Abstract
Although cell death research has progressed rapidly over the two decades with emphasis on the study of apoptosis, non-apoptotic forms of cell death have recently attracted more attention. In the present short review, I will describe how this transition is occurring and emphasize the importance of investigating non-apoptotic forms of cell death as well as apoptosis to fully understand the spectrum of death in eukaryotic cells. The aim is not to list all published forms of cell death, but to indicate the necessity for a conceptual paradigm shift, so I will only introduce a limited number of cell death mechanisms.
Cell death research at the molecular level has made great advances since around 1990, with the discovery of several crucial molecules involved in apoptosis, including the anti-apoptotic oncoprotein Bcl-2 [Citation1,Citation2], the caspase Ced-3 (a Caenorhabditis elegans apoptotic protease) [Citation3], and a death receptor Fas/Apo1/CD95 [Citation4] that transmits apoptotic signals across the plasma membrane. However, research on apoptosis is now reaching maturity. Recently, several mechanisms of non-apoptotic cell death have been reported, and are now undergoing detailed analysis because they are still poorly understood.
In the field of cancer research, apoptosis has long been an important theme, since it was discovered that the oncogene Bc1-2 is activated by chromosome translocation t(14:18) in follicular B cell lymphoma [Citation1], leading to an increase of Bc1-2 oncoprotein that suppresses apoptotic cell death [Citation2]. This discovery had a big impact in the cancer research field, emphasizing the “importance of research on carcinogenesis from a cell death (particularly apoptosis) point of view”, because cancer research had been largely focused on cell proliferation and had not paid much attention to the possibly important role of suppressing the cell death mechanism. Apoptosis is also important in cancer research from the perspective of anti-cancer therapy. Apoptosis could be activated to kill cancer cells, and thus is one of the promising therapeutic strategies for treatment of tumors. However, methods for killing cancer cells do not need to be restricted to apoptosis. If mammalian cells possess mechanisms for other forms of cell death, activation of one or more of these mechanisms could be an alternative strategy. In fact, although many anti-cancer drugs induce apoptosis of cultured cell and are thereby considered to cause apoptosis of tumor cells during clinical use, it is not clear what cell death mechanism(s) has the most critical antitumor effect in vivo because these drugs are also able to induce other forms of cell death under different conditions.
We have recently been investigating programmed cell death in vivo and also non-apoptotic cell death mechanisms in several different cell lines, because we have some doubts about the current dogma that “physiological programmed cell death is mainly mediated by apoptosis”. This assertion is not supported by compelling evidence, and could be merely a “perceived notion”. In fact, we are now obtaining evidence that non-apoptotic cell death makes a larger contribution to programmed cell death in vivo than was previously thought. Therefore, in order to fully understand cell death in mammals, we believe that both apoptosis and non-apoptotic cell death need to be studied. When considering cell death in various diseases, apoptosis seems to be relatively unimportant, so detailed analysis of non-apoptotic cell death is essential to study the pathology and treatment of various diseases.
Definition of non-apoptotic cell death
Non-apoptotic cell death is defined as death mediated by different mechanisms from apoptosis that does not satisfy the criteria for apoptotic death. Therefore, non-apoptotic cell death is defined morphologically as any cell death process that does not feature apoptotic changes (such as chromatin condensation and nuclear fragmentation) and biochemically as death that is not mediated by caspases. Although caspase-independent apoptosis has been reported as cell death mediated by apoptotic molecules released from the mitochondria, such as AIF and endonuclease G, this death process without caspase activation is not well accepted yet, and it may be better classified as one form of non-apoptotic death.
Non-apoptotic programmed cell death in vivo
It has been reported that cells undergoing non-apoptotic death can be observed when programmed cell death occurs during morphogenesis in chick, mouse, and rat embryos. However, these non-apoptotic forms of cell death have mainly been described on a morphological basis only and the molecular mechanisms involved are still largely unknown. Schweichel and Merker originally described three forms of programmed cell death which they called types I, II and III [Citation5]. Subsequently, based on additional observations from other groups, Clark made some modifications to the Schweichel/Merker classification in his review and categorized programmed cell death into types I, II, IIIA and IIIB, with type IIIA being considered as very unusual [Citation6].
Type I (apoptosis)
Many excellent reviews of apoptosis are available for reference.
Type II
Type II programmed cell death is characterized by autophagy (the cell “eating” itself), and is associated with the formation of autophagosomes and autolysosomes. In conjunction with recent rapid progress in the study of autophagy at the molecular level, this form of cell death is currently attracting the attention of many researchers who are performing molecular biological analyses. As described later, death similar to this type of cell death can be induced in cultured cells. Autophagy is activated by starvation to degrade cellular constituents and provide cells with essential nutrients or else functions constitutively to maintain quality control of cellular components by degrading unwanted cellular constituents or removing damaged subcellular organelles, such as mitochondria. These processes are essential for cell survival. Given that cellular constituents are degraded by autophagy, it is not difficult to imagine it also being involved in cell death, although compelling evidence has not been provided yet. To establish a crucial role of autophagy in cell death, molecular markers need to be discovered that can distinguish autophagy involved in cellular survival from that cell death.
Since decreased expression of autophagic proteins such as Beclin 1 is known to enhance tumorigenesis, autophagy has also become an attractive target of cancer research. However, it is still unknown how reduction of Beclin 1 expression enhances tumorigenesis, which may be related to defective quality control of cellular constituents or some defect of autophagic cell death.
Type III
Type III A. This form of cell death was described by Schweichel and Merker [Citation5] and characterized by initial swelling of intracellular organelles followed by the formation of empty spaces in the cytoplasm which fuse with each other and connect with extracellular cavity, and eventual disintegration via extensive fragmentation. This was considered by Clarke to be rather an exceptional case.
Type IIIB. This type of cell death is associated with the destruction of subcellular organelles (mitochondrial swelling, dilation of endoplasmic reticulum, and karyolysis), and morphologically is categorized as necrosis. Some caution needs to be paid with respect to type IIIB cell death, because molecularly different forms of death might result in the type IIIB morphological changes. Once molecules specifically involved in these forms of cell death have been identified, type IIIB cell death may be more precisely classified in the future.
It should be noted that morphological studies suggest that additional mechanisms of non-apoptotic programmed cell death might exist.
Is programmed cell death mainly mediated by apoptosis in vivo?
Some lessons can be learned from several lines of knockout (KO) mice that are deficient in major players in the process of apoptosis, e.g. caspase-9 KO [Citation7,Citation8], Apaf-1 KO [Citation9], and Bax/Bak KO mice [Citation10]. These mice show certain morphological abnormalities, e.g. Apaf-1 KO and caspase-9 KO mice on a 129 background (but not a C57B6 background) develop exencephaly, while Bax/Bak double KO mice have interdigital webs. These morphological abnormalities are considered to be evidence for an important role of apoptosis in programmed cell death in vivo. However, the defects created by changes of embryonic programmed cell death are very limited, and it appears that most programmed cell death still proceeds normally in these mice. This might be explained by the activation of compensatory cell death mechanisms that are usually silent. However, there is also a possibility that much programmed cell death is intrinsically mediated by non-apoptotic death mechanisms ().
Figure 1. Apoptosis vs. non-apoptotic mechanisms of programmed cell death. Mammalian cells seem to possess multiple death mechanisms, but it is unclear how much each of these mechanisms contributes to programmed death in vivo.
In experimental models, there are some examples of non-apoptotic programmed cell death playing a major role in developmental processes. In C. elegans, 131 cells are known to undergo programmed death during development, but this does not seem to have any biological significance. The only known cell death process involved in morphogenesis is the death of linker cells. These cells are located between the gonad (vas deferens) and the cloacal tube that serves as an exit channel for sperm in the adult, so the removal of linker cells facilitates connecting the male reproductive system to the exterior. The death of linker cells is non-apoptotic and is independent of Ced-3 caspase and other apoptotic genes [Citation11], being morphologically characterized by nuclear indentation and swelling of organelles. Thus, apoptotic cell death is not related to developmental morphogenesis in C. elegans and its biological significance is unclear. In Drosophila melanogaster, the programmed death of nurse cells during oocyte maturation is mediated by a non-apoptotic mechanism, although the morphologic features of the dying cells have not been reported [Citation12].
In mammals, there are also cases where non-apoptotic programmed cell death plays the major role. For example, it was recently reported that regression of mammary glands after lactation in mice is mediated by non-apoptotic cell death that involves induction of lysosomal enzymes, including cathepsins B and L, by Stat-1 transcriptional factor [Citation13]. In addition, the turnover of skin epithelial cells is known to be mediated by a non-apoptotic process. Furthermore, although turnover of epithelial cells in the small intestine has been described as both apoptotic and non-apoptotic, we have recently demonstrated that programmed death of small intestinal epithelial cells is mainly mediated by a non-apoptotic process (manuscript in preparation).
Table I. Cell death mechanisms observed in cultured cells.
Non-apoptotic death of cultured cells
Several forms of non-apoptotic cell death have been described in cultured cells, and some of them are listed in . Although there is the possibility that some of these cell death mechanisms might only be activated in certain cell lines under certain conditions and thus have no biological significance, it would not be unreasonable to think that mammalian cells possess mechanisms for multiple forms of cell death with physiological significance. Some forms of cell death that are currently considered to be distinct might come to be classified as the same form of death based on molecular studies. One of the characteristics of necrotic death is disruption of the plasma membrane, so depletion of intracellular ATP leading to plasma membrane disruption might be a common convergence point for necrotic death. The detailed molecular mechanisms and biological significance of these non-apoptotic forms of cell death that have been discovered in cultured cells need to be elucidated in future studies.
Cyclophilin D-dependent necrosis
When isolated mitochondria are treated with Ca2+ or oxidants such as H2O2, the mitochondrial membrane permeability transition (MPT) is initiated with an increase of inner membrane permeability, resulting in loss of the electrical potential difference across the inner membrane, which is followed by mitochondrial swelling and outer membrane rupture eventually leading to mitochondrial dysfunction (). The MPT is inhibited by cyclosporine A. Because it is known that cyclosporine A inhibits cyclophilins, the molecular target through which it blocks the MPT has been suggested to be mitochondrial cyclophilin D. This has been confirmed experimentally by generation of mice lacking cyclophilin D and analysis of mitochondria isolated from these mice [Citation14,Citation15].
Figure 2. Role of the mitochondria in apoptosis and necrosis. Permeabilization of the mitochondrial outer membrane occurs during apoptosis and results in the release of some apoptogenic molecules, including cytochrome c. Subsequently, cytochrome c is assembled into apoptosomes with Apaf-1 and pro-caspase-9, resulting in the activation of caspases. Oxidative stress or an excess of calcium can trigger the mitochondria membrane permeability transition (MPT), which is initiated by an increase of inner mitochondrial membrane permeability that results in loss of the membrane potential. This is followed by mitochondrial swelling and rupture of the outer membrane with mitochondrial dysfunction and depletion of ATP that causes necrotic death.
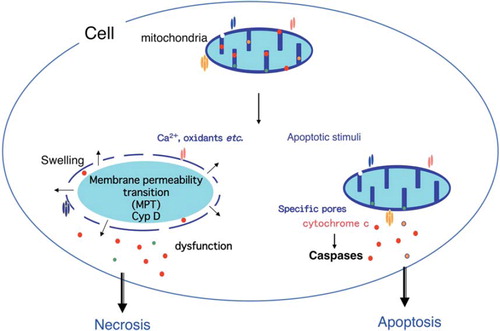
When mouse embryonic fibroblasts (MEF) or mouse hepatocytes are subjected to oxidative stress, these cells undergo MPT-dependent necrotic death. This form of death has been shown to be involved in tissue damage and cell death induced during ischemia-reperfusion injury of the heart and brain, since cyclophilin D-deficient mice show a significant resistance to ischemia-reperfusion injury [Citation14–16]. It has also been reported by crossing the respective disease model mouse with cyclophilin D-deficient mouse that the cyclophilin D-dependent MPT is involved in the pathogenesis of Alzheimer's disease [Citation17], as well as Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy [Citation18]. The latter two are inherited muscular disorders caused by mutations of the gene for Collagen VI (ColVI). ColVI KO mice exhibit a myopathic phenotype associated with ultrastructural changes of the mitochondria and sarcoplasmic reticulum, as well as mitochondrial dysfunction. Disruption of the cyclophilin D gene reverses the disease phenotype of ColVI KO mice. Similarly, treatment with cyclosporine A reverses myofiber alterations in UCMD patients. In an Alzheimer's disease model of mice overexpressing mutant human amyloid precursor protein (J-20 line), cyclophilin D deficiency substantially improves learning, memory, and synaptic function.
Programmed necrosis/necroptosis
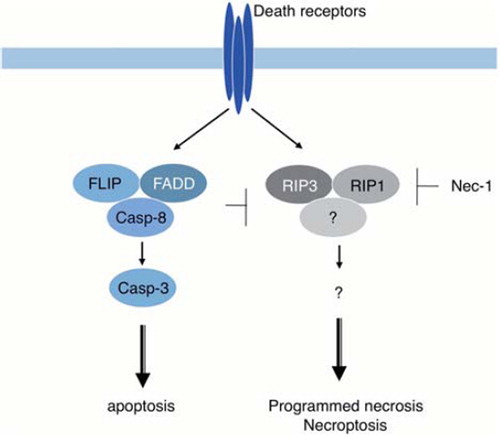
Upon binding with the respective ligand, death receptors, such as TNF receptor and Fas activate apoptotic signal transduction through formation of a protein complex that includes FADD/pro-caspase-8, resulting in the activation of caspase-8. When caspase activation is blocked, for example, by caspase inhibitors (such as zVAD-fmk) or by deficiency of caspase-8 or FADD, cells undergo necrotic death that is called “programmed necrosis” or “necroptosis” (). Certain cell lines such as L929 cells also undergo necroptosis after exposure to zVAD-fmk. Necroptosis is inhibited by a small molecule known as necrostatin-1 (Nec-1) [Citation19]. Electron microscopic observation has revealed that necroptosis is not associated with apoptotic morphological changes and is characterized by non-apoptotic nuclear condensation, swelling of subcellular organelles, and disruption of the plasma membrane. A molecular target of Nec-1 is a kinase called receptor-interacting protein 1 (RIP1) [Citation20]. RIP1 kinase forms a complex with other proteins including RIP3 kinase. An essential role of RIP3 in necroptosis has been shown using cells derived from RIP3 KO mice [Citation21–23]. RIP3 is required for phosphorylation and recruitment of RIP1 to the complex. Although it is not known how the RIP1/RIP3 kinase complex induces necroptosis, a role of ROS has been suggested.
Figure 3. Necroptosis/programmed necrosis. Death receptors can activate two signaling pathways that lead to caspase-8-dependent apoptosis and RIP1/RIP3-dependent necrosis called necroptosis/programmed necrosis. Activated caspase-8 cleaves and inactivates RIP1/3 kinases, shifting the outcome of death receptor activation to apoptosis. When caspase-8 activation is inhibited, for example by a defect of caspase-8 or FADD or by caspase inhibitors, such as zVAD-fmk, cells die of necroptosis instead.
Nec-1, but not Nec-1i (an inactive form of Nec-1), suppresses ischemia/reperfusion injury in mice, suggesting that necroptosis may play a critical role in such injury [Citation19] and providing evidence for a pathological role of necroptosis.
Caspase-8-deficient mice die during the embryonic period, while this is completely prevented by knockout of the RIP3 gene [Citation24,Citation25], suggesting that embryonic death is caused by the abnormal activation of necroptosis. Mice with intestinal epithelial cell-specific knockout of FADD develop epithelial necrosis, enteritis, and severe colitis, all of which are prevented by genetic disruption of RIP3 [Citation26], suggesting that intestinal inflammation can be mediated by necroptosis. Also, RIP3-deficent mice succumb to vaccinia virus infection. It has been suggested that the vaccinia virus induces necroptosis that triggers inflammation and activates the anti-viral response [Citation22]. Thus, necroptosis appears to have an important role in inflammation and innate immunity against viral infection.
iPLA2β-dependent necrosis [Citation27]
Under hypoxic/low glucose conditions, cells undergo a caspase-independent form of death that is morphologically characterized by marked shrinkage of the nucleus. This form of cell death is mediated by the activation of calcium-independent phospholipase A2β (iPLA2β) and is blocked by PLA2 inhibitors, such as bromoenol lactone (BEL). Activation of iPLA2β is mediated by p38 kinase [Citation28].
Autophagy-dependent cell death
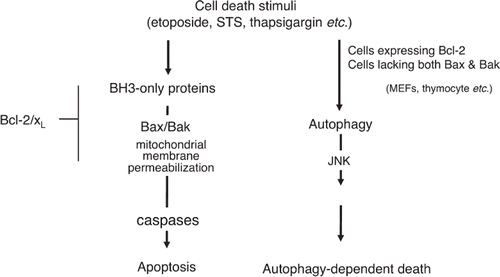
Some apoptosis-resistant cell lines, including MEF cells overexpressing Bcl-2 or Bax/Bak-deficient MEF cells, undergo non-apoptotic death after exposure to apoptotic stimuli, such as genotoxins, etoposide, and the kinase inhibitor staurosporine [Citation29] (). This form of cell death is morphologically characterized by the formation of autophagosomes/autolysosomes and is inhibited by suppression of autophagy via either chemical inhibitors, such as 3-methyadenine (a PI-3 kinase inhibitor) or knockdown of autophagy proteins, such as ATG5 [Citation29], indicating that autophagy is involved in this death process. It was recently shown that JNK is involved in this form of cell death [Citation30]. Since this form of cell death is morphologically similar to type II programmed cell death and there has been no culture system for the study of type II death, the above-mentioned cell lines might be useful for detailed investigation of type II programmed cell death at the molecular level.
Figure 4. Autophagy-dependent death. Depletion of serum, amino acids, or lymphokines, as well as cytotoxic drugs, such as etoposide and staurosporine, induce apoptosis of wild-type MEFs. When apoptosis is blocked by Bax/Bak-deficiency or overexpression of Bcl-2, however, only etoposide and staurosporine (not withdrawal of amino acids, serum, or lymphokines) trigger autophagy-dependent death. Autophagy-dependent death requires the activation of JNK.
Transcriptional repression-induced atypical death (TRIAD) [Citation31]
Based on the observation that transcription is suppressed in the neurons of patients with various neurodegenerative diseases, primary cultures of neurons were treated with a transcription inhibitor (aphidicolin) and were found to undergo non-apoptotic death, which was characterized by the formation of vacuoles and morphologically resemble type IIIB programmed cell death. This form of cell death is known to be associated with downregulation of p73 transcription factor and is inhibited by the neuron-specific isoform of YAP (YAPΔC). However, it is not known yet whether this form of cell death actually occurs in neurodegenerative diseases.
Paraptosis [Citation32]
In the absence of ligands, some cell surface receptors (including neutrophin receptors) can activate cell death signaling pathways, and these are called dependence receptors. Overexpression of Insulin-like growth factor 1 receptor (ILGF1R) triggers non-apoptotic death in various cell lines that has been called “paraptosis”. This is characterized by formation of vacuoles and, thus, might resemble type IIIB programmed death. Paraptosis requires MAPK (MEK-2 and JNK-1) and is inhibited by AIP1/Alix (ALG2-binding protein) via an unknown mechanism.
Killing of cancer cells by DNA-damaging agents [Citation33]
DNA-damaging agents including an anti-cancer drug etoposide (an inhibitor of topoisomerase II) and γ-irradiation kill various cultured tumor cells by apoptosis. However, it is also known that at higher doses, cells die in a caspase-independent manner. Therefore, given that tumor cells in vivo which are under different conditions from in vitro culture (for example, tumor cells in vivo are under hypoxic condition), it is still not clear whether anti-cancer drugs and γ-irradiation really kill tumor cells in vivo mainly by apoptosis. These treatments might activate non-apoptotic death mechanisms in tumor cells in vivo. It might, therefore, be very important to elucidate non-apoptotic death mechanisms of tumor cells.
It is of interest to note that cells, such as MEF undergo non-apoptotic death after treatment with alkylating agents, such as MNNG or nitrogen mustard. This form of cell death requires neither p53 nor Bax/Bak and is not inhibited by Bcl-2. The proposed mechanism is as follows: DNA damage - → activation of NAD-dependent poly ADP ribose polymerase (PARP) - → reduction of cytoplasmic NAD - → suppression of glycolysis (which is NAD-dependent) - → ATP depletion - → necrosis. Pyruvate (methylpyruvate) inhibits this form of cell death by increasing ATP production through mitochondrial oxidative phosphorylation. Since many cancer cells are known to depend mainly on glycolysis (Warburg effect), they are very sensitive to DNA-damaging alkylating agents.
Given the reports of several different forms of cell death in mammalian cell lines, it would seem reasonable to suggest that different cell death mechanisms exist in mammalian cells, and contribute to programmed cell death under various circumstances. Extensive studies of these non-apoptotic cell death mechanisms are required to establish their physiological as well as pathological significance. Once the molecules involved or specific inhibitors are identified, they could be used for classification of cell death mechanisms and for establishing which mechanism is involved in each type of programmed cell death or in various diseases for which the death mechanism has not yet been determined. Furthermore, although there are imaging methods for detecting apoptotic cells in vivo, no method is available to detect cells undergoing non-apoptotic death. Therefore, it would be beneficial to take on the challenge of establishing an imaging method for non-apoptotic cell death. Finally if molecules involved in a variety of non-apoptotic cell death processes can be identified, this would not only make a contribution to fuller understanding of the death mechanisms for mammalian cells but could also assist in the establishment of therapeutic strategies for various diseases, including degenerative diseases and cancer.
Acknowledgments
The studies from my laboratory cited in this article was supported by a Grant-in-Aid for Creative Scientific Research (number 19GS0316 to YT) from the Japan Society for the Promotion of Science (JSPS) and a grant for Global Center of Excellence (COE) Program Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology. This article was an Acta Oncologica Lecture, from the 19th ECDO annual conference with the topic “Metabolism, Epigenetics and Cell Death” on 14th–17th of September, 2011 at Norra Latin, Stockholm.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Tsujimoto Y, Croce CM. Analysis of the structure transcripts and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci USA 1986;83: 5214–8.
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988;335:440–2.
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 1993;75:641–52.
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, . The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991;66:233–43.
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratol 1973;7:253–66.
- Clarke, PGH. Developmental cell death: Morphological diversity and multiple mechanisms. Anat Embryol 1990;181: 195–213.
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, . Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 1998;94:339–52.
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, . Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998;94: 325–37.
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, . Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998;94:739–50.
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, . The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000;6:1389–99.
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell 2007;12:73–86.
- Mazzalupo S, Cooley L. Illuminating the role of caspases during Drosophila oogenesis. Cell Death Differ 2006;13: 1950–9.
- Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, . Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 2011;13:303–9.
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, . Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005;434:652–8.
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, . Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434:658–62.
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, . Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 2005;102:12005–10.
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, . Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 2008;14:1097–105.
- Palma E, Tiepolo T, Angelin A, Sabatelli P, Maraldi NM, Basso E, . Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Human Mol Genet 2009;18: 2024–31.
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, . Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112–9.
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, . Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4: 313–21.
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, . Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137:1100–11.
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, . Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009;137:1112–23.
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, . RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009;325:332–6.
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, . RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011;471: 368–72.
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, . Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471:363–7.
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernández-Majada V, Ermolaeva M, . FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Epub Nature 2011 Jul 31.
- Shinzawa K, Tsujimoto Y. PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J Cell Biol 2003;163:1219–30.
- Aoto M, Shinzawa K, Suzuki Y, Ohkubo N, Mitsuda N, Tsujimoto Y. Essential role of p38 MAPK in caspase-independent, iPLA(2)-dependent cell death under hypoxia/low glucose conditions. FEBS Lett 2009;583:1611–8.
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, . Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004;6:1221–8.
- Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, . Involvement of JNK in the regulation of autophagic cell death. Oncogene 2010;29:2070–82.
- Hoshino M, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, . Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol 2006;172:589–604.
- Sperandio S, Poksay K, de Bell I, Lafuente MJ, Liu B, Nasir J, . Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Diff 2004;11:1066–75.
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Gene Dev 2004;18:1271–82.