
Abstract
Head and neck squamous cell carcinoma (HNSCC) is the fifth most common malignancy worldwide, responsible for approximately half a million new cases every year. The treatment of this disease is challenging and characterised by high rates of therapy failure and toxicity, stressing the need for new innovative treatment strategies. Material and methods. In this study we performed a shRNAmir-based screen on HNSCC cells with the aim to identify tyrosine kinases that are mediating radiotherapy resistance. Results. The receptor tyrosine kinase FLT1 (VEGFR1) was identified as an important driver of cell survival and radioresistance. We show that FLT1 is phosphorylated in HNSCC cells, and document autocrine production of FLT1 ligands VEGFA and VEGFB, leading to receptor activation. Immunohistochemistry on HNSCC patient samples demonstrated FLT1 and VEGFA to be uniformly expressed. Interestingly, FLT1 was selectively overexpressed in tumour tissue as compared to non-cancerous epithelium. Remarkably, we found only membrane permeable FLT1 kinase inhibitors to be effective, which was in agreement with the intracellular localisation of FLT1. Discussion and conclusion. Taken together, we document expression of FLT1 in HNSCC and demonstrate this kinase to modulate radioresistance and cancer cell survival. Given the fact that FLT1 kinase is selectively upregulated in tumour tissue and that its kinase function seems expendable for normal life and development, this kinase holds great promise as a new potential therapeutic target.
The radiotherapy treatment for advanced stage head and neck squamous cell carcinoma (HNSCC) is challenging and characterised by high rates of therapy failure and toxicity. Consequently, the interest for smart molecular tumour targeting strategies is high as they may improve cure rates without adding more toxicity. Therapeutic targeting of the tyrosine kinase EGFR is an example of such an approach, recently proven to be successful in clinical practice [Citation1]. However, the therapeutic potential of other tyrosine kinases in HNSCC remains uncharacterised. In order to tackle this issue we performed an unbiased systematic micro-RNA-based genetic dropout screen to identify additional therapeutic targets in this large family of proteins. In this work, we identify and validate FLT1 (or VEGFR1) as a new target to modify HNSCC survival and radiosensitivity.
FLT1 forms together with FLK (VEGFR2) and FLT4 (VEGFR3) the VEGF-receptor tyrosine kinase family. These receptors consist of seven extracellular immunoglobulin-like domains, a trans-membrane region and a tyrosine kinase domain [Citation2]. FLT1 binds VEGFA, VEGFB and PlGF, whereas FLK binds VEGFA and VEGFC. The role of FLK (and VEGFA) as the primary driving force of angiogenesis is well established. The role of FLT1 in this context is more elusive. The current belief is that FLT1 primarily acts as a decoy receptor, reducing FLK activity by sequestering stimulatory VEGFA. Consistent with this model, FLT1 has been shown to bind VEGFA with much higher affinity than FLK, but shows much less tyrosine kinase activity [Citation2,Citation3]. In accordance, loss of FLT1 expression causes embryonic lethality as a result of vascular overgrowth, while FLT1 kinase dead variants with normal VEGFA binding capacity do show normal vascular development and good health [Citation4,Citation5].
Other evidence, however, indicates that the role of FLT1 in angiogenesis is more complex, as several reports have demonstrated that FLT1-mediated intracellular signalling regulates angiogenesis in several pathological conditions, one of which is cancer [Citation3]. In this context, numerous authors have shown that FLT1 inhibition suppresses tumour growth and metastasis [Citation3].
More recent reports also demonstrate FLT1 expression directly on tumour cells from breast, colon and skin origin, and show it to be an important oncogenic driver in these cells promoting survival, cell proliferation, invasiveness and/or motility, in an angiogenesis-independent manner [Citation6–9].
In this paper we examine the role of FLT1 in HNSCC.
Material and methods
Cell lines
Five HNSCC cell lines were used: SQD9, SCC61, SC263, Cal27 and SCC154. HPV status was confirmed by a PCR reaction using the GP5+/6 + primer set [Citation10] and p16 Western blot (Supplementary Figure 1 available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493). Cells were grown in DMEM containing 10% foetal bovine serum (FBS). SCC154 cells were cultured in MEM containing 10% FBS, 1% MEM non-essential amino acids and 1% L-glutamine. HEK293T cells were used to generate lentiviral vectors.
SiRNA transfection and inhibitors
Sunitinib and axitinib were from Selleck Chemicals [Citation11,Citation12]. Bevacizumab [Citation13] was from Roche. The FLT1 blocking peptide (GNQWFI) [Citation14] was synthesised by Eurogentech. Transfection was performed using Lipofectamine 2000 (Invitrogen). Non-silencing control and FLT1 stealth select siRNAs were obtained from Invitrogen (12935-400, HSS103744 and HSS103745, designated as control siRNA, FLT1 siRNA1 and FLT1 siRNA2, respectively).
Lentivirus-based RNAi screen
A high-throughput shRNAmir dropout screen was performed on SCC61 and SQD9 to identify TK's important for HNSCC cell radioresistance. In short, we used a pooled shRNAmir library containing lentiviral vectors targeting the whole tyrosine kinome. Cells were virally transduced in a way that each cell contained a different shRNAmir, knocking down the expression of a specific tyrosine kinase. The assumption was that when the silenced tyrosine kinase was important for radioresistance or survival in general, cells carrying a shRNA silencing such a kinase would disappear from the cell population when the cell population was irradiated or just cultured. This would result in a dropout of the shRNAmir sequence from the cell cultures. By means of a GS-FLX 454 sequencer (Roche) the relative abundance of each shRNAmir in the cell population was measured at the start of the experiment, after radiotherapy, and after standard culturing. A radiosensitising shRNAmir was defined as a shRNAmir showing at least a reduction of 66% in abundance after radiotherapy as compared to the control condition measured before radiotherapy. Additionally it was required that the shRNAmir loss in the irradiated condition was more pronounced than that of a non-irradiated condition cultured in parallel. ShRNAmirs that already were completely lost in the non-irradiated experimental condition were carried through as kinases affecting survival. Primarily this was done because a potential additional radiosensitising effect could not be evaluated as the shRNAmir already results in complete cell kill even without radiotherapy. Additionally, these shRNAmirs most strongly affecting HNSCC cell survival could reveal interesting therapeutic targets for HNSCC cancer therapy as well. Hits from a first screening round were tested again in the same setup using smaller shRNAmir pools. Consistent hits were then subjected to a second screen setup (follow-up screen). Here shRNAmir-transfected cells were mixed with non-transfected cells. The percentage of shRNAmir expressing cells (also expressing GFP) was measured by flow cytometry in the cultures as a way to determine their capacity to proliferate and survive in the cultures compared to non-transduced cells (without knock-down, GFP negative) with and without radiotherapy. A shRNAmir targeting a particular kinase was identified as important for survival if the rate at which shRNAmir carrying cells disappeared from the mixes was significantly higher than that of the mix containing a non-silencing control shRNAmir in non-irradiated mixes. A shRNAmir was identified as important for radiosensitivity if the same was true for irradiated mixes, but additionally it was required that the overall cell loss of shRNA carrying cells was more pronounced in the irradiated mix as compared to the non-irradiated mix (see Supplementary Appendix, available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493).
Western blotting and (confocal) immunocytochemistry
Western blotting and immunocytochemistry were performed using standard protocols (see Supplementary Appendix available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493).
Real-time qPCR and sequencing
Messenger RNA was extracted from cells using the RNeasy mini Kit (Qiagen) with additional on column DNA digestion. For siRNA knockdown analysis, mRNA was harvested 48 hours after transfection. Experiments were carried out on a Lightcycler 480 II (Roche). Primer sequences and cycling conditions are listed in Supplementary Table I (available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493). FLT1 sequencing was performed at the VIB service facility in Antwerp (Belgium).
Sulphorodamine B (SRB) Survival assays
Cells were seeded one day before treatment. In inhibitor experiments cells were treated for 72 hours at the indicated concentration. In siRNA experiments, cells were trypsinised and replated in 96-well plates 48 hours after transfection and then grown for another 72 hours before analysis. Non-silencing siRNA was used for the control conditions. Readout was performed using a SRB assay, as previously described [Citation15]. In short, cells were fixed after treatment with trichloric acid and stained with sulphorodamine B (SRB). The incorporated SRB was liberated from cells in a TRIS-base solution, and optical densities at 570 nm were determined. Cell survival was determined as the relative absorption of SRB in treated wells in comparison to controls.
Clonogenic assays
Experiments were carried out as previously described [Citation15]. In case of experiments involving siRNA treated cells, radiotherapy was performed 72 hours after transfection and non-silencing siRNA was used for the control conditions. For experiments involving inhibitors, cells were treated (24 hours) and irradiated in culture flasks containing the inhibitor, and subsequently trypsinised, counted and seeded 4–6 hours after radiotherapy. The plating efficiency (PE) was defined as the ratio of cells forming a colony of more than 50 cells over the amount of seeded cells after an incubation period of 2–3 weeks receiving no radiation. The survival fraction was calculated after normalising the amount of colonies formed after having received a certain dose of radiotherapy to the plating efficiency. The dose enhancement factor was calculated as the ratio of irradiation dose needed in the control condition/irradiation dose needed in the experimental condition to obtain a survival fraction of 0.5.
Patient samples, immunohistochemistry and survival analysis
Formalin-fixed paraffin embedded tumour samples were obtained from patients diagnosed with squamous cell carcinoma of the head and neck between 2000 and 2010. Immunohistochemistry was performed according to standard procedures using the anti-FLT1 antibody HPA011740 (Sigma) (see Supplementary Appendix available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493).
FLT1 samples were scored by expression pattern: absent expression, expression within the basal layers or expression within the full epithelial thickness, and alternatively by staining intensity as absent, mild or strong staining intensity. Initially, FLT1 staining was performed on 12 samples of surgically resected squamous cell carcinoma specimen of the larynx. In a second phase FLT1 staining was performed on an additional 139 biopsies of patients’ diagnoses with squamous cell carcinoma of the oropharynx. Of these 139 samples stained in total, 93 patients received curative doses of (chemo)radiotherapy (≥ 60 Gy) on macroscopic tumour, and were selected for survival analysis. The mean follow-up for this group was 618 days. Survival analysis was performed according to the Kaplan-Meier method using STATISTICA11 software.
Results
An shRNAmir screen identifies FLT1, among other tyrosine kinases, as a gene affecting cell survival and radiotherapy resistance in HNSCC
In this work a genetic dropout screen was performed on two HNSCC cell lines (SQD9 and SCC61) to identify tyrosine kinases with potential impact on radiosensitivity and cell survival. The results of these screening experiments are provided as Supplementary Table II (available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493). Our screen setup allowed us to identify FLT1 as a tyrosine kinase affecting radiosensitivity as well as cell survival in both HNSCC cell lines. Further AnneX-eLectro receptor tyrosine kinase (AXL), Bone Marrow tyrosine kinase coded in chromosome X (BMX), and Tyrosine Non-receptor Kinase 1 (TNK1) were identified in SCC61 as potential tyrosine kinase affecting cell survival, whereas Epidermal Growth Factor Receptor (EGFR) was suggested to affect radiosensitivity as well as cell survival. In SQD9 TXK tyrosine kinase (TXK) was identified as another tyrosine kinase potentially affecting cell survival, but not radiosensitivity.
FLT1 is expressed in HNSCC cell lines and its knock-down decreases cell survival and sensitises for the effects of radiation
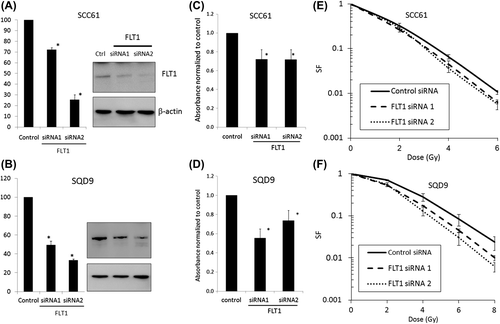
Since FLT1 affected both cell lines, we decided to focus on this kinase. In a first step of validating the role of FLT1, we used a real time quantitative PCR (qPCR) and Western blot to confirm FLT1 expression (). We next determined if siRNA mediated FLT1 knock-down could mimic the effects observed with shRNAmirs. Using two different siRNAs we confirmed decreased survival upon FLT1 knockdown in both cell lines to 52% and 71% in SQD9 and 72% and 72% in SCC61 for FLT1 siRNA1 and 2, respectively (). The radiosensitising potential of FLT1 was also investigated with a clonogenic assay using different siRNAs targeting FLT1 in both cell lines. FLT1 silencing affected PE. The averaged PE's for non-irradiated siRNA treated cells were as follows: 0.12, 0.09 and 0.06 in SCC61 and 0.16, 0.10 and 0.13 in SQD9 for control siRNA, FLT1 siRNA1 and FLT1 siRNA2 respectively. To rule out the possibility that the decreased cell survival seen in the colony assays after FLT1 silencing could be attributable only to direct cytotoxicity of the FLT1 siRNA but not to a synergistic interaction with radiation, we generated the survival curves after normalising to the PE for each siRNA. In this way differences in non-radiotherapy associated toxicity between the different siRNAs were equalised and therefore a difference seen in the survival curves should indicate radiosensitisation. FLT1 silencing did affect radiosensitivity in both HNSCC cell lines () with a dose enhancement factor of the best working FLT1 siRNA being 1.62 and 1.20 in SQD9 and SCC61, respectively.
Figure 1. FLT1 is important for cell survival and radioresistance. (A, B) SiRNA mediated knockdown of FLT1 was confirmed by qPCR (left) and Western blot (right). (C, D) SRB survival assays performed with siRNA transfected SCC61 (C) and SQD9 (D) cells. (E, F) FLT1 silencing causes radiosensitisation in SCC61 and SQD9 as demonstrated by clonogenic assay. Data shown are the averaged means of at least three independent experiments ± SEM and compared to non-silencing control siRNA treated cells. *Significantly different from control siRNA, p < 0.05, students t-test. SF, survival fraction.
FLT1 is overexpressed in primary head and neck carcinoma
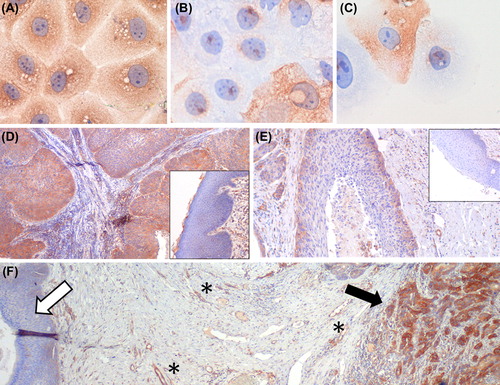
In order to test expression of FLT1 on patients’ samples we validated the specificity of our FLT1 antibody on formalin-fixed HNSCC cells (). Initially, we investigated 12 formalin-fixed paraffin-embedded HNSCC tumour samples by immunohistochemistry (IHC) for expression of FLT1 and ligands. All samples showed FLT1 expression. To confirm these findings, FLT1 stainings were performed on 139 additional samples, finding FLT1 expression in 124 (89%) of these samples. In the whole group of 151 samples 85 sections also contained normal epithelium. To compare FLT1 expression between tumour and normal epithelium, a semi-quantitative scoring system was used: absence of expression (score I), FLT1 expression in the basal layers (score II) and FLT1 expression in the full epithelial thickness (score III) (). FLT1 expression was significantly upregulated in tumour compared to normal epithelia (p < 0.05, Wilcoxon-matched pairs test) as in tumour 15 (10%), 40 (26%) and 96 (64%) of samples were classified as score I, II and III, respectively, while in normal tissue 33 (39%), 49 (58%) and three (3%) samples received score I, II and III (). FLT1 staining pattern in patient samples was not predictive for locoregional control or overall survival (Supplementary Figure 2, available online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493).
Figure 2. FLT1 is overexpressed in human HNSCC. (A–C) The specificity of the FLT1 staining (shown in brown) was examined by immunocytology of formalin-fixed FLT1 silenced SQD9 cells: (A) non-silencing control treated cells (400×); (B, C) FLT1-silenced cells. FLT1 positive cells in the FLT1-silenced conditions are expected since siRNA delivery is not equally effective in all cells. These positive cells prove that the FLT1 staining was performed correctly. (400×) (D–F) Immunohistochemistry on resected HNSCC. (D, E) show images of HNSCC samples with FLT1 expression (brown) in the full epithelial thickness, or expression limited to the basal layers respectively (40×). Corresponding normal epithelia are shown as insets (100×). (F) Immunohistochemistry of a HNSCC resection specimen showing FLT1 expression in the tumour (black arrow) but none in the normal epithelium (white arrow). Blood vessels used as an internal positive control for FLT1 staining are marked by ‘*’ (40×).
FLT1 ligands are produced in HNSCC cell lines and primary tumours and correlate with radioresistance
FLT1 immunoprecipitation experiments documented phosphorylation of the receptor in both HNSCC cell lines used in the screen, suggesting activation of this receptor in these cells (). After sequencing exons 2-30 of the FLT1 receptor in SQD9 and SCC61 we found no evidence of activating mutations. We then determined expression levels of FLT1 ligands which could result in receptor activation [Citation3]. We documented a strong expression of VEGFA and VEGFB in SQD9 and SCC61. PIGF expression was negligible ().
Figure 3. Self-contained activation through autocrine production of FLT1 ligands is documented in HNSCC, and results in radiotherapy resistance. (A) FLT1 immunoprecipitation (FLT1 IP) showing tyrosine phosphorylation (pTyr) of FLT1 in HNSCC cell lines SQD9 and SCC61. The lower panel shows detection of FLT1 in immunoprecipitates using anti-FLT1 antibody. (B) Autocrine production of FLT1 ligands VEGFA, VEGFB and PlGF is examined by qPCR in SCC61 and SQD9. Data are shown as the averaged means of three separate experiments ± SEM after normalisation to 100 000 copies of household gene HPRT. (C) Radiotherapy resistance measured by clonogenic assay as well as basal autocrine VEGFA production measured by qPCR, in a panel of five different HNSCC cell lines. For qPCR data are normalised to 10^5 copies of household gene HPRT. Data are presented as the averaged normalized means ± SEM calculated from three separate experiments. SF, survival fraction. A highly significant correlation was noted between cell survival after radiotherapy (4 Gy) and autocrine VEGFA production (γ = 1, p < 0.01).
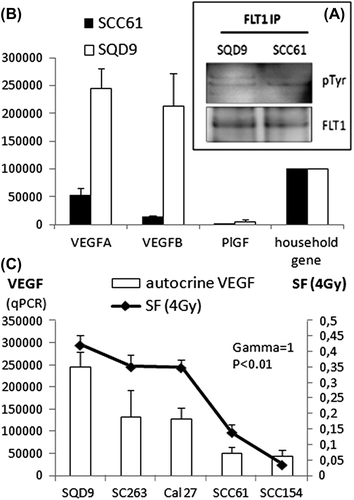
Interestingly, VEGFA, having a FLT1 binding affinity of 10 times higher than the other FLT1- ligands [Citation2], was shown to be the highest expressed. In agreement with this, IHC revealed VEGFA expression in all investigated HNSCC tumour samples (11/11, one sample of 12 not available). VEGFB, however, was detected in only two of 12 investigated tumours. These data suggest that FLT1 activity in HNSCC may be regulated through VEGFA.
We assumed that the level of VEGFA expression per HNSCC cell line could better predict radiotherapy resistance than FLT1 itself in HNSCC cell lines. To test this hypothesis, we examined the level of VEGFA expression in a panel of five HNSCC cell lines. The level of autocrine VEGFA production showed a very tight positive correlation to radioresistance (, γ = 1, p < 0.01). In accordance with these observations, other groups already have shown that higher VEGFA expression in HNSCC patient samples predicts for worse local control after radiotherapy [Citation16,Citation17].
FLT1 is not located on the plasma membrane but on the inside of HNSCC cells
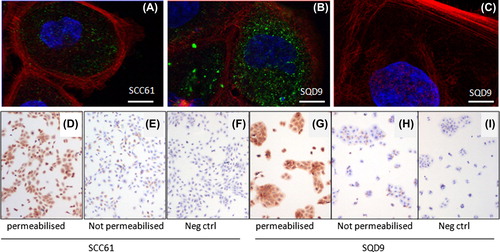
For FLT1, a very unique mechanism of intracrine activation was described earlier in breast cancer cells by Lee et al. [Citation7]. Here the authors showed that the FLT1 receptor was not located at the plasma membrane, where one would normally expect a tyrosine kinase to be, but on the inside of the cell on the nuclear envelope. There FLT1 was shown to mediate cell survival of breast cancer cells, a process which was shown only to be affected by autocrine intracellular (intracrine) VEGFA and not exogenous or extracellular VEGFA. To see if this was also true for HNSCC cells the subcellular localisation of FLT1 was investigated by confocal microscopy using a FLT1 antibody with confirmed specificity on formalin-fixed HNSCC cellular material (). We could indeed confirm intracellular location for FLT1 in HNSCC (). In addition, we used this FLT1-specific antibody, which is raised against the extracellular part of the receptor in another confirmatory assay. If the FLT1 receptor would be localised on the plasma membrane, one would expect FLT1 immunoreactivity to be present even when immunocytochemistry is performed without permeabilising the plasma membrane. We showed this not to be the case since omission of the permeabilising detergent Tween resulted in nearly complete absence of immunoreactivity (). This intracrine signal transduction explains previous observations in HNSCC cells where it was shown that siRNA mediated silencing of VEGF did reduce cell viability, while externally administered VEGF did not have any effect on cell survival [Citation18].
Figure 4. FLT1 has an intracellular localisation. (A–C) Merged colour confocal microscopy images taken from SCC61 (A) and SQD9 (B). FLT1, shown in green is located intracellularly. Fiber-actin (Phalliodin-Alexa555) and nucleus (To-Pro3) are shown in red and blue, respectively. (C) FLT1 negative control staining where only a secondary Alexa 488 coupled Fab fragment was used. Scale bars 10 μm. (D–I) Classical immunocytochemistry using a FLT1 antibody against the extracellular part of the FLT1 receptor (40×). SCC61 (D) and SQD9 (G) show strong immunoreactivity when cells are permeabilised with Tween. However, when Tween permeabilisation is avoided, immunoreactivity in SCC61 (E) and SQD9 (H) becomes comparable to their respective negative controls (F, I).
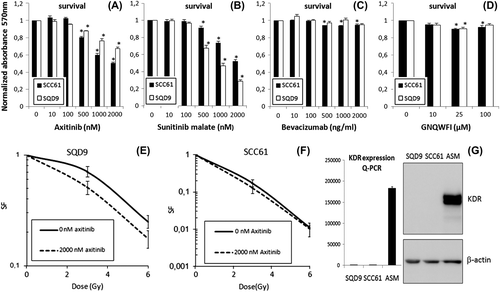
Therapeutic targeting of the FLT1 receptor: a need for membrane permeable inhibitors
If FLT1 driven HNSCC cell survival and radioresistance is an intracellular process one would expect only membrane permeable inhibitors to be useful for therapeutic targeting. Two strategies using membrane impermeable inhibitors were used: a FLT1 blocking peptide (GNQWFI) preventing ligand binding to the FLT1 receptor [Citation14], and the VEGFA blocking monoclonal antibody bevacizumab [Citation13]. None of both strategies was shown to affect HNSCC cell survival (). However, we tested two membrane permeable tyrosine kinase inhibitors with documented FLT1 activity: axitinib and sunitinib [Citation11,Citation12]. Both inhibitors were effective in reducing cell survival of SCC61 and SQD9 cells in a dose dependent manner (). Seventy-two hour treatment with 2uM axitinib resulted in reduction of cell survival to 67% and 50% whereas sunitinib at 2μM concentration reduced cell survival to 39% and 51% in SQD9 and SCC61, respectively (all p < 0.05). Importantly, Western blot and qPCR analysis showed absence of KDR expression in these cell lines, excluding the possibility that these inhibitors induced their cytotoxic effects in a FLT1 independent manner through KDR (). Finally, we tested the effectiveness of axitinib as a radiosensitiser in a clonogenic assay on SCC61 and SQD9 cells. Axitinib was indeed effective in inducing radiosensitisation (). Comparable to what we observed after siRNA mediated FLT1 silencing, the radiosensitising effect of 2 μM axitinib was clearly more pronounced in the more radioresistant HNSCC cell line SQD9 () that produces higher levels of FLT1 activating VEGFA (). The dose enhancement factors we found for axitinib (1.73 for SQD9 and 1.24 for SCC61) were comparable to these found for FLT1 siRNA (1.62 for SQD9 and 1.20 for SCC61).
Figure 5. The effect of different FLT1 targeting strategies on cell survival and radioresistance. (A–D). Cell survival measured with a SRB assay using indicated concentrations of axitinib, sunitinib, bevacizumab and GNQWFI, respectively. Data are presented as averaged normalised means ± SEM calculated from three separate experiments. *Significantly different for the respective controls, p < 0.05, students t-test. (E, F) Clonogenic assays investigating the radiosensitising potential of axitinib in SQD9 and SCC61. (G) Q-PCR (left) and Western blot data (right) demonstrating KDR is not expressed in SQD9 and SCC61, excluding that axitinib and sunitinib induce their cytotoxic or radiosensitising effects through KDR. Angiosarcoma (ASM) cDNA and protein lysates were used as positive controls for KDR expression. For qPCR, expression is shown as the amount of KDR per 10^5 copies of HPRT. β-actin is used as loading control for Western blot. Data shown are the averaged means of at least three independent experiments ± SEM. SF, survival fraction.
Discussion
In this study, we identify FLT1 as an important driver of HNSCC survival and resistance to radiotherapy using a shRNAmir-based dropout screening setup. FLT1 is a tyrosine kinase known as a suppressor of angiogenesis [Citation2,Citation3]. Interestingly, it was shown that FLT1 effectuates this function not through its kinase domain but rather through its extracellular region by trapping VEGFA. In this way FLT1 prevents stimulation of angiogenesis through the VEGFA-VEGFR-2 receptor complex. In HNSCC, however, we believe FLT1 fulfils its function as oncogenic driver through its tyrosine kinase activity, not only by showing that the FLT1 receptor kinase function is activated, and but also by demonstrating that FLT1 tyrosine kinase inhibitors are able to downregulate HNSCC survival and radioresistance.
We believe FLT1 holds great potential as a therapeutic targeting strategy to improve radiotherapy outcome for HNSCC, as we document that FLT1 is selectively overexpressed in HNSCC tumour cells as compared to normal squamous epithelia. Moreover, very specific FLT1 kinase inhibition is unlikely to induce significant toxicity since it was shown that homozygous FLT1-kinase knockout is essentially compatible with normal life and development [Citation4]. In this context, the direct cytotoxic effect of FLT1 kinase targeting over the effect on radiosensitivity would be a valuable advantage.
In breast cancer cells oncogenic FLT1 signalling has been shown to be an intracrine process: FLT1 is located on the inside of the cells where it can only be activated by autocrine produced intracellular VEGFA. The observations we made on HNSCC cells are comparable as we document FLT1 to be located only intracellularly in HNSCC. Further, we document that only plasma membrane permeable FLT1 inhibitors are able to affect HNSCC cell survival and radioresistance. In practice this intracrine signalling severely limits therapeutic options since its means that currently only tyrosine kinase inhibitors can be used. To date, however, no specific FLT1 kinase inhibitor exists. In fact, the currently available candidates are mostly inhibitors designed as anti-angiogenic therapy against KDR with additional anti-FLT1 activity. This is also the case for the inhibitors axitinib and sunitinib used in this study. Although some data are available for FLT1 inhibitor effectiveness from in vitro assays, it is currently not known if tumoural FLT1 can be inhibited using these inhibitors in vivo. Therefore we believe our data should be taken as a proof of concept that encourages the development of better and more specific FLT1 inhibitors.
Finally, we also did interesting observations regarding possible predictive biomarkers for FLT1-targeted therapy. Within a panel of different cell lines derived from primary HNSCC, we document a strong correlation between autocrine VEGFA expression and radioresistance. In accordance with these observations, it has been known that higher VEGFA expression predicts for worse local control in primary HNSCC after radiotherapy [Citation16,Citation17], making VEGFA expression a very promising biomarker.
http://informahealthcare.com/doi/abs/10.3109/0284186X.2013.835493
Download PDF (1.1 MB)Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This work was partly supported by the clinical research fund (KOF) of the UH Leuven (S.N.), the ‘Vlaamse Liga tegen Kanker’ (VLK) (S.N.), by the ‘Stichting tegen Kanker’ (J.C.), the FWO-Vlaanderen (J.C.) and the European Research Council (ERC-Starting grant to J.C.). E.V.L. is a research assistant (aspirant) of the Research Foundation Flanders (FWO). M.P. was supported by the IWT.
References
- Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol 2010; 11:21–8.
- Shibuya M. Structure and dual function of vascular endothelial growth factor receptor-1 (Flt-1). Int J Biochem Cell Biol 2001;33:409–20.
- Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy?. Nat Rev Cancer 2008;8:942–56.
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 1998;95:9349–54.
- Hiratsuka S, Maru Y, Okada A, Seiki M, Noda T, Shibuya M. Involvement of Flt-1 tyrosine kinase (vascular endothelial growth factor receptor-1) in pathological angiogenesis. Cancer Res 2001;61:1207–13.
- Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 2005;24:2647–53.
- Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, et al. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med 2007; 4:e186.
- Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010;140:268–79.
- Naik S, Dothager RS, Marasa J, Lewis CL, Piwnica-Worms D. Vascular endothelial growth factor receptor-1 is synthetic lethal to aberrant {beta}-catenin activation in colon cancer. Clin Cancer Res 2009;15:7529–37.
- de Roda Husman AM, Walboomers JM, van den Brule AJ, Meijer CJ, Snijders PJ. The use of general primers GP5 and GP6 elongated at their 3’ ends with adjacent highly conserved sequences improves human papillomavirus detection by PCR. J Gen Virol 1995;76:1057–62.
- Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res 2008;14:7272–83.
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008;26:127–32.
- Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 2004;7:335–45.
- Bae DG, Kim TD, Li G, Yoon WH, Chae CB. Anti-flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin Cancer Res 2005;11:2651–61.
- De Schutter H, Kimpe M, Isebaert S, Nuyts S. A systematic assessment of radiation dose enhancement by 5-Aza- 2’-deoxycytidine and histone deacetylase inhibitors in head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys 2009;73:904–12.
- Parikh RR, Yang Q, Haffty BG. Prognostic significance of vascular endothelial growth factor protein levels in T1-2 N0 laryngeal cancer treated with primary radiation therapy. Cancer 2007;109:566–73.
- Smith BD, Smith GL, Carter D, Sasaki CT, Haffty BG. Prognostic significance of vascular endothelial growth factor protein levels in oral and oropharyngeal squamous cell carcinoma. J Clin Oncol 2000;18:2046–52.
- Tong M, Lloyd B, Pei P, Mallery SR. Human head and neck squamous cell carcinoma cells are both targets and effectors for the angiogenic cytokine, VEGF. J Cell Biochem 2008; 105:1202–10.