To the Editor,
With an average annual incidence rate of only 0.8 per 100 000 person-years [Citation1], pheochromocytoma (PHEO) and paraganglioma (PGL) are rare neuroendocrine tumors (NET). The tumors arise from chromaffin tissue in the adrenal medulla (PHEO) or from extra-adrenal chromaffin cells (PGL) [Citation3]. Malignancy is confirmed only if metastases are present in non-chromaffin sites [Citation7], or when invasion of neighboring tissue is seen [Citation8,Citation9]. Approximately 5% of PHEO and 33% of PGL are malignant. About 20–30% of PHEO/PGL are familial, with a higher malignancy percentage in familial PGL, making genetic testing an integral part of diagnosis [Citation6]. The plasma-free or urinary-fractionated metanephrines are the most sensitive biochemical tests [Citation6].
PHEO/PGL express human norepinephrine transporter (hNET) and tumor cells also often have a relatively high density of somatostatin receptors. Thus, specific functional imaging with, e.g. radio-labeled catecholamine precursor ligands as well as non-specific functional imaging techniques are used to ascertain the extent of disease and the feasibility of radionuclide treatment [Citation3,Citation21].
Treatment of metastatic disease is usually palliative. Debulking surgery is often the first step followed by systemic treatment modalities with 123I-MIBG (123I-metaiodobenzylguanidine) or somatostatin analogs [Citation6,Citation7]. Objective response to chemotherapy is rarely observed [Citation3]. New types of anti-neoplastic drugs with effect on neuroendocrine (NET) tumors may be useful, although experience is still very limited [Citation7,Citation19]. The following cases from a single institution illustrate the complexity of this rare disease and the heterogeneous effect of established and new treatment modalities.
Case 1
A 27-year-old male with no familial disposition presented with a 10-year anamnesis of intermittent hypertension, headache, blushing and sweating. Abdominal US and MRI showed a 6-cm cystic, well-vascularized, retroperitoneal tumor, technically inoperable due to involvement of large vessels. 123I-MIBG SPECT/CT revealed abnormal uptakes in the right suprarenal region and in the skeletal system, and a cerebral magnetic resonance image (MRI) disclosed metastases in the dura mater. The patient had a 20-fold increase in 24 h urinary noradrenaline excretion. Blood DNA sequencing demonstrated a mutation in SDHB. Three treatment rounds with 131I-MIGB (total dose 22.8 GBq) resulted in biochemical and clinical remission. One and a half years later, urinary catecholamines rose sharply, and 123I-MIBG and 111In-octreotide SPECT/CT revealed new tumor growth in the calvarium. This metastasis was surgically removed and histology showed PHEO, immunohistochemically positive for cytokeratine AE 1/3, synaptophysine and chromogranin A, with a Ki-67 proliferation rate of 7%. Two years after metastasectomy, new tumors evolved in the abdomen and in the orbita. The patient completed two treatments with 90Y[DOTA-Tyr3]octreotide (90Y-DOTATOC). However, two and a half months later, progression was demonstrated with involvement of multiple organs. Chemotherapy with temozolamide 200 mg/m² for five days every four weeks was offered. Toxicity consisted of fatigue and ileus CTC grade II. The disease, however, progressed already at the first assessment after three cycles, and the patient died shortly after, six years and four months after the diagnosis.
Case 2
A 31-year-old male presented with a left-sided neck mass. No paraneoplastic symptoms or familiar dispositions were present. Computed tomography (CT) and MRI revealed a 10 × 15 cm paravertebral mass in the thoracic-lumbar region. Biochemical tests were normal. An 111In-octreotide SPECT/CT revealed increased activity in the apex of the left lung and in the retroperitoneum. Histology from a cervical lymph node showed mPGL since the tumor cells were of neuroendocrine type, with a majority of cells being strongly positive for synaptophysin, CD56, chromogranin A, and a minority S-100 positive sustentacular cells. The Ki-67 proliferation rate was 20%. There were no somatic mutations found. The patient was treated with one course of cyclofosfamide, adriamycine and vincristine, followed by resection of the paravertebral tumor. Postoperatively, the 123I-MIBG, 111In-Octreotid SPECT/CT and 18F-FDG PET/CT showed persistent abnormal uptake at the left side of the neck, and 25 lymph nodes were removed, all containing metastases, however, with a proliferation rate of only 6–8%. One year after, the patient developed symptoms of spinal cord compression due to progressing tumor at Th 8 and Th 11. A successful decompressive laminectomy/metastasectomy was performed followed by two 90Y-DOTATOC sessions. Three months after, however, a new relapse in the right pleura and retroperitoneum was disclosed. Treatment with sunitinib was started (50 mg daily for four weeks on, two weeks off). Toxicity was significant and consisted of CTC grade II fatigue, rash and grade III neutropenia, requiring treatment interruption. At the first evaluation after two courses, stable disease was found, whereas a CT after four courses showed progression, and treatment was terminated. Symptoms of spinal cord compression recurred and decompression/metastasectomy was repeated, resulting in full recovery. Two additional courses of 90Y-DOTATOC were administered and the disease was stable for 10 months. Thereafter, symptomatic spinal cord compression reoccurred and a third decompression/metastasectomy at Th 8 and Th 12 was performed. Radiotherapy in palliative doses (20 Gy in 4 fractions) was given to progressing cervical lymph node metastases and resulted in slight regression of these. Treatment with temozolamide was initiated, interrupted by 90Y-DOTATOC. The treatment was well tolerated, and despite the advanced stage of disease, the patient is in perfect shape almost four years after diagnosis.
Case 3
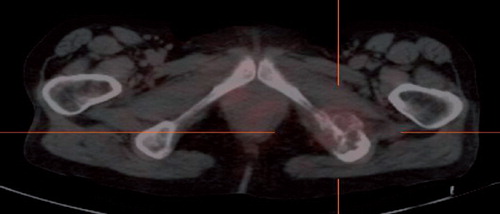
A 54-year-old woman, operated for a right-sided PHEO 14 years previously, presented with thoracic pain due to a metastasis in Th3. A surgical biopsy presented clear cells, suggestive of renal cell carcinoma. The patient was offered postoperative irradiation with 20 Gy in 4 fractions. The 24 h urinary noradrenaline excretion level was twice the upper limits of normal values. Genetic testing did not reveal gene mutations. An MRI delineated a 4 cm tumor at the site of the right adrenal gland, nodal metastases at the liver hilum and metastasis in L5. The 111In-octreotide SPECT/CT revealed abnormal uptake in a soft tissue process in thorax, and in the left side os ileum and femur. Six series of chemotherapy with cyclofosfamide, vincristine, and dacarbazine was given and led to clear biochemical response and radiological stabilization of the disease. After eight years with no clinical evidence of disease, urinary catecholamines rose again. An 111In-octreotide SPECT/CT revealed a new focus in the left tuber ischiadicum (). 123I MIBG SPECT/CT and MRI subsequently confirmed progression of bone and intraabdomianl metastases. At the age of 63, she commenced sunitinib treatment (six week courses of 50 mg daily for four weeks on, two weeks off). Sunitinib caused skin toxicity, neutropenia, hypertension, and myxoedema CTC grade III. Supportive treatment was initiated and sunitinib dose was reduced to 37.5 mg and further to 25 mg daily. The MRI after two courses revealed regression of the adrenal tumor (from 48 mm to 35 mm), but otherwise stable disease. Moreover, the 111In-Octreotid SPECT/CT showed significantly diminished receptor density in the left tuber ischiadicum (). After four months on sunitinb, U katecholamine excretion was normalized. At the 10th month on sunitinib, the MRI showed progression of the metastasis in os ischium, but otherwise stable disease. High dose radiotherapy with 60 Gy in 30 fractions was applied to this metastasis. After radiation, the patient proceeded with sunitinib at a further reduced dose of 12.5 mg daily due to severe neutropenia, however, 17 months after initiating treatment with sunitinb, the abdominal metastases progressed, and sunitinib was replased by temozolamide in 75% dosis. Toxicity consisted of neutropenia grade II. Stable disease was observed at the first assessment. The patient is still in good performance (WHO PS 1), 25 years after adrenalectomy and 11 years after relapse of mPHEO.
Figure 1. A 111In-octreotide SPECT/CT in Case 3 patient, as baseline before sunitinib treatment. The patient have a focus of markedly increased receptor density in the left tuber ischiadicum, showing relaps of disease after eight years remission.
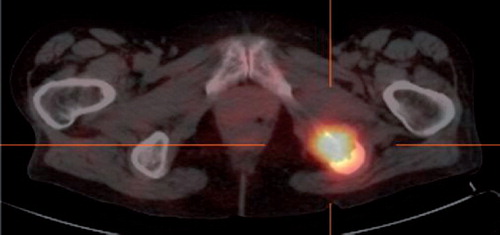
Figure 2. A 111In-octreoitide SPECT/CT in Case 3 patient, after two courses of sunitinib treatment, showing clear regression of 111In-octreoitide uptake in osseous lesion, and corresponding to PR of disease.
Discussion
As demonstrated by the three cases above, a multimodal approach in diagnosis and treatment of patients with mPHEO/PGL is mandatory.
A suspicion of mPHEO/PGL is virtually always first risen by demonstration of excessive catecholamines and metanephrines production in urine or plasma [Citation5,Citation6].
Diagnostic work up includes CT and/or MRI, which have high sensitivity for adrenal disease, but lower for example, extra-adrenal metastatic or recurrent disease [Citation21]. Additionally, specific functional imagining modalities, for example PET with 18F-fluorodopamine (18F-FDA), 18F-fluoroDOPA (18F-DOPA), 11C-hydroxyephedrine (HED), and 123I-MIBG SPECT, or non-specific functioning imaging methods visualizing tumor cell glucose metabolism (18F-Flourodeoxyglucose (FDG) PET) or the expression of somatostatin receptors (111In-DTPA-octreotide scintigraphy), often provide accurate information on disease extent and biology [Citation10]. 123I-MIBG SPECT is especially useful for the diagnosis because of its high affinity uptake in chromaffin cells. Still only about two thirds of metastases are avid for 123I-MIBG, and some patients can harbor both MIBG-negative and positive lesions [Citation4,Citation10,Citation21]. Somatostatin receptor scintigraphy has high sensitivity in mPGL/PHEO. Positron emission tomography (PET) with different tracers is also useful for establishing the extent of disease: 11C-labeled HED was the first PET tracer developed specifically for imagining the sympatic nervous system, but other tracers are now more widely used for practial reasons. 18F-FDA PET has high sensitivity and is excellent to localize mPHEO, whereas PET with 18F-DOPA may be superior in extra-adrenal disease and neck PGL. It is recommended that primary mPGL/PHEO preferably should be detected with 18F-FDA PET/CT or equal alternatives such as 123I-MIBG or 18F-DOPA, whereas the use of other radiopharmaceuticals depends on desired information [Citation21].
The treatment of mPHEO/PGL includes advanced surgery, nuclear medicine, chemotherapy, radiotherapy and, experimentally, new biologically targeted drugs. Treatment with high dose 131I-MIGB in patients with MIGB avid metastases may often result in sustained remission or stable disease [Citation4] as showed in our Case 1. However, it is not useful if MIGB-scintigraphy is negative, and toxicity may sometimes limit the use of this modality [Citation4,Citation11]. Treatment with radio-labeled somatostatin receptor ligands may be of benefit to patients showing a high uptake on octreotide scintigraphy [Citation12] as demonstrated in our Case 2, for example by using 90Y-DOTATOC.
Chemotherapy is generally considered of limited effect and is usually reserved to treat advanced or aggressive disease. Several chemotherapeutic regimens have been used [Citation2]. Among these is cyclophosphamide, vincristine and dacarbazine (CVD) which have shown to produce symptomatic and hormonal improvement, but often transient and with minimal tumor shrinkage [Citation3,Citation5,Citation6,Citation9]. In our Case 3, a clear biochemical response to CVD was observed, and the period of stable disease lasted exceptionally long for eight years. Temozolamide alone or in combination with thalidomide has produced promising response rates in NET [Citation13] and in mPHEO/PGL [Citation14], although the effect has not been verified in randomized studies.
Some new, molecular targeted drugs are of interest in mPHEO/PGL, as these tumors express several dysregulated molecular pathways [Citation2,Citation15]. Sunitinib, a multi tyrosine kinase inhibitor, has demonstrated effect in pancreatic NET tumors [Citation19], and cases demonstrating response to sunitinib treatment in mPHE/PGL are emerging [Citation16–18]. Two of three patients with mPGL treated with sunitinib had ongoing response 40 weeks after initiation of treatment [Citation16], one patient maintained clinical and radiological response for 16 weeks [Citation17], and one patient had a clinical and metabolic response [Citation18]. In our additional two cases of mPHEO/PGL treated with sunitinib, no major objective tumor shrinkage was observed, but disease progression was apparently stabilized for 24 and 68 weeks, respectively, and in Case 3 a biochemical and scintigraphic response was seen. Other newly developed biological agents may have implications for patients with mPHEO/PGL, for example, the mTOR inhibitors (everolimus, temsirolimus) [Citation20], and some of these targeted drugs, including sunitinib, are currently under investigation in clinical trials (16, http://www.ClinicalTrials.gov.).
mPHEO/PGL represent a rare entity of NET. We present three cases that illustrate the heterogeneity and complexity of this disease and contribute to the very limited knowledge regarding the effect of new drugs, including sunitinib and temozolamide. A precise diagnosis depends on a fusion of advanced imaging techniques, which also allows for individualization of radio-ligand therapy, based on the molecular expression profile of the tumor. A multitarget approach can result in significant and long-lasting biochemical and symptomatic response, and in some cases, in sustained tumor regression. Clinical trials of new drugs, with stratification according to tumor characteristics are greatly needed and, fortunately, under way.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc 1983;58:802–4.
- Adjallé R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res 2009;41: 687–96.
- Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2007;14: 569–85.
- Fitzgerald PA, Goldsby RE, Huberty JP, Price DC, Hawkins RA, Veatch JJ, . Malignant pheochromocytomas and paragangliomas: A phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG). Ann N Y Acad Sci 2006;1073:465–90.
- Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, . Pheochromocytoma: Current approaches and future directions. Oncologist 2008;13:779–93.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Pheochromocytoma. Lancet 2005;366(9486):665–75.
- Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, . Malignant pheochromocytoma: Current status and initiatives for future progress. Endocr Relat Cancer 2004;11:423–36.
- Disick GI, Palese MA. Extra-adrenal pheochromocytoma: Diagnosis and management. Curr Urol Rep 2007;8:83–8.
- Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, . Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab 2009;94:2850–6.
- Shulkin BL, Ilias I, Sisson JC, Pacak K. Current trends in functional imaging of pheochromocytomas and paragangliomas. Ann N Y Acad Sci 2006;1073:374–82.
- Gedik GK, Hoefnagel CA, Bais E, Olmos RA. 131I-MIBG therapy in metastatic phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2008;35:725–33.
- Forrer F, Riedweg I, Maecke HR, Mueller-Brand J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging 2008;52:334–40.
- Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, . Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 2007;13:2986–91.
- Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, . Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006;24:401–6.
- Santarpia L, Habra MA, Jiménez C. Malignant pheochromocytomas and paragangliomas: Molecular signaling pathways and emerging therapies. Horm Metab Res 2009;41: 680–6.
- Joshua AM, Ezzat S, Asa SL, Evans A, Broom R, Freeman M, . Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab 2009;94:5–9.
- Hahn NM, Reckova M, Cheng L, Baldridge LA, Cummings OW, Sweeney CJ. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J Clin Oncol 2009;27:460–3.
- Park KS, Lee JL, Ahn H, Koh JM, Park I, Choi JS, . Sunitinib, a novel therapy for anthracycline- and cisplatin-refractory malignant pheochromocytoma. Jpn J Clin Oncol 2009;39:327–31.
- Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, . Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 2008;26: 3403–10.
- Dimou AT, Syrigos KN, Saif MW. Neuroendocrine tumors of the pancreas: What's new haven highlights from the “2010 ASCO Gastrointestinal Cancers Symposium”. Orlando, Florida, USA. January 22–24, 2010 JOP. J Pancreas [Internet] 2010 Mar 5; 11:135–8.
- Carrasquillo JA, Chen CC. Molecular imaging of neuroendocrine tumors. Semin Oncol 2010;37:662–79.