Abstract
Background. Intracranial haemangiopericytoma (HPC), a rare malignant tumour, should be distinguished from meningioma and solitary fibrous tumour, which have been considered as separate entities since 1993, according to histopathology and clinical characteristics. Methods. A PUBMED search for “Intracranial Haemangiopericytoma” yielded 176 articles, where 26 were of particular interest for this review article. Case report. Our patient, a 27-year-old man with HPC of grade III according to WHO, presents with an acute intracerebral haematoma, which is extremely rare. Results. Surgery (total resection) is the primary treatment. Long-term close clinical and radiological follow-up is crucial due to the high rate of recurrence and tendency for development of metastasis. Discussion. The effects of postoperative radiotherapy need further investigation. Besides neurosurgery, radiotherapy should always be considered in both patients with these highly malignant tumours (WHO grade III) and in patients with partial resection or inoperable cases (WHO grade II).
Intracranial haemangiopericytoma (HPC) is a rare tumour with malignant features. It is locally aggressive with a tendency to recur and even to metastasise extracranially if not adequately treated. To this date, 563 patients with intracranial HPC have been reported in the literature [Citation1]. Intracranial HPC represent about 2% of all meningeal tumours and 1% of intracranial tumours [Citation2–6]. Common presenting symptoms include: headache 50%, epileptic seizures 33% or focal signs [Citation2,Citation4,Citation7–9]. HPCs are associated with an early symptom debut, most likely due to the fast growth rate of the tumour. Intracerebral haematoma occurs in around 5% of cases with intracranial neoplasm [Citation4,Citation10]. Only very few cases of HPC related to intracerebral haematoma have been reported in the literature, in total 13 cases from 1969 to 2006 () [Citation4,Citation10].
Table I [Citation4,Citation10].
This paper includes a case report of a haemorrhagic HPC and a literature review on HPC emphasising clinical history, histopathology, radiological appearance, treatment and prognosis.
Methods
A search was made in PUBMED using the following Keywords: “Intracranial Haemangiopericytoma”. This search yielded 176 articles but only 26 were of particular interest for this article. There were no time limits in the search.
Results
In 1923 Zimmermann described a modified smooth muscle cell (leiomyoblastic cell) later named Zimmermann's pericytes, which is located around capillaries and post-capillary venules [Citation2–5,Citation10,Citation11].
Solitary fibrous tumour (SFT) was described in pleura by Klemperer and Rabin in 1931 [Citation12].
The first case of meningeal HPC, then called angioblastic meningioma, was reported in 1938 by Cushing and Eisenhardt [Citation2,Citation4].
Stout and Murray were the first to introduce the term HPC for the extracranial tumours derived from Zimmermann's pericytes in 1942 [Citation3,Citation12–14].
Begg and Garret described an intracranial meningeal HPC in 1954, similar to the HPCs described by Stout and Murray in soft tissues [Citation3,Citation5]. At that time, HPC was still considered to be a special, angioblastic form of meningioma.
WHO decided to separate HPC from meningiomas in 1993 [Citation3,Citation6]. Subsequently, a unique grading scheme for HPC was introduced where the tumour corresponds histologically to grades II or III. The anaplastic form (grade III) shows a high degree of mitotic activity (a minimum of five mitoses per 10 HPF) and/or necrosis plus at least two of the following: haemorrhage, moderate to high nuclear atypia and cellularity [Citation13].
According to the “WHO classification of tumours of the central nervous system” HPC and SFT have been considered to be separate entities since 2007 [Citation13].
HPC is a meningeal tumour although the cells of origin are not meningeal (pericytes). Though theoretically they can arise from pericytes anywhere in the intracranial compartment, they have been mostly dural based masses. Purely intra-axial, non-dural based intracranial HPC is very unusual [Citation5].
In conclusion, the differential diagnoses to HPC are meningioma and SFT, which both display some similarities when it comes to location, radiology and clinics, but differ in terms of histology.
HPCs tend to occur more frequently in males in contrast to meningiomas [Citation1,Citation2,Citation5,Citation9,Citation11]. The male to female ratio is highly variable (1.1/1–1.5/1) among different series [Citation3].
HPCs also tend to have an earlier presentation in life (average age 41, with a range of 38–44 years) compared to meningiomas, which occur at least a decade later [Citation2,Citation5].
Some radiological features of HPCs can also differentiate them from meningiomas. HPCs are multilobulated tumours, most of which are large at presentation (more than 4 cm in the greatest dimension). They have a narrow base of dural attachment, a feature not typically seen in meningiomas. Unlike meningiomas, which frequently show hyperostosis and intratumoural calcifications, HPCs show bone erosion and lack of hyperostosis and calcifications [Citation15].
Histopathological examination is required for a correct diagnosis. Histologically, the tumours are composed of closely packed cells, which are randomly oriented. There is little fibrosis. The tissue is highly vascular with numerous vascular channels, many of which appear compressed and slit-like. Some thin-walled blood vessels are gaping and ramified in “staghorn patterns” [Citation10,Citation13]. Immunohistochemically, the tumours are diffusely positive for vimentin. A patchy positivity for CD 34 is seen in 33–100% of the cases in contrast to SFT, in which the CD34 expression is generally diffuse [Citation13]. Further, a positive staining is seen for reticulin and BCL2. Negative staining is seen for EMA, cytokeratin, GFAP, S-100 and CD31 [Citation2–4,Citation11,Citation12,Citation16].
Haemorrhage from HPC is not associated with any particular feature such as age, sex or size/location of the tumour [Citation10]. The mechanism favouring bleeding from HPCs remains unknown, but proposed hypotheses include: coagulatory changes in HPC, tumour growth causing erosion/distorsion/distension of blood vessels, tumour vascularity, change in the structure of vessel walls, endothelial proliferation which cases obliteration of vessels resulting in distal necrosis followed by bleeding [Citation4]. The bleeding is usually located to the dural attachment since HPCs often are dural based and supplied by the internal carotid artery [Citation5,Citation11].
Primary treatment is surgical resection [Citation1,Citation2,Citation5]. HPC is associated with relatively long survival time () [Citation1,Citation17]. Total resection gives an average survival time of 18.6 years, compared to subtotal resection with an average survival time of 9.75 years [Citation1]. The mean overall survival was 60 months longer in patients undergoing complete versus incomplete resection [Citation5].
Table II. The average survival time for patients with HPCs [Citation1].
HPCs have higher rates of recurrence and metastasis compared to meningiomas [Citation2,Citation12,Citation14,Citation18,Citation19]. Recurrence is seen in 50–80% of HPC cases [Citation8], rates as high as 91% is seen in some studies [Citation20]. Average time for recurrence is 39 months [Citation7]. High-grade (III) tumours recurred at an average of 36 months earlier than low-grade (II) tumours [Citation20]. Metastases are seen in 14–30% of HPC cases [Citation4]. Common localisations are bone, lung, liver, rib and neck [Citation6,Citation12,Citation21]. Average time for metastasis to occur is 79.2 months [Citation21]. In 10 years 33% of all HPCs show metastatic disease, and in 15 years the frequency of metastasis is 64% [Citation6]. In one case report metastasis appeared 22 years after initial diagnosis [Citation6]; therefore, long-term close clinical and radiological follow-up is crucial [Citation7,Citation19].
Postoperative radiotherapy is generally recommended to reduce the risk of local recurrence, improve survival rate and delay recurrence [Citation2,Citation4]. Despite been given postoperative radiotherapy, HPC cases may develop peripheral metastasis, suggesting HPC have a poor radiosensitivity [Citation4,Citation14]. A general recommendation may be harmed by the fact that most of the published literature does not include a sufficient number of patients (due to the rareness of the diagnosis) to give statistically significant results. Moreover, earlier studies which give support for postoperative radiotherapy are based on patient materials collected between 1955 and 1992, a period when the diagnosis of intracranial HPC was not generally established. According to a recent literature review, including 277 patients, the effect of postoperative radiotherapy could be questioned: “In total resection, addition or absence of radiation does not seem to confer a survival benefit. The results in the same study revealed that patients receiving more than 50 Gy of fractionated irradiation had worse survival outcomes, four years vs. 18.6 years. In subtotal resection postoperative radiotherapy decreases survival time from 9.75 to 6 years” [Citation1]. The same author (2012), however, states that there was a trend in their material to a prolonged time to recurrence among patients who received adjuvant irradiation after primary surgical resection [Citation22,Citation23]. Many writers also find signs of prolonged survival and postponed time for recurrence after adding irradiation postoperatively and recommend an aggressive approach with total doses of fractionated radiotherapy to 50 Gy and more [Citation24–26].
Case report
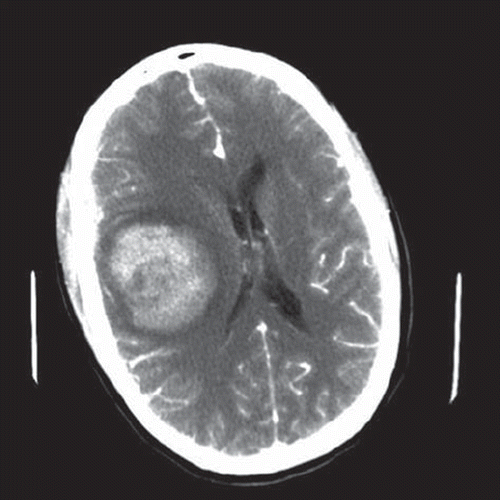
A 27-year-old man with no history of trauma, arterial hypertension or anticoagulant therapy was admitted to the emergency ward due to sudden headache and vomiting followed by impaired consciousness. It later turned out that he had experienced severe headache for the last six months. A complete AV-block III was obvious from the electrocardiogram. Further physical examination revealed decreasing consciousness and left side hemi-paresis. A computed tomography (CT)-scan was obtained, which showed a right-sided frontoparietal haemorrhagic lesion (5 × 6 cm) with surrounding edema, a right-sided temporofrontal subdural haematoma of 7 mm and a marked midline shift (12 mm), (). CT-angiography ruled out any arteriovenous malformations. Subsequently, the decision was made to emergently take the patient to the operating room. Mannitol 300 ml was administrated preoperatively. An acute evacuation of the haematoma was performed and en passant a tumour suspicious component invading the dura was found. Peroperatively, two diagnoses were mainly suspected: glioma due to the gross appearance of the mass and malignant melanoma due to the patient's young age and profuse bleeding.
Figure 1. First CT-scan performed after i.v. contrast injection shows a right-sided frontoparietal haemorrhagic lesion (5 × 6 cm) with surrounding oedema, a right-sided temporofrontal subdural haematoma of 7 mm and a marked (12 mm) shift of the midline structures.
Tumour characterisation
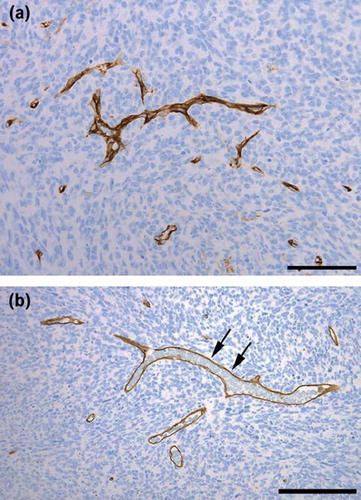
Light microscopy revealed a highly cellular tumour consisting of polygonal or spindle-shaped cells, which tended to form fascicles. There was considerable nuclear polymorphism, numerous mitoses and necroses ( and ). The tumour tissue was richly vascularised with numerous slit-like compressed blood vessels as well as wide, thin-walled blood vessels, arranged in staghorn patterns, ( and ). The tumour tissue showed a general expression of vimentin and BCL-2. Some minor, focal expression of CD34, CD31, AE1/AE3, NSE, and NFP was also noticed.
Figure 2. Highly cellular tumour tissue, displaying nuclear polymorphism and mitoses (arrows) (Haematoxylin-eosin. Bar: 100 μm).
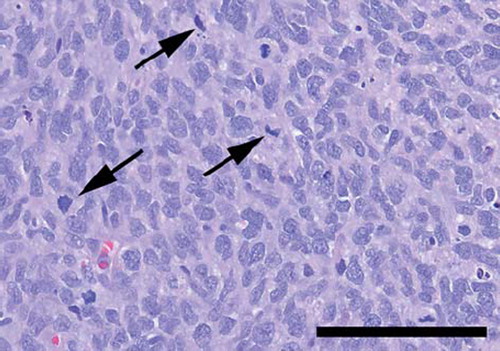
Figure 3. There were numerous necroses throughout the tumour (Haematoxylin-eosin. Bar 500 μm).
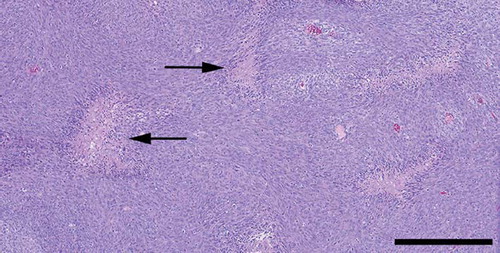
Figure 4. The tumour tissue was rich in compressed, thin-walled blood vessels. (Anti CD34 – haematoxylin. Bar: 100 μm). (b) Thin-walled sinusoidal blood vessels, forming staghorn patterns were found throughout the tumour (Anti CD34 – haematoxylin. Bar: 200 μm).
The immunophenotype gave no support for differential diagnoses such as malignant melanoma, anaplastic meningioma, solitary fibrous tumour, parenchymal brain tumour or Ewing sarcoma. FISH analysis gave no support for synovial sarcoma. Thus, based on the general histopathological appearance, the vascular pattern, immunocytochemistry and the exclusion of a number of malignant tumours, the final diagnosis was haemangiopericytoma of grade III according to WHO.
Course after surgery
Postoperatively, the development of a left side hemi-paresis was evident. A magnetic resonance imaging (MRI) examination was performed one week postoperatively, which demonstrated only remnants of the haematoma. A CT-scan four weeks later revealed no signs of remaining tumour and the haematoma was totally absorbed (). Postoperative radiotherapy was given in 2 Gy fractions, 5 fractions per week for 5½ weeks, with a total dose of 56 Gy.
The patient recovered well with help of physical therapy and three months after surgery the left side hemi-paresis was slowly improving.
Figure 5. Five weeks postoperatively, CT-scan performed after i.v. contrast injection, showing no signs of remaining tumour and the haematoma is totally absorbed.
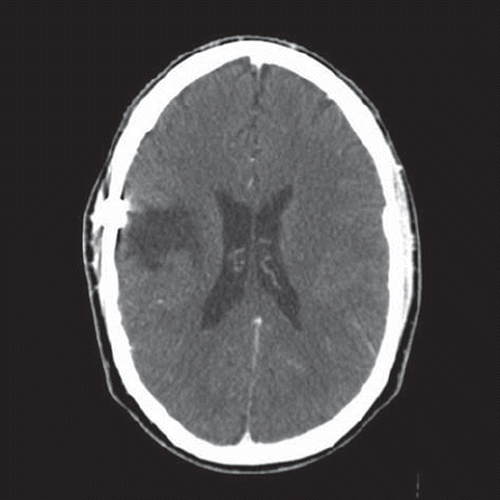
At the one year follow-up MRI showed gliotic-postoperative changes in one area measuring 5.5 × 4.5 × 5 cm. No pathological contrast uptake of residual tumour was observed, no signs of a mass effect and only a slight compensatory dilation of the right lateral horn.
At one year follow-up, there were no residual clinical deficits, on the contrary an improvement of the left side spastic paresis was evident. The patient used his left arm with some difficulties in daily practice and walked with support in stairs. Besides the physical improvement the patient was both feeling better mentally and making plans for the future.
The patient and his family have given their written permission to publish the case report.
Discussion
There are only 13 cases reported of meningeal haemangiopericytoma associated with subdural haematoma, subarachnoid haematoma, intratumoural haematoma or intracerebral haematoma () [Citation4,Citation10]. Although quite rare, HPC carries the risk of life-threatening bleeding, and can be included in the differential diagnosis of intracranial haemorrhage.
In other cases of intracerbral haematoma of unknown origin (not too old patient, no hypertension) a follow-up MRI before and after contrast injection is helpful if it is desirable to exclude or detect tumour as the aetiology of the haemorrhage.
The rarity of HPC, only recently established as a separate diagnosis, and the clinical and radiological similarities with meningioma and SFT makes it practically impossible to reach a prompt diagnosis without surgery. It is essential to reach a correct HPC diagnosis due to differences from meningiomas and SFTs when it comes to treatment and prognosis. A histopathological examination is needed. In our case, a number of differential diagnoses were excluded, based on immunocytochemistry and FISH. Still, the immunoprofile of haemangiopericytoma is diverse, and no antibody is 100% sensitive or specific. There is generally a diffuse expression of vimentin. The reactivity for CD34 is usually patchy which is in contrast to the diffuse CD34 expression typical for solitary fibrous tumours. Also, a patchy positivity for cytokeratin may be seen. Moreover, the present tumour showed sparse multifocal expression of NSE and NFP. This may not be controversial since CNS micovascular pericytes exhibit multipotential stem cell activity. In cell culture, fully differentiated cells of neuronal lineage may develop [Citation27].
HPC has a high rate of recurrence and metastasis, sometimes occurring several years after initial diagnosis. Therefore it is important to have a long-term close clinical and radiological follow-up. For follow-up, MRI is the method of choice, due to the higher sensitivity for detection of meningeal lesions than CT. In addition, if a long-term follow-up of relatively young patients is performed, a method without ionising radiation should always be preferred. Furthermore, total surgical resection is crucial, since radicality correlates with better prognosis.
Radiation therapy in HPC in general is still a matter of debate as the studies that we have found all are retrospective in scope. The radiotherapy, when given, varies between different institutions in target definitions, treatment techniques and total doses, which makes interpretation and conclusions difficult.
In our mind we find radiotherapy well motivated in HPC, WHO grade II, if there is a tumour left after surgery or the patient is not operated on. In patients with macroscopically removed tumour we will always consider postoperative radiotherapy to a total fractionated dose of 50 Gy. For HPC, WHO grade III, we recommend total doses of 56–60 Gy, given in 2.0 Gy fractions.
Our patient had a histological HPC grade III, which is rarely seen compared with grade II tumours. HPC grade III is associated with an increased recurrence rate, which in our case motivated a more aggressive treatment, similar to the one given to a malignant glioma. Future multicentre prospective trials, controlling for low- and high-grade HPC, might give new information regarding strategies and the benefit of radiation therapy in the treatment of this type of tumour.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Rutkowski MJ, Sughrue ME, Kane AJ, Aranda D, Mills SA, Barani IJ, et al. Predictors of mortality following treatment of intracranial hemangiopericytoma. J Neurosurg 2010;113:333–9.
- Binello E, Bederson JB, Kleinman GM. Hemangiopericytoma: Collision with meningioma and recurrence. Neurol Sci 2010;31:625–30.
- Fountas KN, Kapsalaki E, Kassam M, Feltes CH, Dimopoulos VG, Robinson JS, et al. Management of intracranial meningeal hemangiopericytomas: Outcome and experience. Neurosurg Rev 2006;29:145–53.
- Maruya J, Seki Y, Morita K, Nishimaki K, Minakawa T. Meningeal hemangiopericytoma manifesting as massive intracranial hemorrhage – two case reports. Neurol Med Chir (Tokyo) 2006;46:92–7.
- Shetty PM, Moiyadi AV, Sridhar E. Primary CNS hemangiopericytoma presenting as an intraparenchymal mass – case report and review of literature. Clin Neurol Neurosurg 2010;112:261–4.
- Suzuki H, Haga Y, Oguro K, Shinoda S, Masuzawa T, Kanai N. Intracranial hemangiopericytoma with extra cranial metastasis occurring after 22 years – case report. Neurol Med Chir (Tokyo) 2002;42:297–300.
- Bassiouni H, Asgari S, Hübschen U, König HJ, Stolke D. Intracranial hemangiopericytoma: Treatment outcomes in a consecutive series. Zentralbl Neurochir 2007;68:111–8.
- Heiser MA, Waldron JS, Tihan T, Parsa AT, Cheung SW. Temporal fossa hemangiopericytoma: A case series. Otol Neurotol 2009;30:985–9.
- Kim JH, Jung HW, Kim YS, et al. Meningeal hemangiopericytomas: Long term outcome and biological behaviour. Surg Neurol 2003;59:47–54.
- Kaen A, Arrese I, Lagares A, Cabello A, Lobato RD. Short illustrated review haemangiopericytoma presenting with acute intracerebral haemorrhage. Acta Neurochir 2007;149:415–8.
- Sibtain NA, Butt S, Connor SEJ. Imaging features of central nervous system haemangiopericytomas. Eur Radiol 2007; 17:1685–93.
- Ambrosini-Spaltro A, Eusebi V. Meningeal hemangiopericytomas and hemangiopericytoma/solitary fibrous tumours of extracranial soft tissues: A comparison. Virchows Arch 2010;456:343–54.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97–109.
- Thanni LOA. Extremity haemangiopericytoma, a case report from Nigeria. Afr Health Sci 2005;5:261–4.
- Chiechi MV, Smirniotopoulos JG, Mena H. Intracranial hemangiopericytomas: MR and CT features. Am J Neuroradiol 1996;17:1365–71.
- Hori E, Kurimoto M, Fukuda O, Takahashi C, Nagai S, Oya T, et al. Recurrent intracranial solitary fibrous tumor initially diagnosed as hemangiopericytoma. Brain Tumour Pathol 2007;24:31–4.
- Spatola C, Privitera G. Recurrent intracranial hemangiopericytoma with extracranial and unusual multiple metastases: Case report and review of the literature. Tumori 2004;90:265–8.
- Hayashi Y, Uchiyama N, Hayashi Y, Nakada M, Iwato M, Kita D, et al. A reevaluation of the primary diagnosis of hemangiopericytoma and the clinical importance of differential diagnosis from solitary fibrous tumour of the central nervous system. Clin Neurol Neurosurg 2009;111:34–8.
- Olson C, Yen CP, Schlessinger D, Sheehan J. Radiosurgery for intracranial hemangiopericytomas: Outcomes after initial and repeat gamma knife surgery. J Neurosurg 2010;112:133–9.
- Schiariti M, Goetz P, El-Maghraby H, Tailor J, Kitchen N. Hemangiopericytoma: Long-term outcome revisited. J Neurosurg 2010;114:747–55.
- Kano H, Niranjan A, Kondziolka D, Flickinger J, Lunsford D. Adjuvant stereotactic radiosurgery after resection of intracranial hemangiopericytomas. Int J Radiat Oncol Biol Phys 2008;5:1333–9.
- Rutkowski MJ, Bloch O, Jian BJ, Chen C, Sughrue ME, Tihan T, et al. Management of recurrent intracranial hemangiopericytoma. J Clin Neurosci 2011;18:1500–4.
- Rutkowski MJ, Jian BJ, Bloch O, Chen C, Sughrue ME, Tihan T, et al. Intracranial hemangiopericytoma: Clinical experience and treatment considerations in a modern series of 40 adult patients. Cancer 2012;118:1628–36.
- Zweckberger K, Jung CS, Mueller W, Unterberg AW, Schick U. Hemangiopericytomas grade II are not benign tumors. Acta Neurochir 2011;153:385–94.
- Schirmer CM, Heilman CB. Hemangiopericytomas of the skull base. Neurosurg Focus 2011;30:E10.
- Schiariti M, Goetz P, Maghraby El, Tailor J, Kitchen N. Hemangiopericytoma: Long-term outcome revisited. J Neurosurg 2011;114:747–55.
- Dore-Duffy P, Katychev A, Wang X, Van Buren E. CNS microvascular pericytes exhibit multipotential stem cell activity. Rapid Communication. J Cereb Blood Flow Metab 2006;26:613–24.