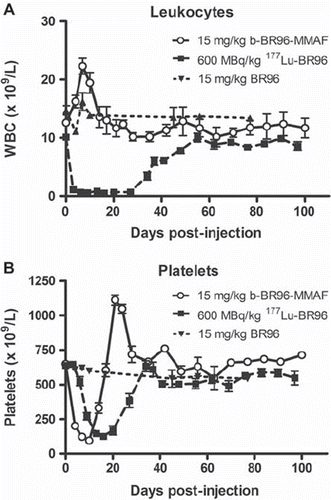
The effects of the treatments on the number of leukocytes were remarkably different. The WBC increased after injection of b-BR96-MMAF (group 1; ), whereas it decreased in animals given the radioimmunoconjugate (group 2; ). No decrease was seen in erythrocytes in any of the groups (data not shown).
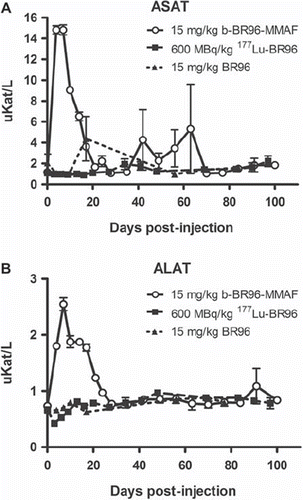
Hepatotoxicity. All rats injected with b-BR96-MMAF (group 1) showed increased levels of ASAT (grade 3 toxicity, n = 6) and ALAT (grade 2 toxicity, n = 5; grade 3 toxicity, n = 1) during the first three weeks after injection (, ). The increase was detected already on day 4 (the first day of sampling p.i.) and peaked on day 7. The rat surviving for only eight days had the highest ALAT value (3.0), and the rat surviving for ten days had a value of 2.4, which is comparable to the other rats in the group (range 2.2–2.7). No increase in ALAT was observed in the group injected with 177Lu-BR96 (group 2) or the unconjugated BR96 (group 3).
Figure 4. Serum levels of ASAT (A) and ALAT (B) in animals given b-BR96-MMAFconjugates (group 1), 177Lu-labeled BR96 (group 2), and unconjugated BR96 (group 3).
Rats treated with b-BR96-MMAF (group 1) displayed an increase in the levels of G-GT during the first three weeks. The two rats surviving for eight and ten days reached a level corresponding to grade 2 toxicity, whereas the other four rats reached grade 3. The peak values were reached after day 10 in these rats. Most of the rats given 177Lu-BR96 (group 2) displayed a mild increase in G-GT; 2/6 rats reached grade 2 toxicity whereas 4/6 rats were below grade 1. No increase in G-GT was observed in group 3.
The levels of ALP fluctuated considerably in the group given the drug-immunoconjugate. No bone metastases were seen at subsequent autopsies, which indicates that ALP represents liver toxicity. Serum bilirubin was not influenced by the treatment in any of the groups.
Nephrotoxicity. Treatment with the conjugates did not have any major influence on the serum creatinine levels of the rats. The rats treated with 177Lu-BR96 (group 2) showed a slow increase over the observation period, reaching only a level corresponding to grade 1toxicity. Three of six rats in this group had creatinine levels corresponding to grade 1 toxicity already prior to administration of the radioimmunoconjugate.
Therapeutic effect
All animals injected with conjugated BR96 (groups 1 and 2) showed complete local response within ten days p.i. No local recurrence was observed in these two groups. Four of six rats injected with unconjugated BR96 (group 3) showed transient complete local response but three rats were sacrificed due to local recurrence before day 45 p.i., and the fourth died due to distant metastases on day 77 p.i.
Two animals treated with b-BR96-MMAF (group 1) died due to toxicity within ten days p.i. One of these rats had the highest ALAT value; otherwise the laboratory parameters did not differ for these rats compared to the other rats in the group. All late deaths (i.e. after day 45) were attributed to metastatic disease. Only one rat treated with b-BR96-MMAF developed metastases, whereas metastases were observed in three of six rats given 177Lu-BR96 (group 2). Metastases were often observed at multiple locations in the liver, kidneys, lungs, abdominal lymph nodes, and peritoneum. Only two of six rats in the control group (unconjugated BR96) developed metastases, probably due to local tumor growth and the associated short survival.
Discussion
The present study demonstrates very different toxicity profiles in the same immunocompetent syngeneic rat tumor model when comparing radiolabeled (177Lu) BR96 mAb with drug-conjugated (monomethylauristatin F) BR96 mAb at doses corresponding to the maximal tolerable dose for each of the conjugates based on our previous studies. The administered dose of BR96-MMAF in present study resulted in toxicity somewhat above MTD, which probably does not change the pattern of observed toxicity.
Myelotoxicity is dose limiting for low-energy beta-emitting 177Lu conjugated BR96 (average beta energy 133 keV, range 0.67 mm). The dose-limiting factor is the prolonged suppression of leukocytes (>28 days) with increased risk of infections. We have previously evaluated the high-energy beta 90Y-conjugated BR96 (average beta energy 935 keV, range 3.9 mm) in the same animal model with a similar toxicity profile. Studies with short-ranged alpha emitters and very short-ranged Auger emitters have not been performed, since they are still rarely used in the clinic.
For monomethylauristatin-conjugated BR96, the dose-limiting toxicity was liver toxicity. The grade of short-term thrombocytopenia or loss of body weight in the present study with auristatin conjugates was not regarded as dose limiting or correlated to reduced survival, based on previous experience with radiolabeled BR96 in the same syngeneic tumor model [Citation8]. The catabolic breakdown of the auristatin conjugates in the liver resulted in severe hepatotoxicity (grade 3 toxicity, according to the NCI toxicity criteria) in all rats treated with the conjugate. Bilirubin levels were not affected, but this is only seen when almost all the liver parenchyma is involved.
No difference was seen in the therapeutic efficacy of the two conjugates on local disease. All the late deaths (after day 45) were attributed to metastatic disease. It is well recognized that long-ranged beta-emitting radionuclides are better suited for the treatment of macroscopic tumors as most of the radiation is delivered as “cross-fire” from adjacent cells, thus also eliminating tumor cells not expressing the target-antigen. However, these long-ranged beta-emitting radionuclides are not suitable for the treatment of microscopic tumors as most of the radiation energy is absorbed far from the location of the antibody conjugate, resulting in a low absorbed dose in the tumor cells. One advantage of drug-conjugated mAbs is that they also efficiently kill single cells. In the clinical setting drug conjugates are generally administered repeatedly over a number of weeks. In this study we administered the drug conjugate only once. The use of antigen-targeted therapy might result in the selection of antigen-negative cell clones that finally develop into metastatic disease.
By combining these two conjugates of BR96, with partially overlapping toxicity, it could be possible to increase the therapeutic ratio and hence also the efficacy. Therefore, it would be interesting to investigate combined treatment with these two conjugates by evaluating various sequencing and dosing patterns. The dose-limiting toxicity of the combined treatment would probably be the decrease in platelet number. As the platelets recover more rapidly from the drug-conjugated antibody treatment it might be advantageous to start the treatment with the administration of drug-immunoconjugates followed by radioimmunoconjugates. Using this sequence it might be possible to treat microscopic metastatic disease at an early stage, thus preventing the growth of distant metastases, since drug conjugates also have a better effect on cell clusters than do long-range beta-emitting radionuclides. Conversely, through the bystander effects, the long ranged beta emitting radio nuclides have a better effect on macroscopic tumors by compensating for poor penetration of the targeting molecule into the tumor mass and overcome possible heterogeneity of target expression.
No kidney toxicity was seen, which also indicates that large amounts of radiolabeled or nephrotoxic fragments were not filtrated through the kidneys.
In rats treated with unconjugated BR96 transient complete remission were observed in four of six rats, however most of these rats developed rapidly local recurrences that were not observed in the conjugate treated groups. It might still be of interest to investigate unconjugated antibody as consolidation therapy after therapy with radionuclide-conjugates or drug-conjugates as naked antibody might be efficient against cell clusters and small tumors. Prolonged disease free survival was seen when anti-CD20 radioconjugate therapy of non-Hodgkin lymphoma in mice was followed by unlabeled anti-CD20 consolidation therapy [Citation16].
Although toxicity depends on the species and the conjugates applied, this study demonstrates the different toxicity profiles very distinctly when a beta-emitting radionuclide is compared to a drug-conjugate of the same mAb in the same tumor model. This should make it possible to improve the therapeutic window without significantly increasing the toxicity.
Acknowledgements
We would like to thank Dr. Peter Senter (Seattle Genetics, Seattle, WA, USA) for kindly providing the monoclonal antibody BR96 and the auristatin-BR96 conjugate. The research was supported by grants from the Swedish Cancer Society, Mrs. Berta Kamprad's Foundation, Gunnar Nilsson's Foundation, Lund University Medical Faculty Foundation, and The Lund University Hospital Fund. The authors have no affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this publication.
References
- Chari RV. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc Chem Res 2008;41:98–107.
- Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol 2009;13:235–44.
- Vaishampayan U, Glode M, Du W, Kraft A, Hudes G, Wright J, . Phase II study of dolastatin-10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clin Cancer Res 2000;6:4205–8.
- Doronina SO, Mendelsohn BA, Bovee TD, Cervemy CG, Alley SC, Meyer DL, . Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug Chem 2006;17:114–24.
- Younes A, Forero-Torres A, Bartlett NL, Leonard JP, Rege B, Kennedy DA, . Objective responses in a phase I dose-escalation study of SGN-35, a novel antibody-drug conjugate (ADC) targeting CD30, in patients with relapsed or refractory Hodgkin lymphoma. J Clin Oncol 2008;26(May 20 Suppl) ASCO Annual Meeting Abstract 8526.
- Hwu P, Sznol M, Kluger H, Rink L, Kim KB, Papadopoulos NE, . A phase I/II study of CR011-vcMMAE, an antibody toxin conjugate drug, in patients with unresectable stage III/IV melanoma. J Clin Oncol 2008;26(May 20 Suppl) ASCO Annual Meeting Abstract 9029.
- Martensson L, Nilsson R, Ohlsson T, Sjogren HO, Strand SE, Tennvall J. High-dose radioimmunotherapy combined with extracorporeal depletion in a syngeneic rat tumor model – evaluation of toxicity, therapeutic effect and tumor model. Cancer 2010;116(4 Suppl):1043–52.
- Martensson L, Wang Z, Nilsson R, Ohlsson T, Senter P, Sjogren HO, . Determining maximal tolerable dose of the monoclonal antibody BR96 labeled with 90Y or 177Lu in rats: Establishment of a syngeneic tumor model to evaluate means to improve radioimmunotherapy. Clin Cancer Res 2005;11(19 Pt 2):7104s–8s.
- Nilsson R, Martensson L, Eriksson SE, Sjogren HO, Tennvall J. Toxicity-reducing potential of ECAT in combination with the auristatin-conjugated mAb BR96 in a syngeneic rat tumor model. Cancer 2010;116(4 Suppl):1033–42.
- Sjogren HO, Isaksson M, Willner D, Hellstrom I, Hellstrom KE, Trail PA. Antitumor activity of carcinoma-reactive BR96-doxorubicin conjugate against human carcinomas in athymic mice and rats and syngeneic rat carcinomas in immunocompetent rats. Cancer Res 1997;57:4530–6.
- Ross HJ, Hart LL, Swanson PM, Ratrick MU, Figlin RA, Jacobs AD, . A randomized, multicenter study to determine the safety and efficacy of the immunoconjugate SGN-15 plus docetaxel for the treatment of non-small cell lung carcinoma. Lung Cancer 2006;54:69–77.
- Wilbur DS, Chyan MK, Hamlin DK, Kegley BB, Nilsson R, Sandberg BE, . Trifunctional conjugation reagents. Reagents that contain a biotin and a radiometal chelation moiety for application to extracorporeal affinity adsorption of radiolabeled antibodies. Bioconjug Chem 2002; 13:1079–92.
- Wang Z, Martensson L, Nilsson R, Bendahl PO, Lindgren L, Ohlsson T, . Blood pharmacokinetics of various monoclonal antibodies labeled with a new trifunctional chelating reagent for simultaneous conjugation with 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid and biotin before radiolabeling. Clin Cancer Res 2005;11(19 Pt 2): 7171s–7s.
- Pass D, Freeth G. The rat. ANZCCART 1993;6:1–4.
- Sun MM, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, . Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 2005;16:1282–90.
- Sharkey RM, Karacay H, Goldenberg DM. Improving the treatment of non-Hodgkin lymphoma with antibody-targeted radionuclides. Cancer 2010;116(4 Suppl): 1134–45.