Abstract
Purpose. To assess the efficacy of imatinib for different tumor genotypes in Korean patients with advanced gastrointestinal stromal tumors (GIST). Material and methods. Clinical data were collected from 370 consecutive patients with locally advanced unresectable, metastatic, or recurrent GIST treated with imatinib 400 mg/day between August 2001 and December 2007 at 20 Korean institutions. Tumor genotypes were determined for 290 patients by direct DNA sequencing of KIT exons 9, 11, 13, and 17, and PDGFRA exons 12, 14, and 18. Results. Of 290 patients assessed for genotype, 261 (90.0%) had mutations in KIT: 222 (76.6%) in exon 11, 35 (12.1%) in exon 9 and two each (0.7%) for exons 13 and 17. Four patients (1.4%) had mutations in the PDGFRA gene: one in exon 12, and three in exon 18. Twenty-five patients (8.6%) had no detectable mutations. The best responses of the 235 patients with measurable lesions were: 15 complete response (6.4%), 126 partial response (53.5%), 86 stable disease (36.6%), and eight progressive disease (3.4%). Patients with KIT exon 9 mutations, compared with patients with KIT exon 11 mutations, had a lower objective response rate (36.7% vs. 63.6%, p = 0.007) and a shorter progression-free survival (median 28.7 months vs. 49.4 months, p = 0.001). No statistical difference in overall survival was observed between these genotypes. Conclusion. This study confirms that imatinib efficacy is dependent on genotype in Korean GIST patients, consistent with results demonstrated by Western patients with GIST.
Gastrointestinal stromal tumors (GIST) are mesenchymal neoplasms that arise mainly in the gastrointestinal tract wall, and are characterized by the expression of the receptor tyrosine kinase (RTK) KIT (CD117) [Citation1,Citation2]. Activating mutations of KIT are present in up to 92% of GIST, and these mutations are likely to play a fundamental role in the development of these tumors [Citation3–10]. For GIST with no detectable KIT mutations, activating mutations of the RTK platelet-derived growth factor receptor alpha (PDGFRA) have also been identified [Citation11,Citation12]. Furthermore, a subset of GIST possesses no detectable mutations in either RTK [Citation11,Citation12].
Imatinib mesylate (Glivec®/Gleevec®, Novartis Pharma AG, Basel, Switzerland) is a competitive inhibitor of the RTKs BCR-ABL, ARG, KIT, PDGFRA, and PDGFRB [Citation13–16]. The activity of imatinib in GIST was initially demonstrated in a single-patient pilot study of a heavily pretreated patient with bulky advanced-stage metastatic GIST [Citation17]. Thereafter, results from the US-Finland B2222 Phase II study confirmed that imatinib was safe and effective at doses of 400 mg and 600 mg daily, and led to imatinib being widely accepted as standard first-line treatment for patients with advanced GIST [Citation18,Citation19]. Further analysis from the B2222 study by Heinrich and colleagues revealed that the efficacy of imatinib was dependent on GIST primary genotype, that is patients with KIT exon 11 mutations had a higher objective response rate compared with patients with KIT exon 9 mutations (83.5% vs. 47.8%, p = 0.0006) [Citation20].
Two large-scale, randomized, Phase III trials (European Organization for Research and Treatment of Cancer [EORTC] 62005 and Southwest Oncology Group [SWOG] S0033) were subsequently initiated to compare the clinical benefits of high-dose [800 mg daily (400 mg twice daily)] to standard-dose imatinib (400 mg daily) [Citation21,Citation22]. In the EORTC 62005 trial, a significant improvement in progression-free survival (PFS) was initially observed for patients administered imatinib 800 mg/day [Citation22]. However, the difference observed for PFS between the two dose groups became statistically insignificant with longer follow-up, and no differences in PFS were observed in the SWOG S0033 trial [Citation21,Citation23]. Moreover, no significant differences in objective response or disease control rates (DCRs; defined as percentage of patients with an objective response or stable disease) were observed between the two dose groups in either the EORTC 62005 or SWOG S0033 trial.
Although no significant differences in PFS were observed between the dose groups overall, a subanalysis from the EORTC 62005 trial revealed that patients with KIT exon 9 mutations had superior PFS when initially treated with high-dose imatinib [Citation24]. A pooled meta-analysis of the SWOG S0033 and EORTC 62005 trials (MetaGIST) further revealed that the estimated risk of progression or death was reduced by 42% in the high-dose cohort compared with the standard dose cohort (p = 0.017, Wald test) for patients with KIT exon 9 mutations [Citation25]. These results spurred the European Society for Medical Oncology and the National Comprehensive Cancer Network to update their guidelines to include the use of high-dose imatinib in patients with KIT exon 9 mutations; this development represented the first major incorporation of mutational status in treatment decision making for GIST [Citation26,Citation27].
To date, no large-scale prospective studies have compared the clinical benefit of imatinib treatment for different GIST genotypes in Asian patients [Citation28–30]. However, in two small retrospective studies of imatinib-treated Korean and Taiwanese patients with GIST, no significant differences were observed in clinical outcomes with regard to primary genotype [response rates, PFS, and overall survival (OS)] [Citation29,Citation30]. Genotype-specific differences in the efficacy of imatinib may not have been observed due to the small sample sizes in these studies and/or potentially higher imatinib plasma levels in Asian patients compared with Western patients as a result of a smaller body size. Therefore, to further investigate if treatment outcome is dependent on tumor genotype in Asian patients with GIST, we initiated a large retrospective analysis that evaluated whether a potential relationship between clinical response and genotype exists in a cohort of consecutive Korean patients with advanced GIST who were treated with imatinib.
Material and methods
Patients
This retrospective study collected clinical data from 370 patients administered imatinib 400 mg daily for the treatment of locally advanced unresectable, metastatic, or recurrent GIST from August 2001 to December 2007 at 20 institutions in Korea. The study protocol was approved by the institutional review board of each participating institution. Patients with confirmed KIT expression (KIT+) by immunohistochemical staining (IHC; A4502; DAKO, Glostrup, Denmark) and KIT-negative (KIT−) patients who did not express KIT but were clinically and histologically confirmed to have GIST and were negative for desmin and S-100 by IHC were included in the study. Unfortunately, 80 of the 370 patients were excluded from the final analysis because paraffin-embedded tumor specimens were not available, genomic DNA extraction from specimens failed, or the amount of DNA extracted was inadequate for genotype determination.
Patients were evaluated every three months by computed tomography (CT). Both objective response and progressive disease (PD) were radiologically assessed with respect to lesion size by Response Evaluation Criteria In Solid Tumors (RECIST) [Citation31]. However, an increase in the sum of greatest diameters of target lesions, which corresponds to PD by RECIST, was not regarded as such when accompanied by definite cystic changes suggestive of necrosis [Citation32]. In addition, the development of new, small cystic lesions in the liver during early imatinib treatment was not regarded as PD [Citation33]. In contrast, detection of a new enhancing nodule within a mass without change in overall tumor size, so called “nodule in nodule”, was considered as PD even though RECIST was not met [Citation34]. Three medical oncologists (Min-Hee Ryu, Byung Woog Kang, Yoon Hee Choi) with considerable experience of GIST, assessed best overall response and PD for all study patients using the above mentioned response criteria, and independently reviewed CT images, excluding those from their own centers.
Analysis of KIT and PDGFRA mutations
Genotyping was performed centrally at five centers: the Asan Medical Center, Chonnam National University Hospital, Kyungpook National University Hospital, the Samsung Medical Center, and Seoul National University Hospital. Amplification (by polymerase chain reaction), and direct DNA sequencing of KIT exons 9, 11, 13, and 17, and PDFGRA exons 12, 14, and 18 were performed according to previously described procedures [Citation35,Citation36]. Mutational analysis for KIT exon 11 was performed in all cases. Patient samples negative for exon 11 mutations were subsequently analyzed for KIT exons 9, 13, and 17 and PDGFRA exons 12, 14, and 18. The order in which these exons were sequenced was dependent on the primary tumor site.
When no mutations were detected, or unknown, double, or unusual mutations were identified, mutational analysis was repeated at one of the other four centers.
Statistical analyses
Statistical analysis was performed using the χ2 test or Fisher's exact test for the comparison of means, and the binary logistic regression test was used to assess the relationship between tumor genotype and imatinib response. Co-factors investigated in the analysis included age, gender, Eastern Cooperative Oncology Group (ECOG) performance status, body surface area, primary site of tumor, tumor size, baseline laboratory parameters, and tumor genotype. All statistical tests were two-sided. PFS was measured from the first day of imatinib treatment to disease progression or death due to any cause. OS was measured from the first day of imatinib treatment to death due to any cause. Patients still alive and progression free at the time of analysis (30 September 2010) were censored at their last follow-up dates. Kaplan-Meier analysis was used to construct survival curves, which were compared using the log-rank test. Multivariate analysis was performed by Cox proportional hazards regression modeling. P-values of <0.05 were considered statistically significant.
Results
Characteristics of patients
The baseline characteristics of the 290 patients assessed for PFS by tumor genotype are summarized in . Median patient age was 60 years (range 24–82 years). The most common primary sites were the small intestine (52.4%) and stomach (34.5%). The most common sites of metastasis were the liver (60.3%) and peritoneum (42.1%).
Table I. Patients’ characteristics and tumor genotypes (n = 290).
Genotype analysis
We identified KIT or PDGFRA-activating mutations in 265 of 290 (91.4%) of all analyzed patients. Of these 290 patients, 261 (90.0%) had mutations in KIT, with 222 (76.6%) in exon 11; 35 (12.1%) in exon 9; and two each (0.7%) for exons 13 and 17 ().
The most common type of KIT exon 11 mutations were deletions (151 of 222, 68.0%), followed by substitutions (33, 14.9%), complex-type mutations (31, 14.0%), and insertions (7, 3.1%). Most KIT exon 11 deletion mutations occurred between codons 550 and 570 (the most commonly involved codons were 557–558). Isolated single-point mutations in KIT exon 11 occurred most frequently in codon 559 (Appendix Table A1,online only).
The most common KIT exon 9 mutations were AY 502–503 duplications (31 of 35 patients). The remaining KIT exon 9 mutations were duplications of codons 506–508 (2), and deletion mutations (KHNGT484–488T; 2) identical to mutations previously described [Citation37,Citation38].
Substitutions of K642E were found in both cases of GIST with mutations in KIT exon 13. The two cases of GIST with mutations in KIT exon 17 were A794T and G812D point mutations. Four patients (1.4%) harbored mutations in the PDGFRA gene. One patient had a D583N point mutation in PDGFRA exon 12. The mutations found in PDGFRA exon 18 were two cases of substitutions of D842V and one case of deletion mutation of codon 842. Twenty-five patients (8.6%) had no detectable mutation in the KIT or PDGFRA gene [referred to hereafter as wild type (WT)].
Response to imatinib by genotype
Response assessments by RECIST to imatinib 400 mg/day were made for 235 of the 290 patients with a determined genotype; 55 patients were not evaluable due to resection of metastatic disease or possession of target lesions too small to quantify. There were 15 complete responses (CRs; 6.4%), 126 partial responses (PRs; 53.6%), 86 patients with stable disease (SD; 36.6%), and eight with PD (3.4%), resulting in an overall response rate (ORR) of 60.0% [95% confidence interval (CI): 53.7%–66.3%]. Moreover, the DCR in this study was 96.6% (227 of 235), including 86 SDs (36.6%). Median time to best response was 2.9 months (range 1.0–59.0 months). Median duration of response was 27.3 months (range 2.0–88.2 months). As shown in , ORR associated with imatinib 400 mg/day was 63.6% (112 of 176) in patients with KIT exon 11 mutations, 36.7% (11 of 30) in those with KIT exon 9 mutations, and 69.6% (16 of 23) in those with WT GIST. Stepwise logistical regression analysis was performed to identify clinical factors that predict disease control (i.e. CR/PR/SD at 24 weeks from start of treatment) or an objective response to imatinib. The following clinical factors were included in the analysis: age, gender, performance status, body surface area, primary site of tumor, tumor size, baseline laboratory parameters, and tumor genotype. Tumor genotype was found to be the only significant predictor of response. Patients with GIST harboring KIT exon 11 mutation were more likely to respond to imatinib therapy than those with KIT exon 9 mutations (in terms of ORR, 63.6% vs. 36.7%, respectively; p = 0.007) (Appendix Table A2, online only).
Table II. Responses to imatinib 400 mg daily by genotype (n = 235).
Of the 30 patients with KIT exon 9 mutations evaluable for response, ORR was 37.9% (11 of 27) in patients with AY 502-503 duplications and no with other than AY 502-503 duplications in KIT exon 9 responded to imatinib (There were three SDs). However, this difference in response between mutational subtypes of KIT exon 9 was statistically insignificant (p = 0.239).
For the two assessable patients with a PDGFRA exon 18 mutation, one patient with a deletion mutation of the 842 codon achieved SD, but the other patient with a D842V mutation had PD.
Progression-free and overall survival by genotype
All 290 patients with a determined genotype were assessed for PFS. and show overall and genotype-associated PFS and OS curves. With a median follow-up of 42.8 months, the median PFS for all 290 patients was 43.3 months (95% CI: 37.7–49.0 months). The median OS was not reached for all 290 patients, or for any of the genotype-specific subpopulations. Patients with KIT exon 11 mutations had significantly longer PFS than those with KIT exon 9 mutations (median 49.4 months vs. 28.7 months, respectively; p = 0.001). However, no significant difference in OS was observed between patients with KIT exon 11 and KIT exon 9 mutations (p = 0.884). A proportional hazards model for PFS and OS was constructed using the potential prognostic factors described above (Appendix Table A3, online only). In addition to tumor genotype, univariate analyses identified several other factors associated with poor PFS (poor performance status, high neutrophil count) and with OS (poor performance status, high neutrophil count). As shown in , tumor genotype [KIT exon 9 vs. KIT exon 11: hazard ratio (HR) = 2.427, p = 0.003; WT vs. KIT exon 11: HR = 2.187, p = 0.018] was found to be an independent prognostic factor of PFS, as was performance status (ECOG 2–3 vs. 0–1: HR = 2.385, p = 0.005), and neutrophil count (>7500/μl vs. ≤7500/μl: HR = 3.756, p = 0.003). For OS, tumor genotype (WT vs. KIT exon 11: HR = 3.819, p = 0.011) and neutrophil count (>7500/μl vs. ≤7500/μl: HR = 5.534, p = 0.034) were found to be independent prognostic factors.
Figure 1. Progression-free survival for all patients (A) and by tumor genotype (B). N, number of patients.
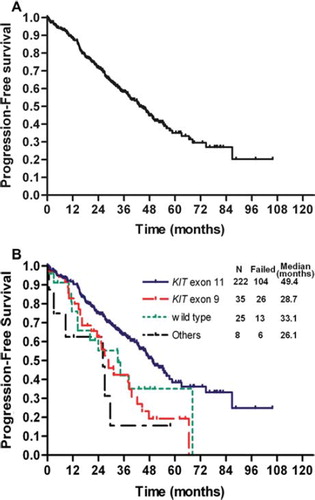
Figure 2. Overall survival for all patients (A) and by tumor genotype (B). N, number of patients; NR, not reached.
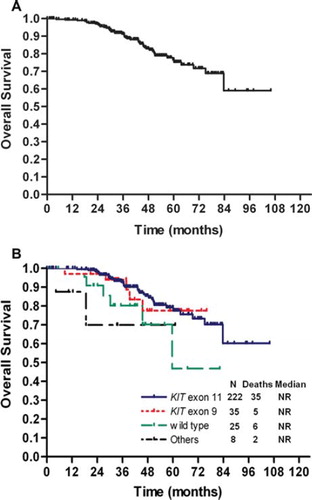
Table III. Multivariate analyses of the prognostic factors of progression-free and overall survival (n = 290).
PFS and OS were not found to differ significantly between KIT exon 11 mutation types or locations (W557 or K558 vs. others; codon 565 or 579 vs. others) or by the presence of a long deletion (>15 codons; data not shown).
Salvage treatments after progression on imatinib 400 mg/day
One hundred and thirty-seven (47.2%) of the 290 patients received salvage treatments after progression on imatinib 400 mg/day. During the period from first progression to death or the end of this analysis, 108 (78.8%) patients were treated with imatinib at a higher dose (600 mg and/or 800 mg). In addition, 92 (67.2%) and 26 (19.0%) patients were exposed to sunitinib or nilotinib, respectively.
Discussion
To the best of our knowledge, this retrospective study is the largest study to evaluate the relationship between treatment outcome and tumor genotype in Asian patients with advanced GIST. Here we show that in Korean GIST patients treated with standard-dose imatinib (400 mg/day), those with KIT exon 9 mutations had a lower ORR and inferior PFS compared with KIT exon 11-mutant patients, as has previously been reported in Western patients with GIST. These results suggests that the lack of a significant difference between different genotypes in regard to clinical outcomes reported in two previous studies conducted in Korean and Taiwanese GIST patients [Citation29,Citation30] may have been related to insufficient sample sizes.
The incidence and spectrum of RTK mutations found in the present study are consistent with those found during two previous Phase III trials [Citation24,Citation38], and are similar to those of the previous report from Korean GIST patients [Citation29].
The response rate for Korean patients treated in this study is comparable to that previously reported for Western patients. In the B2222 study of patients treated with 400 or 600 mg/day imatinib, an ORR of 68.1% (95% CI: 59.8%–75.5%) was observed. The ORR in this study was 60.0% (95% CI: 53.7%–66.3%) with 15 CRs (6.4%) and 126 PRs (53.6%). However, when also considering patients achieving SD, a higher DCR (96.6%) was observed in this study compared with 83.6% for the B2222 trial. This difference may be due in part, to how progression was defined in these two studies. For B2222, progression was based solely on bidimensional Southwest Oncology Guidelines tumor size criteria. For this study, progression was evaluated based on size by RECIST, however new small cystic lesions or size increase with internal necrosis were not regarded as progression [Citation32].
Also consistent with results from B2222 reported by Heinrich et al., patients with KIT exon 9 mutations showed significantly lower ORR than those with KIT exon 11 mutations (36.7% vs. 63.6%, p = 0.007) [Citation38]. In contrast, the Korean patients with WT GIST treated with imatinib in our study achieved higher ORRs (69.6%) compared with previously reported ORRs of 0.0% [Citation38], 23.1% [Citation24], and 44.6% [Citation21] for patients with WT GIST, in other studies. It remains unclear why higher ORRs were observed for Korean patients. This may relate to the fact that ‘WT’ usually includes all patients for which no detectable mutations are found, which is largely dependent on methodology used to determine genotype. As such, the nonuniform manner in which GIST genotype is determined makes comparison of results for ‘WT’ GIST patients across trials problematic. Nonetheless, the results of this study suggest that a majority of Korean patients with ‘WT’ GIST derive clinical benefit from imatinib. However, in terms of DCR at 24 weeks, ‘WT’ GIST patients had poorer results than KIT exon 11-mutant patients (73.9% vs. 95.4%, respectively; p = 0.001) (Appendix Table A2, online only). This means ‘WT’ GIST patients can achieve a good response on imatinib, but that good initial response is not lastly maintained otherwise in KIT exon 11-mutant patients.
The Korean patients in this study had a longer median PFS compared with patients from the B2222 study (43.3 months vs. 24 months, respectively). This may relate to the fact that many patients with a very high tumor burden and more advanced disease waiting for effective treatment were enrolled in B2222, while patients with less tumor burden are usually included in recent studies including this study. Related to this, a large number of patients on B2222 received chemotherapy (n = 75, 51%; median 2 regimens, range 1–7) and/or radiotherapy (n = 22, 15%) prior to enrollment in B2222, whereas all patients included in this study received imatinib as first-line therapy.
No significant difference in OS between patients with KIT exon 11 and KIT exon 9 mutations were observed (), even though patients with KIT exon 11 mutations had longer PFS than those with KIT exon 9 mutations (median PFS 49.4 months vs. 28.7 months, respectively, after a median of 42.8 months follow-up). A possible explanation for this discrepancy is that patients with KIT exon 9 mutations may derive greater clinical benefit from salvage therapy, which for the majority of patients receiving salvage therapy in this study (78.8%) included imatinib dose escalation. Indeed, the EORTC 62005 trial found that patients with KIT exon 9 mutations derived greater clinical benefit after crossover from 400 mg to 800 mg daily imatinib compared with those with KIT exon 11 mutations (responses to the dose increase assessed using a growth modulation index: 57% vs. 7%, p = 0.0017) [Citation24]. In addition, the majority of patients receiving salvage therapy were treated with sunitinib (67.2%), and previous studies have shown that patients with KIT exon 9 mutations or WT GIST had better outcomes (PFS and OS) compared with patients with KIT exon 11 mutations in response to second-line sunitinib treatment [Citation39,Citation40]. Related to this, acquisition of secondary mutations occurs more frequently in KIT exon 11-mutant GIST compared with KIT exon 9-mutant GIST or WT GIST (73%, 17%, and 0%, respectively). However, our analysis regarding the significance of genotype on the impact of salvage therapy is limited, as the study was not designed to address this issue. Therefore, we suggest that further studies be conducted to explore this topic. In particular we anticipate that Korean patients with KIT exon 9 mutations would stand to benefit from treatment with high-dose imatinib, similar to Western patients with GIST, although this needs to be rigorously tested.
http://www.informahealthcare.com/doi/abs/10.3109/0284186X.2011.636753
Download PDF (34.3 KB)Acknowledgments
We thank co-investigators (Yeul Hong Kim, Seong Hoon Shin, Hun-Mo Ryoo, Young Jin Choi, Seok Yun Kang, Sung Sook Lee, Hyeok Shim, Dae Young Zang) for collection of clinical data and provision of study materials. And we thank Ms Sun-young Hwangbo who is an administrative secretary of the KGSG. The authors acknowledge Robert Gillespie, PhD, of Chameleon Communications International, who provided editorial assistance with funding from Novartis Pharmaceuticals. Supported in part by Novartis Pharma AG, Basel, Switzerland.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, . Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol 2002;33:459–65.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors – definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1–12.
- Buchdunger E, Zimmermann J, Mett H, Meyer M, Müller M, Druker BJ, . Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 1996;56:100–4.
- Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol 2002;160:1567–72.
- Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999;154:53–60.
- Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, . Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000;157:1091–5.
- Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, . KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000;156: 791–5.
- Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF Jr. Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999;18:1897–902.
- Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Matsuda H, . Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999;59:4297–300.
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, . Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279: 577–80.
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, . PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299: 708–10.
- Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, . Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660–7.
- Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, . Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther 2000;295:139–45.
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, . Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561–6.
- Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000;96:925–32.
- Okuda K, Weisberg E, Gilliland G, Griffin D. ARG tyrosine kinase activity is inhibited by STI571. Blood 2001;97: 2440–8.
- Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, . Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344: 1052–6.
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, . Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620–5.
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, . Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–80.
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, . Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9.
- Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, . Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–32.
- Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, . Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet 2004;364:1127–34.
- Casali PG, Verweij J, Kotasek D, LeCesne A, Reichardt P, Blay JY, . Imatinib mesylate in advanced gastrointestinal stromal tumors (GIST): Survival analysis of the intergroup EORTC/ISG/AGITG randomized trial in 946 patients. Eur J Cancer 2005;3:201(abstr 711).
- Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, . KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42: 1093–103.
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1,640 patients. J Clin Oncol 2010;28:1247–53.
- National Comprehensive Cancer Center. Clinical practice guidelines in oncology. Soft tissue sarcoma. V.2.2010.http://www.nccn.org/professionals/physician_gls/PDF/sarcoma.pdf
- Casali PG, Blay JY. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21(Suppl 5):v98–102.
- Du CY, Shi YQ, Zhou Y, Fu H, Zhao G. The analysis of status and clinical implication of KIT and PDGFRA mutations in gastrointestinal stromal tumor (GIST). J Surg Oncol 2008;98:175–8.
- Kim TW, Ryu MH, Lee H, Sym SJ, Lee JL, Chang HM, . Kinase mutations and efficacy of imatinib in Korean patients with advanced gastrointestinal stromal tumors. Oncologist 2009;14:540–7.
- Yeh CN, Chen TW, Lee HL, Liu YY, Chao TC, Hwang TL, . Kinase mutations and imatinib mesylate response for 64 Taiwanese with advanced GIST: Preliminary experience from Chang Gung Memorial Hospital. Ann Surg Oncol 2007;14:1123–8.
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, . New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205–16.
- Ryu MH, Lee JL, Chang HM, Kim TW, Kang HJ, Sohn HJ, . Patterns of progression in gastrointestinal stromal tumor treated with imatinib mesylate. Jpn J Clin Oncol 2006;36:17–24.
- Linton KM, Taylor MB, Radford JA. Response evaluation in gastrointestinal stromal tumours treated with imatinib: Misdiagnosis of disease progression on CT due to cystic change in liver metastases. Br J Radiol 2006;79:e40–4.
- Shankar S, van Sonnenberg E, Desai J, Dipiro PJ, van Den Abbeele A, Demetri GD. Gastrointestinal stromal tumor: New nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylate. Radiology 2005;235: 892–8.
- Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, . Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007;31:224–32.
- Kim TW, Lee H, Kang YK, Choe MS, Ryu MH, Chang HM, . Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer Res 2004; 10:3076–81.
- Keun PC, Lee EJ, Kim M, Lim HY, Choi DI, Noh JH, . Prognostic stratification of high-risk gastrointestinal stromal tumors in the era of targeted therapy. Ann Surg 2008; 247:1011–8.
- Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, . Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360–7.
- Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, . Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9.
- Van Glabbeke M, Verweij J, Casali PG, Le Cesne A, Hohenberger P, Ray-Coquard I, . Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: A European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group study. J Clin Oncol 2005;23:5795–804.