Abstract
Purpose. This pre-clinical study was designed to investigate the effect of various vascular disrupting agents (VDAs) that have undergone or are in clinical evaluation, had on the oxygenation status of tumours and what effects that could have on the combination with radiation. Material and methods. The tumour model was a C3H mammary carcinoma grown in the right rear foot of female CDF1 mice and treated when at 200 mm3 in size. The VDAs were the flavenoid compounds flavone acetic acid (FAA) and its more recent derivative 5,6-dimethylxanthenone-4-acetic acid (DMXAA), and the leading tubulin binding agent combretastatin A-4 phosphate (CA4P) and the A-1 analogue OXi4503. Oxygenation status was estimated using the Eppendorf oxygen electrode three hours after drug injection. Radiation response was determined following single or fractionated (10 fractions in 12 days) irradiations with a 240 kV x-ray machine using either a tumour re-growth or local tumour control assay. Results. All VDAs significantly reduced the oxygenation status of the tumours. They also influenced radiation response, but the affect was time and sequence dependent using single radiation schedules; an enhanced effect when the VDAs were injected at the same time or after irradiating, but no or even a reduced effect when given prior to irradiation. Only OXi4503 showed an increased response when given before the radiation. CA4P and OXi4503 also enhanced a fractionated radiation treatment if the drugs were administered after fractions 5 and 10. Conclusions. VDAs clearly induced tumour hypoxia. This had the potential to decrease the efficacy of radiation. However, if the appropriate timing and scheduling were used an enhanced effect was observed using both single and fractionated radiation treatments.
An adequate supply of oxygen and nutrients are necessary for tumour cells to remain viable and for the tumour to increase in size. Initially these factors come from the host normal vascular supply, but when tumours reach a few millimeters in size the cells can exceed the diffusion distance of oxygen and other essential nutrients [Citation1,Citation2]. Further tumour development is then only possible if the tumour forms its own functional vascular system from the host vessels by the process of angiogenesis [Citation3,Citation4]. This tumour neo-vasculature not only provides tumours with oxygen and nutrients, it also allows for the removal of waste products, and is the principal vehicle for metastatic spread [Citation5].
This importance of the tumour vascular supply makes it a potential therapeutic target and two major groups of vascular targeting agents (VTAs) have now emerged [Citation6,Citation7]. One is based on controlling blood vessel development by inhibiting the angiogenesis process; these agents are collectively referred to as angiogenesis inhibitors (AIs). The other approach involves compromising the function of the already existing blood vessels using so called vascular disrupting agents (VDAs). Numerous examples of both AIs and VDAs are currently undergoing clinical evaluation [Citation8–11]. Regardless of the VTA approach used there is no evidence that when used alone they will actually result in tumour control [Citation7] and thus for their full clinical potential to be realised they must be combined with more conventional therapies, especially radiation [Citation7,Citation12]. However, the tumour vascular supply has a significant influence on the microenvironmental conditions within tumours [Citation13] and since the tumour microenvironment plays a major role in determining the outcome to radiation therapy, any treatment that targets the tumour vasculature, has the potential to alter the microenvironmental conditions and thus change the tumour response to the radiation with which it is combined.
The aim of this pre-clinical study was to investigate this issue using a variety of VDAs. These include the two major classes of small molecule drugs [Citation6,Citation7]. The first are the flavenoid compounds, which have a complex mechanism of action that is poorly understood, but their main effect on vascular endothelial cells is thought to involve a cascade of direct and indirect effects, the latter involving the induction of cytokines, especially tumour necrosis factor alpha (TNF-α), leading to the induction of haemorrhagic necrosis [Citation14,Citation15]. These include the historical compound flavone acetic acid (FAA) and its more recent derivative 5,6-dimethylxanthenone-4-acetic acid (DMXAA; ASA404; vadimezan) which is currently in clinical evaluation [Citation16,Citation17]. The second group includes tubulin binding agents that are primarily believed to selectively disrupt the cytoskeleton of proliferating endothelial cells, resulting in endothelial cell shape changes and subsequent thrombus formation and vascular collapse [Citation15]. They include the leading small molecule VDA combretastatin A-4 phosphate (CA4P; fosbretabulin; zybrestat) and the newer A-1 analogue OXi4503 [Citation18,Citation19].
Material and methods
Animal and tumour model
A C3H mammary carcinoma grown in the right rear foot of 10–14-week-old female CDF1 mice was used in all experiments. The derivation and maintenance of this tumour have been described previously [Citation20]. Experimental tumours were produced following sterile dissection of large flank tumours. The excised tumour tissue was minced with a pair of scissors and 5–10 μl of this material injected into the foot of experimental animals. This location ensured easy access to the tumour for treatment without involvement of critical normal tissues in the radiation treatment field. Experiments were preformed when tumours had reached approximately 200 mm3 in size, which typically occurred three weeks after inoculation; tumour volume being calculated from the formula D1*D2*D3*π/6, where the D values represent the three orthogonal diameters. Additional studies have demonstrated that when this tumour model is treated at this size in the foot the results obtained with vascular disrupting drugs are identical to those seen in patients [Citation21]. All experiments were conducted in accordance with National and European Union approved guidelines for animal welfare, with the Danish Animal Experiments Inspectorate's approval.
Drug preparation
The VDAs used in this study included CA4P and its A-1 derivative OXi4503 (supplied by Oxigene Inc., South San Francisco, CA, USA), flavone acetic acid (FAA; obtained from Lyonnaise Industrielle Pharmaceutique, Lyon, France) and its derivative DMXAA (provided by Dr. William Denny at the University of Auckland, New Zealand). All drugs were prepared fresh before each experiment by dissolving either in saline (CA4P, OXi4503, and DMXAA) or in a 1% Na2CO3 solution (FAA). They were injected intraperitoneally (i.p.) in a volume of 0.02 ml/g mouse body weight; the final concentrations being CA4P (250 mg/kg), OXi4503 (50 mg/kg), FAA (150 mg/kg), and DMXAA (20 mg/kg).
Eppendorf electrode measurements
Tumour oxygenation measurements were made as previously described [Citation22]. Basically, tumour bearing mice were transferred to a specially made Lucite jig which restrained the non-anaesthetised animal but allowed for the tumour bearing leg to be exposed and the foot attached to the jig with tape. A fine needle autosensitive electrode probe (Eppendorf, Hamburg, Germany) was then inserted about 1 mm into the tumour, and the needle then moved automatically through the tissue in 0.7 mm increments, followed each time by a 0.3 mm backward step prior to measurement. Response time was 1.4 s. After making measurements along one track in the tumour the probe was removed and repeated parallel insertions were made. The number of tracks per tumour was 3–6 with the total number of measurements being 35–90. The relative frequency of the oxygen partial pressure (pO2) measurements was automatically calculated and displayed as a histogram. From the raw data various parameters may be selected, but we chose to use the percentage of pO2 values ≤ 5 mmHg and the median pO2, because the former is likely to include all the radiobiologically hypoxic cells in the tumour, while the latter is indicative of the overall oxygenation status. Measurements were made either three hours after VDA injection or after applying a clamp to achieve total hypoxia. Clamping involved tightening a rubber tube around the leg proximal to the tumour for five minutes before the start of measurement and maintaining the clamp in position during the entire measurement period.
Radiation treatments
Irradiations were given using a conventional therapeutic x-ray machine (240 kV, 15 mA, 2-mm Al filter, 1.1-mm Cu half-value layer, dose rate of 2.3 Gy/min) as previously described [Citation22]. Dosimetry was accomplished by using an integrating chamber. All treatments to tumour-bearing feet were given locally to the tumours of non-anesthetised mice placed in the restraining jigs as described for the Eppendorf measurements. The tumour-bearing legs were again exposed and loosely attached with tape to the jig, without impairing the blood supply to the foot. Only tumours were irradiated; the remainder of the mouse was shielded by 1 cm of lead. To secure homogeneity of the radiation dose, the tumours were immersed in a water bath with about 5 cm of water between the x-ray source and the tumour. Radiation was administered either as single irradiations or given in a fractionated schedule of 10 fractions in 12 days (5 daily irradiations – 2 day gap – 5 daily irradiations). Tumour response to radiation treatment was assessed using either a regrowth delay or local tumour control assay. The tumour regrowth experiments involved determining tumour volume five times each week after treatment, and the tumour growth time (TGT3 – time in days to reach three times the treatment volume) was determined. For the tumour control studies mice were observed on a weekly basis and the percentage of animals in each treatment group showing local tumour control 90 days after treatment recorded.
Data and statistical analysis
Results with the Eppendorf oxygen electrode are shown as mean (± 1 S.E.) and statistically significant differences between the values were determined using a Student's t-test. For the radiation studies the data was analysed either by determining the slope of the tumour regrowth dose response curve or calculating the TCD50 dose (radiation dose producing tumour control in 50% of treated animals) following logit analysis of the dose response curve for tumour control. Statistical comparison of the TCD50 values was made using a χ2-test. For all statistical tests the significance level was p < 0.05.
Results
Typical histograms showing the results obtained in our C3H mammary carcinoma with the Eppendorf oxygen electrode under control or clamped conditions are illustrated in . Clamping tumours substantially shifted the oxygen partial pressure distributions to the left and this was reflected by a significant decrease in the median pO2 and a corresponding significant increase in the percentage of pO2 values ≤ 5 mmHg (). also shows the results for these two parameters obtained three hours after injection of the different VDAs. Although variability was seen with the percentage of pO2 values ≤ 5 mmHg between the various VDAs, all were significantly higher than that found in control animals. All VDAs significantly reduced the median pO2 values and here there was less variability between the drugs. Several of the VDAs actually resulted in levels of hypoxia (percentage of pO2 values ≤ 5 mmHg) that were close to those found under clamped conditions.
Figure 1. Representative histograms showing the oxygen partial pressure (pO2) profiles obtained in C3H mammary carcinomas with the Eppendorf oxygen electrode. Results show the relative frequency of the various pO2 values measured in 3–6 mice under either control or clamped conditions.
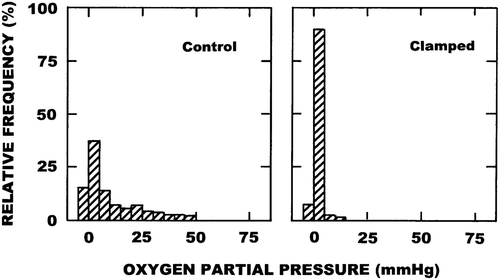
Table I. The effect of VDA treatment on tumour oxygenation.
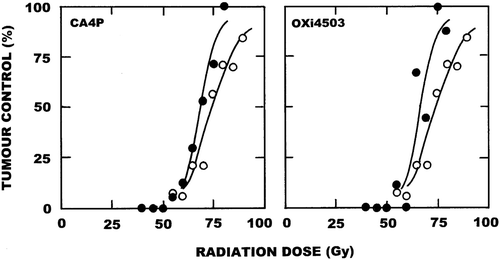
Representative examples showing how VDAs affect the radiation response of the C3H mammary carcinoma are shown in the top panels of . The VDA used in these examples was the newest combretastatin analogue OXi4503. For this tumour model a linear relationship exists between the TGT3 and radiation dose, with a slope value of 0.75. OXi4503 alone significantly influenced the mean (± 1 S.E.) TGT3; going from a value of 3.3 days (3.1–3.5) for control tumours to 7.7 days (7.3–8.1) following OXi4503 treatment. Injecting OXi4503 two hours after irradiating increased the TGT3 at all radiation doses, resulting in a slope of 1.0 and an enhancement ratio (ER; ratio of the slopes obtained for radiation alone and radiation + OXi4503) of 1.33. Similar results were seen when using a local tumour control assay (). Here the TCD50 dose (± 95% confidence intervals) for radiation alone was 54 Gy (52–57). Administering OXi4503 two hours following irradiation caused a parallel shift in the radiation dose response curve, with the TCD50 value being significantly reduced to 41 Gy (39–45). Comparing these two TCD50 values again resulted in an ER of 1.32. From these tumour growth and tumour control results, and from additional similar studies, we calculated the ERs when a variety of VDAs were injected at different times before or after irradiating and those results are summarised in the bottom panel of . Clearly, injecting the VDAs at the same time or after irradiating always enhanced the radiation response of this C3H mammary carcinoma. This effect was generally independent of the VDA used, or the time interval between radiation and VDA treatment. When the VDAs preceded the radiation a different response was observed. For CA4P and DMXAA there was either no enhancement of radiation response or, when irradiating within a few hours of injecting the VDA, there was even a decreased effect. This was not observed with OXi4503 in which a similar degree of enhancement was obtained regardless of whether the drug was given before after irradiating.
Figure 2. The effect of VDAs on the radiation response of C3H mammary carcinomas. Left panel top: Tumour growth time (time for tumours to grow to three times treatment volume) following local tumour irradiation. Results are the means (± 1 S.E.) from 6–8 mice per group with the lines fitted following linear regression analysis using individual data points. Right panel top: The percentage local tumour control obtained 90 days after irradiation. Results are from 8–19 mice per group, with the curves fitted following logit analysis. For both figures the results are for radiation alone (○) or radiation followed by an injection with OXi4503 (50 mg/kg) two hours later (●). Bottom panel: Enhancement ratios (ER; ratio of the effect of radiation alone and radiation plus VDA) as a function of varying the time interval and schedule between radiation and VDA treatment. These ERs were calculated from the data shown in the top panels, plus similar unpublished and published data [Citation23–26]. Radiation was always given at time zero and no enhancement of radiation response is represented by an ER of 1.0. Points are for FAA (▲), DMXAA (∆), CA4P (○), and OXi4503 (●).
![Figure 2. The effect of VDAs on the radiation response of C3H mammary carcinomas. Left panel top: Tumour growth time (time for tumours to grow to three times treatment volume) following local tumour irradiation. Results are the means (± 1 S.E.) from 6–8 mice per group with the lines fitted following linear regression analysis using individual data points. Right panel top: The percentage local tumour control obtained 90 days after irradiation. Results are from 8–19 mice per group, with the curves fitted following logit analysis. For both figures the results are for radiation alone (○) or radiation followed by an injection with OXi4503 (50 mg/kg) two hours later (●). Bottom panel: Enhancement ratios (ER; ratio of the effect of radiation alone and radiation plus VDA) as a function of varying the time interval and schedule between radiation and VDA treatment. These ERs were calculated from the data shown in the top panels, plus similar unpublished and published data [Citation23–26]. Radiation was always given at time zero and no enhancement of radiation response is represented by an ER of 1.0. Points are for FAA (▲), DMXAA (∆), CA4P (○), and OXi4503 (●).](/cms/asset/8df5e952-8b1e-43b3-8a51-19401dad97bc/ionc_a_825050_f0002_b.gif)
illustrates how VDAs could be combined with a fractionated radiation schedule. In these experiments radiation was administered as 10 fractions in 12 days (5 daily irradiations – 2 day gap – 5 daily irradiations) and the resulting TCD50 value was found to be 76 Gy (73–79). The VDAs were then injected one hour after giving radiation fractions 5 and 10 and the resulting TCD50 values were found to be 69 Gy (66–72) when using CA4P and 67 Gy (63–71) with OXi4503. These TCD50 values obtained with CA4P and OXi4503 were significantly lower than the radiation only value.
Figure 3. The effect of VDAs on the response of the C3H mammary carcinoma when irradiated in a fractionated schedule of 10 fractions in 12 days. Local tumour control was determined 90 days after completion of irradiation. Results are from an average of 18 mice per group, with the curves fitted following logit analysis. For both figures the results are for radiation alone (○) or radiation followed by an injection with the VDA one hour after radiation fractions number 5 and 10 (●); the VDAs being either CA4P (250 mg/kg; left panel) or OXi4503 (50 mg/kg; right panel).
Discussion
This current study, using representative examples of the two leading groups of small molecule VDAs, demonstrated that within three hours following injection they all significant increased the level of hypoxia in this C3H mammary carcinoma. Similar findings have been reported by us in this murine tumour model using other small molecule VDAs, including tubulin binding agents like ZD6126 [Citation27] and plinabulin [Citation28]. Increases in tumour hypoxia have also been observed by others using various VDAs [Citation29–32]. Such effects are not entirely surprising since all these agents are known to significantly reduce tumour blood flow as a result of the induced vascular damage, especially in this C3H mammary carcinoma [Citation27,Citation28,Citation33–35]. The time at which the nadir in flow reduction occurs is drug dependent, being reached within three hours for the tubulin binding agents [Citation27,Citation28,Citation34], but taking up to six hours for the flavenoid compounds [Citation33,Citation34]. Thus, while starting the oxygenation measurements in our current study three hours after drug injection may have been idea for CA4P and OXi4503, an even larger decrease may have been obtained with FAA and DMXAA if we had waited for an additional three hours. But, even at the time we used, a large significant increase in hypoxia was observed. The effects seen with VDAs are in contrast to those seen with the other principle group of VTAs, the AIs. These generally reduce tumour vascular density [Citation7], which as expected often results in an increased tumour hypoxia [Citation7]. However, this is not a universal effect, since numerous studies also report no change in oxygenation status and even an improvement [Citation7].
Regardless of whether one uses AIs or VDAs, targeting tumour vasculature can certainly inhibit tumour growth; this is illustrated by the increase in TGT3 for OXi4503 alone (). However, there are no examples in the literature showing VTAs to be capable of inducing tumour control. As such, all VTAs must be combined with other more conventional therapies and such a combination with radiation has been extensively investigated in pre-clinical studies [Citation7]. However, tumour response to radiation is strongly dependent on the microenvironmental conditions within the tumour. Oxygen is critical for the efficacy of radiation to damage cells [Citation36] and numerous pre-clinical and clinical studies have clearly demonstrated that the greater the degree of tumour hypoxia the worse the response to radiation [Citation36]. So any VTA that induces hypoxia has the potential to reduce the cell killing potential of radiation. This was illustrated with the ER values in , where injecting CA4P or DMXAA prior to radiation not only failed to enhance radiation response, but with short time intervals between drug injection and irradiation they could also result in a reduced level of tumour control. Obviously there were cells in the tumour that were probably sufficiently oxygenated to be killed by the radiation treatment, that survive the vascular shut-down and because of the induced hypoxia actually now become radiation resistant. Of course this issue can be avoided by injecting the VDAs after irradiating and in fact this was the schedule used in those pre-clinical studies reporting an enhancement of radiation response using single radiation treatments [Citation7]. The ER data of clearly illustrates the benefit obtained using this approach. The problem arises when one has to fractionate the radiation treatment as is done clinically; administering the drug after one radiation fraction may reduce the effectiveness of the next fraction. In our study, we tried to overcome this problem by only giving the VDAs after the final radiation treatment each week (). Our assumption was that the three-day interval between the last radiation treatment in one week and the first treatment of the following week would allow sufficient time for either the induced hypoxic cells to reoxygenate or even die if the oxygen and nutrient starvation was prolonged. The results of using this procedure in our limited fractionated study clearly demonstrated a benefit, supporting the suggestion of using such an approach if these agents are to be applied clinically in combination with radiation therapy.
Interestingly, the effect with OXi4503 in a fractionated schedule was exactly the same as CA4P, yet OXi4503 did not show the same time and schedule dependency; OXi4503 enhanced radiation response to the same degree regardless of whether it was administered before or after irradiating (ER data of ). OXi4503 is an analogue of CA4P, which is superior to CA4P because it induces greater vascular damage [Citation37] and also undergoes oxidative activation to a quinine intermediate, thereby, giving it direct cell killing properties [Citation38]. Thus, the enhancement seen when OXi4503 is administered prior to irradiating is probably a result this drug killing any hypoxic cells it induces. Since both OXi4503 and CA4P were identical in their enhancement of the fractionated radiation treatment it also suggests that the hypoxic cells induced by CA4P actually die during the three day recovery period rather than reoxygenate. The fact that OXi4503 does not show a time-sequence dependency probably means that it could be given more often during a fractionated radiation schedule than is possible with other VDAs, but the question is how often should it be given? Once vascular damage has been induced it would seem counter-productive to administer OXi4503 again until the vasculature has shown significant recovery. One study that investigated the time course for recovery suggested that a second treatment should be delayed for 72 hours [Citation37]. This would suggest the optimal application would be giving OXi4503 only twice per week rather than more often.
Apart from influencing the local tumour response to radiation, hypoxia has also been shown to have an effect on malignant progression, especially the development of distant metastases [Citation39–41]. Recent studies with AIs have now shown that prolonged treatment with these agents, while inhibiting the growth of primary tumours, significantly increased tumour hypoxia and this subsequently led to an increase in metastatic burden [Citation42,Citation43]. Whether the same effect occurs after VDA treatment, has never been investigated. It may not be such a significant issue because unlike the studies with AIs using continuous and prolonged exposure times, treatment with VDAs is intermittent and would only be during the relatively short radiation schedule. In addition, if the induced hypoxic cells are being killed following the drug treatment, and this is likely to be the case with OXi4503 and perhaps with other VDAs, it is unlikely that these cells will be able to give rise to metastases. Nevertheless, it is an important issue that needs investigation prior to extensive clinical application of such VDAs.
In conclusion, our current study has clearly demonstrated that VDAs induce tumour hypoxia. While this suggests a potential negative factor for the combination of VDAs with radiation therapy, it is clearly not a problem if VDAs are administered after, rather than prior, to irradiation. In a clinical fractionated schedule this would mean giving the VDA after the final radiation treatment each week. Using VDAs like OXi4503 which have dual functional activity and can kill any hypoxic cells that it may induce as a result of the vascular shut-down such concerns about timing become a less relevant issue.
Acknowledgements
The authors would like to thank Ms. Dorthe Grand, Ms. Inger Marie Horsman, Ms. Pia Schjerbeck, Ms. Marianne Verner Bjerre, and Ms. Marianne Kristiansen for excellent technical assistance.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by grants from the Danish Cancer Society and the Danish Council for Independent Research: Medical Sciences.
References
- Brem S, Brem H, Folkman J, Finkelstein D, Patz A. Prolonged tumor dormancy by prevention of neovascularization in the vitreous. Cancer Res 1976;36:2807–12.
- Folkman J. How is blood vessel growth regulated in normal and neoplastic tissue?Cancer Res 1986;46:467–73.
- Hahnfeldt P, Panigrahy D, Folkman J, Hlatky L. Tumor development under angiogenic signaling: a dynamic theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res 1999;59:4770–5.
- Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 2003;3:401–10.
- Stoeltzing O, Ellis LM. The role of microvasculature in metastasis formation. In: Siemann DW, editor. Vascular- targeted therapies in oncology. Chichester: John Wiley & Sons, Ltd.; 2006. p. 31–62.
- Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, et al. Differentiation and definition of vascular-targeted therapies. Clin Cancer Res 2005;11: 416–20.
- Horsman MR, Siemann DW. Pathophysiological effects of vascular targeting agents and the implications for combination with conventional therapies. Cancer Res 2006;66:11520–39.
- National Cancer Institute Website: www.cancer.gov/clinicaltrials.
- Hofer S, Elandt K, Greil R, Hottinger AF, Huber U, Lemke D, et al. Clinical outcome with bevacizumab in patients with recurrent high-grade glioma treated outside clinical trials. Acta Oncol 2011;50:630–5.
- Cao C, Wang J, Bunjhoo H, Xu Y, Fang H. Risk profile of bevacizumab in patients with non-small cell lung cancer: a meta-analysis of randomized controlled trials. Acta Oncol 2012;51:151–6.
- Patterson DM, Rustin GJS. Vascular damaging agents. Clin Oncol 2007;19:443–56.
- Siemann DW, Warrington KH, Horsman MR. Targeting tumor blood vessels: an adjuvant strategy for radiation therapy. Radiother Oncol 2000;57:5–12.
- Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol 2012;9:674–87.
- Baguley BC. Antivascular therapy of cancer: DMXAA. Lancet Oncol 2003;4:141–8.
- Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer 2005;5:423–35.
- Lara PN Jr, Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Onol 2011;29:2965–71.
- Früh M, Cathomas R, Siano M, Tscherry G, Zippelius A, Mamot C, et al. Carboplatin and paclitaxel plus ASA404 as first-line chemotherapy for extensive-stage small-cell lung cancer: A multicenter single arm phase II trial (SAKK 15/08). Clin Lung Cancer 2013;14:34–9.
- Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin Investig Drugs 2009;18:189–97.
- Patterson DM, Zweifel M, Middleton MR, Price PM, Folkes LK, Stratford MR, et al. Phase I clinical and pharmacokinetic evaluation of the vascular-disrupting agent OXi4503 in patients with advanced solid tumors. Clin Cancer Res 2012;18:1415–25.
- Overgaard J. Simultaneous and sequential hyperthermia and radiation treatment of an experimental tumor and its surrounding normal tissue in vivo. Int J Radiat Oncol Biol Phys 1980;6:1507–17.
- Nielsen T, Murata R, Maxwell RJ, Stødkilde-Jørgensen H, Østergaard L, Horsman MR. Preclinical studies to predict the efficacy of the vascular changes induced by combretastatin A-4 disodium phosphate in patients. Int J Radiat Oncol Biol Phys 2008;70:859–66.
- Horsman MR, Nordsmark M, Khalil AA, Hill SA, Chaplin DJ, Siemann DW, et al. Reducing acute and chronic hypoxia in tumours by combining nicotinamide with carbogen breathing. Acta Oncol 1994;33:371–6.
- Murata R, Siemann DW, Overgaard J, Horsman MR. Interaction between combretastatin A-4 disodium phosphate and radiation in murine tumours. Radiother Oncol 2001; 60:155–61.
- Murata R, Siemann DW, Overgaard J, Horsman MR. Improved tumor response by combining radiation and the vascular damaging drug 5,6-dimethylxanthenone-4-acetic acid. Radiat Res 2001;156:503–9.
- Horsman MR, Murata R. Combination of vascular targeting agents with thermal and radiation therapy. Int J Radiat Oncol Biol Phys 2002;54:1518–23.
- Murata R, Horsman MR. Tumour specific enhancement of thermoradiotherapy at mild temperatures by the vascular targeting agent 5,6-dimethylxanthenone-4-acetic acid. Int J Hyperthermia 2004;20:393–404.
- Horsman MR, Murata R. Vascular targeting effects of ZD6126 in a C3H mouse mammary carcinoma and the enhancement of radiation response. Int J Radiat Oncol Biol Phys 2003;57:1047–55.
- Bertelsen LB, Shen YY, Nielsen T, Stødkilde-Jørgensen H, Lloyd GK, Siemann DW, et al. Vascular effects of plinabulin (NPI-2358) and the influence on tumour response when given alone or combined with radiation. Int J Radiat Biol 2011;87:1126–34.
- Wachsberger PR, Burd R, Marero N, Daskalakis C, Ryan A, McCue P, et al. Effect of the tumor vascular-damaging agent, ZD6126, on the radioresponse of U87 glioblastoma. Clin Cancer Res 2005;11:835–42.
- Eikesdal HP, Bjerkvig R, Raleigh JA, Mella O, Dahl O. Tumor vasculature is targeted by the combination of combretastatin A-4 and hyperthermia. Radiother Oncol 2001; 61:313–20.
- El-Emir E, Boxer GM, Petrie IA, Boden RW, Dearling JL, Begent RH, et al. Tumour parameters affected by combretastatin A-4 phosphate therapy in a human colorectal xenograft model in nude mice. Eur J Cancer 2005;41: 799–806.
- Zhao D, Jiang L, Hahn EW, Mason RP. Tumor physiologic response to combretastatin A4 phosphate assessed by MRI. Int J Radiat Oncol Biol Phys 2005;62:872–80.
- Horsman MR, Sampson LE, Chaplin DJ, Overgaard J. The in vivo interaction between flavone acetic acid and hyperthermia. Int J Hyperthermia 1996;12:779–89.
- Murata R, Overgaard J, Horsman MR. Comparative effects of combretastatin A-4 disodium phosphate and 5,6-dimethylxanthenone-4-acetic acid on blood perfusion in a murine tumour and normal tissues. Int J Radiat Biol 2001; 77:195–204.
- Horsman MR, Ehrnrooth E, Ladekarl M, Overgaard JThe effect of combretastatin A-4 disodium phosphate in a C3H mouse mammary carcinoma and a variety of murine spontaneous tumors. Int J Radiat Oncol Biol Phys 1998;42:895–8.
- Horsman MR, Wouters BG, Joiner MC, Overgaard J. The oxygen effect and fractionated radiotherapy. In: van der Kogel AJ, Joiner M, editors. Basic clinical radiobiology, 4th ed. Hodder Arnold: London; 2009. p 207–16.
- Salmon HW, Siemann DW. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res 2006;12:4090–4.
- Folkes LK, Christlieb M, Madej E, Stratford MRL, Wardman P. Oxidative metabolism of combretastatin A-1 produces quinine intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem Res Toxicol 2007;20:1885–94.
- Rofstad EK, Galappathi K, Mathiesen B, Ruud EBM. Fluctuating and diffusion limited hypoxia in hypoxia-induced metastasis. Clin Cancer Res 2007;13:1971–8.
- Lunt SJ, Chaudary N, Hill RP. The tumor microenvironment and metastatic disease. Clin Exp Metastasis 2009;26:19–34.
- Ellingsen C, Hompland T, Mathiesen B, Rofstad EK. Microenvironment-associated lymph node metastasis of human cervical carcinoma xenografts. Acta Oncol 2012;51:465–72.
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15:220–31.
- Ebos JML, Lee CR, Cruz-Munoz W, Bjamason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009;15:232–9.