
Abstract
Background. The Warburg phenotype identified decades ago describes tumor cells with increased glycolysis and decreased mitochondrial respiration even in the presence of oxygen. This particular metabolism also termed ‘aerobic glycolysis’ reflects an adaptation of tumor cells to proliferation in a heterogeneous tumor microenvironment. Although metabolic alterations in cancer cells are common features, their impact on the response to radiotherapy is not yet fully elucidated. This study investigated the impact of cellular oxygen consumption inhibition on the tumor response to radiotherapy.
Material and methods. Warburg-phenotype tumor cells with impaired mitochondrial respiration (MD) were produced and compared in respect to their metabolism to the genetically matched parental cells (WT). After characterization of their metabolism we compared the response of MD cells to irradiation in vivo and in vitro to the genetically matched parental cells (WT).
Results. We first confirmed that MD cells were exclusively glycolytic while WT cells exhibited mitochondrial respiration. We then used these cells for assessing the response of WT and MD tumors to a single dose of radiation and showed that the in vivo tumor growth delay of the MD group was increased, indicating an increased radiosensitivity compared to WT while the in vitro ability of both cell lines to repair radiation-induced DNA damage was similar.
Conclusion. Taken together, these results indicate that in addition to intrinsic radiosensitivity parameters the tumor response to radiation will also depend on their metabolic rate of oxygen consumption.
Aerobic glycolysis in cancer has long been described as relying mainly on impaired mitochondrial metabolism [Citation1]. However, recent findings have revealed that this metabolic phenotype is not only a consequence of mitochondrial dysfunction but also the result of fine-tuned oncogene-driven metabolic reprogramming required to support anabolic growth and microenvironmental adaptations [Citation2].
Hypoxia is a common feature of the tumor microenvironment. So far, it is believed that hypoxia selects for cells performing glycolysis and that the acquisition of a constitutive glycolytic phenotype, also termed Warburg phenotype, provides an advantage for tumor development [Citation3]. Hypoxia is defined as limited oxygen availability due to an imbalance between oxygen consumption by tumor cells and supply from blood vessels. Clinically, hypoxia is predictive of metastasis and poor outcome, and is associated with resistance to treatments, especially radiation therapy [Citation4]. A mathematical model has suggested that reducing oxygen consumption rate (OCR) may be more effective than elevating blood flow or oxygen content as a method to reduce tumor hypoxia [Citation5]. Data on the modulation of oxygen consumption and its impact on radiation response have been published in the past [Citation6]. However, most of these data were created from inhibition of mitochondrial respiration through non-selective pharmacological treatment. More recently, Sonveaux and collaborators [Citation7] have shown that inhibition of lactate uptake in tumors resulted in delayed tumor growth and rendered the remaining cells sensitive to irradiation due to inhibition of oxygen consumption. Indeed, the authors suggest that lactate previously considered as a waste product, can be used as a fuel for the oxidative metabolism of oxygenated tumor cells [Citation7]. Therefore, we investigated the impact of mitochondrial oxygen consumption inhibition on the tumor response to radiotherapy.
For this purpose, we produced an isogenic model of mitochondrial dysfunction (MD) and compared their response to ionizing radiation to that of their genetically matched parental cells (WT). We showed that while the ability of both cells to repair radiation-induced DNA damage in vitro was similar, the in vivo tumor growth delay of the MD group was increased, therefore indicating an increased radiosensitivity of MD tumors compared to WT.
Material and methods
Cells and culture conditions
Hela (a kind gift of Prof. C Michiels, UNamur) and SiHa (WT) tumor cells (purchased from American Type Culture Collection, ATCC, Manassas, USA) were cultured in DMEM containing 4.5 g/L glucose (Gibco, Lifetechnologies, Paisley, UK) with 10% FBS, 2 mM Glutamax and 1% antibiotic mix (Penicillin-streptomycin, Gibco). SiHa mitochondrial dysfunction (MD) cells were obtained by chronic exposure (8 weeks) to low concentrations of ethidium bromide (50 ng/μl) and subsequent clonal selection as detailed in [Citation8] (Appendix: Material and methods and Supplementary Figure 1 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006).
UPCI-SCC154 and UPCI-SCC90 tumor cells were purchased from DSMZ (German collection of microorganisms and cell culture, Germany) and were cultured in MEM (Gibco) supplemented with non-essential amino acids and 10% FBS. For some experiments, cells were treated with 2.5 μM of Antimycin A (Sigma-Aldrich, St Louis, MO, USA). Hypoxia was achieved in an InViVO2 system (Ruskinn Technology, Bridgend, UK) maintained at 5% CO2, 1% O2 and 37°C. All cell lines were regularly verified to be mycoplasma-free and were authenticated by DNA fingerprinting (Health Protection Agency, London, UK).
Extracellular metabolite determination
To determine extracellular metabolite levels, supernatants were collected and filtered on 10-kDa cut-off centrifugation columns. L-Lactate and glucose concentrations were quantified using specific enzymatic assays on a CMA600 analyzer (CMA Microdialysis, Kista, Sweden). Cells were lysed in 50 μl of ice-cold RIPA buffer (50 mM Tris HCL pH 7.4, 150 mM NaCl, 1% Triton-X 100, 0.05% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM Na3VO4 and protease inhibitors) and protein levels were determined by bicinchoninic acid (BCA) assay (Thermo-Fisher Scientific Inc., Rockford, IL, USA).
Measurements of oxygen consumption and extracellular acidification rates
The determination of cell oxygen consumption and extracellular acidification rates was carried out using the fluorescent O2-sensitive and pH-sensitive probes Mito-Xpress and pH-Xtra (Luxcel Bioscience, Cork, Ireland), respectively, as described previously [Citation9]. Briefly, cells in 96-well plates were cultured overnight in a standard CO2 incubator and then maintained in a CO2-free incubator at 37°C for three hours before measurements. Measurements were performed after addition of the probes in 150 μl of measurement buffer and 100 μl of mineral oil as recommended by manufacturer. Fluorescence decay was measured kinetically on a multilabel plate reader (Victor2) at 37°C for a minimum of 120 minutes in time-resolved fluorescence (TR-F) mode using a standard filter set as follows, 340 nm excitation and 642 nm emission filters, delay times 30 and 70 μs, and measurement window 30 μs: integrator capacitors, 3; integrator reference level, 6; flash area, high; flash energy level, 220.
Cell irradiation and clonogenic assay
Cells in exponential growth phase were irradiated at room temperature using a 137Cs γ-irradiator at a dose-rate of 0.80 Gy/min for a total absorbed dose of 2, 4, 6, 8 or 10 Gy. Immediately after irradiation cells were detached using trypsin-EDTA, plated at multiple densities (from 100 to 1000) into six-well plates and allowed to attach. After 10–12 days of incubation, the medium was discarded and cells were fixed and stained with 1% formalin/crystal violet solution for 30–60 minutes, washed with tap water, air-dried, and counted. Colonies of at least 50 cells were counted. The survival fraction was expressed as (number of colonies after treatment divided by the number of cells seeded) × plating efficiency. The oxygen enhancement ratio (OER) was determined by calculating dose in hypoxic cells (closed wells)/dose in aerated (open wells) cells for killing 90% of the cells [Citation10].
Flow cytometry for γH2AX, cell cycle analyses and apoptosis
Samples at the indicated recovery times after irradiation (5 Gy) were fixed in 70% ethanol and stored at −20°C. The day of analysis, cells were rehydrated in PBS for 10 minutes, centrifuged and suspended in 200 μl of PBS + 0.1% Triton-X 100 with Alexa Fluor 488 mouse anti-phospho-H2AX (pS139) antibody (1:500 dilution, BD Biosciences, San Jose, CA, USA). Cells were incubated for one hour at room temperature, then rinsed and suspended in PBS containing 2.5 μg/ml of propidium iodide and 0.5 mg/ml of RNase A (Sigma-Aldrich). Acquisition of fluorescence intensity was performed with FACScalibur (BD Biosciences) and samples were gated on DNA content and time of flight to eliminate debris and cell doublets before the analysis of γH2AX antibody staining intensity. Apoptosis was determined as the percentage of cells in sub-G1 24 hours after irradiation (5 Gy). Analysis of the data was performed with FlowJo software (TreeStar, OR, USA) by gating the populations of γH2AX-labeled cells according to control histograms to determine the percentage of positive cells versus time.
Mouse models and in vivo experiments
For in vivo experiments, 20 (10/group) athymic female nude mice (Harlan Laboratories Inc., Indianapolis, IN, USA) were injected subcutaneously in the thigh with 100 μl of Matrigel (BD Biosciences) containing 106 SiHa WT or MD cells. Tumor size was monitored with a caliper according to three different axes and expressed as mean diameter. Tumors were allowed to grow up to 7 mm in mean diameter before experimentation. On the day of irradiation (Day 0), the tumor bearing leg was locally irradiated (137Cs γ-irradiator) with a single dose of 16 Gy. Mice were anesthetized (isoflurane in 21% oxygen), and the tumor was centered in a 3-cm diameter circular irradiation field. Treated tumors were measured every day until they reached a diameter of 16 mm, time at which the mice were sacrificed.
MicroPET imaging and image analysis
For PET studies, mice were injected intravenously with 12.5 ± 5.1 MBq [18F]-FDG and 16.3 ± 4.2 MBq [18F]-FAZA that was produced as previously described [Citation11]. For [18F]-FDG injection, animals were fasted overnight and kept under anesthesia on a heated (35°C) bed (Minerve, France) before and during acquisition. PET scans were acquired one hour for [18F]-FDG and three hours for [18F]-FAZA after tracer injection on a dedicated small-animal PET scanner (MOSAIC, Philips, Eindhoven, The Netherlands [Citation12]) with a spatial resolution of 2.5 mm (FWHM). Mice anesthetized with 1.5% isoflurane underwent first a 10 minute emission scan followed by a 10 minute transmission scan using a 370-MBq 137Cs source. After the correction with attenuation factors obtained from the transmission scan, images were reconstructed using a fully three-dimensional (3D) iterative algorithm (3D-RAMLA) in a 128 × 128 × 120 matrix, with a voxel size of 1 mm3. The tumor volume was segmented using a fix threshold defined as background + 1SD. The background volume was determined manually as a box of defined size placed in the muscle of the contra-lateral leg. The result of tracer distribution in tumors was expressed as SUV Max, calculated as the FDG uptake normalized to injected dose/unit weight of mice or as tumor to background (T/B) ratio, calculated as the mean FAZA activity in the tumor volume divided by the mean activity in the background volume.
Pimonidazole staining and fluorescence microscopy
Tissue hypoxia was assessed ex vivo by immunohistochemistry using Hypoxyprobe™-1 Omni Kit (Hypoxyprobe, Burlington, MA, USA) as described in Appendix: Material and methods. The hypoxic fraction (%) was defined as the tumor area positive for pimonidazole relative to the total tumor area (nucleus staining with DAPI). Both areas were extracted from the images using interactive threshold segmentation. Areas of necrosis (determined by histological observation of nuclei stained by DAPI) and of tissue artifacts (wholes), if any, were excluded from the analysis of hypoxic fraction. For the zonal analysis of distance of hypoxic areas from the nearest vessel, vessels were identified using CD31 staining (dilution 1/500, rat anti-mouse CD31, BD Bioscience). Quantification of microregional pimonidazole intensities as a function of blood vessels was achieved using methods previously described (Appendix: Material and methods).
Ethical statement
All in vivo experiments were performed with approval of the Université Catholique de Louvain authorities (Comité d’Ethique Facultaire pour l’Experimentation Animale) according to national animal care regulations.
Data analysis
Results are expressed as mean± SEM. Student's t-test, one-way ANOVA (Bonferroni post-hoc test) and two-way ANOVA were used where indicated (Prism Graph Pad, La Jolla, CA, USA). For OCRs, the slopes of individual linear regression lines were compared. p-Value < 0.05 was considered to be statistically significant for all experiments.
Results
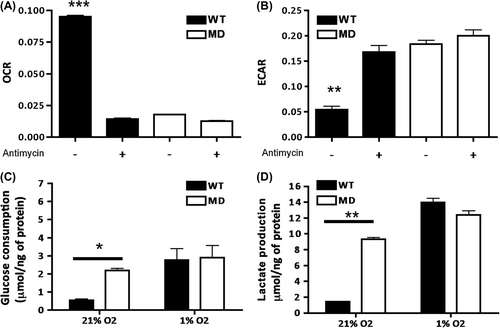
After selection of MD monoclonal population, we confirmed the constitutively glycolytic phenotype of MD cells by analyzing OCR in parallel with extracellular acidification rate (ECAR) (). Compared to WT, MD cells showed a significantly decreased OCR () and increased ECAR (). A comparable glycolytic profile was obtained upon inhibition of mitochondrial respiration with antimycin A in WT cells, as evidenced by a drop in OCR whereas ECAR increased. The metabolic profiles of the cells were further confirmed by measuring extracellular metabolites under normoxia (21% O2): compared to WT, MD cells had a glycolytic phenotype with significantly higher glucose consumption and lactate production rates (). Under hypoxia (1% O2), WT cells increased their glucose consumption and lactate production to reach a rate similar to that of MD cells.
Figure 1. Metabolic characterization of SiHa WT and MD cells. (A) Oxygen consumption rates (OCR) expressed as oxygen-sensitive probe fluorescence decay (lifetime in μs)/minute were measured in the absence or presence of antimycin (2.5 μM) a mitochondrial complex III inhibitor. ***, p < 0.001 ANOVA1 (n = 4). (B) Extracellular acidification rates (ECAR) expressed as pH-sensitive probe fluorescence decay (lifetime in μs)/minute were measured in the absence or presence of antimycin (2.5 μM). ***, p < 0.001 ANOVA1 (n = 4). (C, D) Extracellular glucose and lactate concentrations were determined enzymatically in the culture media and expressed as a delta metabolite concentration after 24 hours of culture and after normalization to protein content. *, p < 0.05 and **, p < 0.01 ANOVA2 and Bonferroni post-test (n = 3).
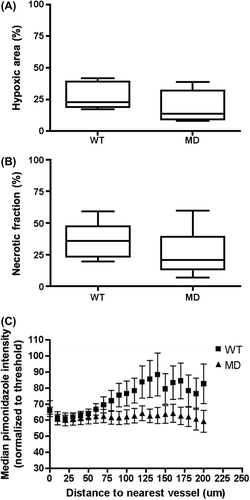
After confirmation that MD cells exhibited a glycolytic metabolic profile with reduced oxygen consumption in vitro, we characterized both models in vivo. Assessment of tumor growth before treatment indicated no difference between WT and MD tumors when cells were implanted in Matrigel. When the tumors reached a mean diameter of 7.5 mm, mice were first injected with [18F]-FDG in order to image glucose uptake with a positron emission tomography (PET) scanner. Although there was no statistical difference in [18F]-FDG uptake ratios (SUV Max) in MD group compared to WT, the latter group exhibited a higher variability than MD (Supplementary Figure 2A to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). Analysis of [18F]-FAZA, a tracer of hypoxia [Citation11], indicated a trend for increased uptake for WT compared to MD (p = 0.06) (Supplementary Figure 2B to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). To confirm these results, we also analyzed the hypoxic fraction in tumor sections following injection of pimonidazole. Here, no statistical difference in hypoxic fraction was observed between the two groups ( and Supplementary Figure 3 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006) although a similar trend than with [18F]-FAZA was observed. We also evaluated the extent of necrotic areas in both groups and found larger necrotic fraction in WT tumors ( and Supplementary Figure 3 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). We then analyzed the microregional differences in pimonidazole staining as a function of distance to nearest blood vessel ( and Supplementary Figure 4 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). The results indicate that WT tumors exhibited increased values of pimonidazole staining as function of distance, whereas this was not observed in MD tumors (ANOVA2; hypoxia vs. distance p < 0.001).
Figure 2. In vivo characterization and micro regional differences of hypoxia distribution in WT and MD tumors. (A, B) Quantification of hypoxic and necrotic fraction for both groups after staining of WT and MD tumor sections with hypoxyprobe. © Zonal analysis of pimonidazole intensities as a function of distance from the nearest total blood vessel. Each point represents the median pimonidazole intensity within a selected zone for at least four different areas/tumor (n = 3). ***, p < 0.001 ANOVA2 for hypoxia.
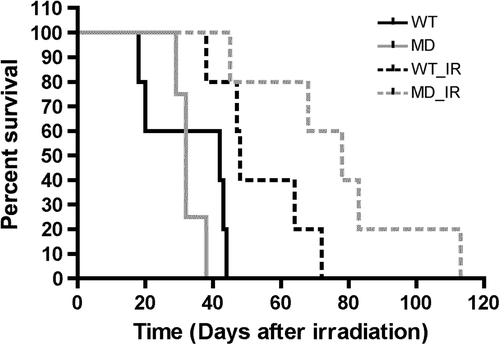
Next, we irradiated tumors from half of the mice/group with a single dose of 16 Gy and followed their growth rate compared to non-irradiated countermates (). Results expressed as the average day for reaching the endpoint (16-mm mean diameter) showed a statistically significant increase in regrowth delay of irradiated MD tumors, suggesting an increased sensitivity to radiotherapy (Supplementary Figure 5 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006).
Figure 3. Growth delay after irradiation of WT and MD tumors. (A) Kaplan-Meier survival curves were obtained by introducing individual dates for reaching mean tumor diameter of 16 mm, determined as endpoint (n = 4–5).
To test whether the increased radioresponse of MD versus WT tumors could result from intrinsic differences in cell radiosensitivity, we investigated the radioresponse of the corresponding cell lines in vitro. We first assessed the clonogenic survival of irradiated cells under normoxic (21% O2) or hypoxic (1% O2) conditions (). Under normoxia, MD cells showed a slight but not significant increase in radioresponse compared to WT. Under hypoxia, the level of acquired radioresistance was not different between cell lines (). Although apoptotic rates 24 hours after irradiation (5 Gy) were statistically different between both groups, overall less than 5% of cells were apoptotic (WT 1.11 ± 0.02% vs. MD 0.81 ± 0.09% of cells; p < 0.05). We next examined the kinetics of H2AX histone phosphorylation, a marker of double strand-breaks repair. Half an hour after a single 5-Gy dose of γ-rays, 41 ± 6% of WT and 27 ± 2% (NS; p-value = 0.11) of MD cells were positive for γH2AX, followed by a progressive decline. We further measured the kinetics of γH2AX dephosphorylation relative to the maximum reached half an hour after irradiation. The half-life of γH2AX was 3.2 ± 0.3 hours for WT and 2.6 ± 0.2 hours for MD; this difference was not statistically significant (). We also monitored cell cycle arrest after irradiation (). Twenty-four hours after a single dose of 5 Gy, both WT and MD showed a significant increase of cells in G2/M phase, indicating a cell cycle arrest at this checkpoint for both cell lines.
Figure 4. Assessment of response to irradiation of WT and MD cells in vitro. (A) Survival curves after irradiation of WT and MD cells in normoxia (plain curves) and hypoxia (dashed curves). (B) DNA damage repair after a single dose irradiation (5 Gy) expressed relative to the maximum (1 hour post-irradiation) (n = 3).
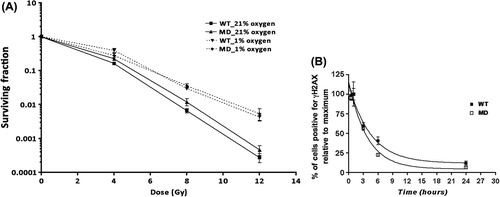
Table I. Analysis of cell cycle distribution in WT and MD after irradiation (5 Gy).
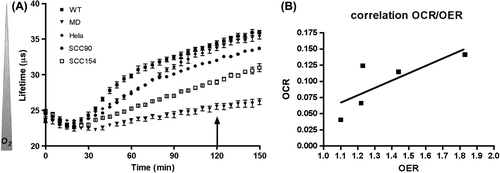
Finally, because our results showed a decreased rate of oxygen consumption in MD compared to WT cells (), we wondered if this metabolic difference in oxygen consumption could be translated into radiosensitivity and whether this observation could be extended to other cell lines with different OCR. However influence of OCR will only be detectable in vivo since classic radiosensitivity assessment in vitro is done through clonogenic assay where cells are either cultivated in an open system (atmospheric 21% O2) either under artificial hypoxic conditions (atmosphere at 1% O2). So, we plated several cell lines in a closed system (the same used for the measurement of OCR), allowed the cells to consume oxygen for two hours and then irradiated the cells with 0–2–4 Gy and compared their survival fraction to the one obtained in an open system (Supplementary Figure 6 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). The difference in survival fraction in open versus closed wells was expressed in terms of OER. illustrates oxygen consumption curves for different cell lines as a function of time. When cells were irradiated in an open system, their survival curves reflected their intrinsic radioresponse, while irradiation of these cells in a closed system, two hours after sealing the wells, resulted in radioresistance of most of the cells except for SiHa_MD cells (Supplementary Figure 4 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). As expected, OCR correlated positively (, R2 = 0.77) with acquired radioresistance expressed as OER.
Figure 5. Oxygen consumption and radioresistance of WT, MD, Hela, SCC90 and SCC154 cell lines. (A) Oxygen consumption measured by fluorometry. The oxygen-sensitive probe fluorescence decay (lifetime in μs) is inversely correlated to oxygen concentration in the wells. The arrow illustrates the time point at which cells were irradiated (0–2–4 Gy). (B) Oxygen enhancement ratio (OER) was calculated as the ratio between survival fraction for hypoxic cells (closed wells)/aerobic cells (open wells) through a linear quadratic model and was then correlated to oxygen consumption rate (OCR). Spearman's correlation coefficient = 0.77.
Discussion
It was previously believed that most cancer cells presented mitochondrial impairment, therefore relying mainly on aerobic glycolysis to meet their energetic demand. However, it has been shown that mitochondrial respiration persists in most cancer cells and that increased glucose uptake and aerobic glycolysis serve to support biomass accumulation and redox maintenance [Citation13]. Wild-type SiHa cells have been shown to exhibit a mitochondrial respiration that can be fueled by diverse metabolites including glucose [Citation7,Citation14]. However, MD SiHa cells presented increased glucose uptake () and increased lactate production () accompanied by increased ECAR (). Mitochondria play a critical role in several cellular processes including apoptosis and their impairment might be linked to radioresistance due to a decreased apoptotic rate through p53, as suggested by Compton [Citation15]. Here, although 24 hours after irradiation WT cells presented increased apoptotic rate compared to MD (overall less than 5%), there was no significant difference in clonogenic survival (). As SiHa cells are known to be HPV positive, they have impaired p53 function due to the viral E6 oncoprotein that targets p53 to ubiquitin degradation [Citation16]. In addition, apoptosis is just one mode of cell death that contributes to the loss of clonogenicity after irradiation, as demonstrated by experiments performed in p21 knockout cells [Citation17]. Assessing the presence of γH2AX foci at different time points after irradiation was demonstrated to be a highly sensitive method for detecting the presence and/or repair of DNA double-strand breaks in irradiated cells [Citation18]. Here, there were no differences in γH2AX clearance between both cell lines and our cell cycle analysis indicated a clear arrest of WT and MD cells in G2/M phase 24 hours after irradiation.
Surprisingly, when the response to radiation was assessed, in vivo results showed that MD tumors were more radiosensitive compared to WT. In addition, since the growth rate for both groups was similar, we can assume that there was no growth impairment in MD tumors due to their metabolic profile. Here, metabolic profiling in vivo through molecular imaging with PET showed in WT tumors a trend for increased uptake of [18F]-FAZA and similar [18F]-FDG uptake compared to MD (Supplementary Figure 2 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). Yet our in vitro data indicated that under hypoxia (1% O2), WT cells were able to adapt to their environment by increasing their glucose consumption () supporting the hypothesis that [18F]-FDG SUV Max values similar to the one observed in MD group reflects a decrease in tumor oxygen availability within WT tumors. Analysis of hypoxic fraction was performed in vivo and ex vivo using [18F]-FAZA and pimonidazole staining, respectively. Both agents are nitroimidazoles derivatives, a class of molecules that accumulate in hypoxic cells [Citation19]. Although PET imaging still suffers some weaknesses, such as the voxel size analyzed by PET scans being much larger than the scale of hypoxia heterogeneity [Citation20], it has been recently shown by correlation to pO2 measurement that [18F]-FAZA is an effective surrogate of hypoxic fractions [Citation21]. However, systems used here for quantification of hypoxic fraction are dependent of a threshold in partial oxygen (< 10 mmHg) and global quantification might not be sensitive enough to detect subtle changes in intracellular oxygen concentration [Citation22]. In addition, results from experiments correlating pimonidazole labeling with radiobiological hypoxia of clonogenic cells in different tumor models are controversial [Citation23,Citation24]. Here, the spatial distribution of hypoxic fraction in WT and MD tumors showed different patterns with heterogeneous distribution in WT compared to a core area in MD (Supplementary Figure 3 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006). Furthermore, quantification of necrotic areas indicated necrosis in WT tumors (). These results suggest that WT tumors have less available oxygen and feature areas of diffusion-limited hypoxia (positive for pimonidazole) and areas of perfusion-limited hypoxia (necrotic areas) whereas hypoxic fraction in MD tumors would only reflect perfusion-limited hypoxia. Furthermore in MD tumors analysis of pimonidazole staining as a function of distance to vessels () shows that hypoxia is found closer to blood vessels in WT tumors compared to MD. This suggests that the absence of mitochondrial respiration in these cells increase the cellular oxygen availability. Indeed, tissue O2 concentration is determined by oxygen supply and oxygen demand and mathematical models of tumor oxygenation predict that hypoxia may be abolished by reductions in OCR. The underlying reason for this is that reduction of consumption rate not only increases the pO2 of blood in tumor microvessels, but also reduces pO2 gradients within the tumor tissue [Citation5]. Furthermore, several pharmacological treatments that inhibit cell oxygen consumption have also been linked to increased radiosensitivity although differences in hypoxic fraction of the treated tumors were not always demonstrated [Citation6,Citation25–29]. Our data suggest that subtle changes in O2 availability, determined by cellular oxygen consumption will modulate tumor response to radiation. To support this hypothesis, we analyzed the oxygen consumption of several cell lines and correlated it to their survival after irradiation in a closed system. As expected, this experiment indicated a positive correlation between the OCR and radioresistance expressed as OER. Data on the modulation of oxygen consumption and its impact on radiation response have been published in the past [Citation6]. However, our study provides data from an isogenic model of mitochondrial dysfunction with no alteration in intrinsic radiosensitivity neither on cell proliferation in comparison to pharmacological treatments or decreased metabolism [Citation6,Citation30]. Compared to a pharmacological treatment affecting global cellular oxygen consumption, our WT as well as MD model might suffer from a lack in perfusion that could explain limited influence in hypoxic fraction between both tumors. In addition, although in vitro experiments show no difference between both models in intrinsic radioresponse, respiration rates might not be the only mitochondrial process to be disrupted in MD cells and further investigations on MD model might lead to discovery of other defective mechanisms [Citation31].
In conclusion, this study highlights the impact of tumor metabolic oxygen consumption on the response to irradiation in a syngeneic model of impaired mitochondrial respiration. In addition, our results provide a rationale for combining anti-metabolic therapeutic strategies to radiotherapy in particular if such therapies focus on decreasing the oxygen consumption rate of tumor cells.
Supplementary material available online
Supplementary Appendix: Material and methods
Supplementary Figures 1–6 to be found online at http://informahealthcare.com/doi/abs/10.3109/0284186X.2014.932006
ionc_a_932006_sm8226.pdf
Download PDF (1 MB)Acknowledgments
This work was supported by the Fond Joseph Maisin, the European Research Council (FP7/2007-2013 European Research Council Independent Researcher Starting Grant No. 243188 TUMETABO to P.S.), Interuniversity Attraction Pole (IAP) grant #UP7-03 from the Belgian Science Policy Office (Belspo), an Action de Recherche Concertée from the Communauté Française de Belgique (ARC 09/14-020), and the Fonds National de la Recherche Scientifique (F.R.S.-FNRS). V.B. is a Télévie post-doctoral fellow, O.F. is a Honorary Research Director and P.S. is a Research Associate of the F.R.S.-FNRS. No potential conflicts of interest were disclosed.
References
- Warburg O. On respiratory impairment in cancer cells. Science 1956;124:269–70.
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–74.
- Kim J-W, Dang CV. Cancer's molecular sweet tooth and the Warburg effect. Cancer Res 2006;66:8927–30.
- Vaupel P. Hypoxia and aggressive tumor phenotype: Implications for therapy and prognosis. Oncologist 2008; 13(Suppl 3):21–6.
- Secomb TW, Hsu R, Ong ET, Gross JF, Dewhirst MW. Analysis of the effects of oxygen supply and demand on hypoxic fraction in tumors. Acta Oncol 1995;34:313–6.
- Biaglow JE, Manevich Y, Leeper D, Chance B, Dewhirst MW, Jenkins WT, et al. MIBG inhibits respiration: Potential for radio- and hyperthermic sensitization. Radiat Oncol Biol Phys 1998;42:871–6.
- Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930–42.
- De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, Sonveaux P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One 2012;7:e46571.
- Hynes J, O’Riordan TC, Zhdanov AV, Uray G, Will Y, Papkovsky DB. In vitro analysis of cell metabolism using a long-decay pH-sensitive lanthanide probe and extracellular acidification assay. Anal Biochem 2009;390:21–8.
- Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1:2315–9.
- Reischl G, Ehrlichmann W, Bieg C, Solbach C, Kumar P, Wiebe LI, et al. Preparation of the hypoxia imaging PET tracer [18F]FAZA: Reaction parameters and automation. Appl Radiat Isot 2005;62:897–901.
- Huisman MC, Reder S, Weber AW, Ziegler SI, Schwaiger M. Performance evaluation of the Philips MOSAIC small animal PET scanner. Eur J Nucl Med Mol Imaging 2007;34:532–40.
- DeBerardinis RJ. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med 2008;10:767–77.
- Busk M, Horsman MR, Jakobsen S, Bussink J, van der Kogel A, Overgaard J. Cellular uptake of PET tracers of glucose metabolism and hypoxia and their linkage. Eur J Nucl Med Mol Imaging 2008;35:2294–303.
- Compton S, Kim C, Griner NB, Potluri P, Scheffler IE, Sen S, et al. Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J Biol Chem 2011; 286:20297–312.
- Vozenin M-C, Lord H-K, Hartl D, Deutsch E. Unravelling the biology of human papillomavirus (HPV) related tumours to enhance their radiosensitivity. Cancer Treat Rev 2010;36:629–36.
- Wouters BG, Giaccia AJ, Denko NC, Brown JM. Loss of p21Waf1/Cip1 sensitizes tumors to radiation by an apoptosis-independent mechanism. Cancer Res 1997;57:4703–6.
- Banáth JP, MacPhail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res 2004;64:7144–9.
- Haubner R. PET radiopharmaceuticals in radiation treatment planning – synthesis and biological characteristics. Radiother Oncol 2010;96:280–7.
- Christian N, Deheneffe S, Bol A, De Bast M, Labar D, Lee JA, et al. Is (18)F-FDG a surrogate tracer to measure tumor hypoxia? Comparison with the hypoxic tracer (14)C-EF3 in animal tumor models. Radiother Oncol 2010; 97:183–8.
- Tran L-B-A, Bol A, Labar D, Jordan B, Magat J, Mignion L, et al. Hypoxia imaging with the nitroimidazole (18)F-FAZA PET tracer: A comparison with OxyLite, EPR oximetry and (19)F-MRI relaxometry. Radiother Oncol 2012;105:29–35.
- Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer 1995; 61:567–73.
- Raleigh JA, Chou SC, Arteel GE, Horsman MR. Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors. Radiat Res 1999;151:580–9.
- Yaromina A, Hölscher T, Eicheler W, Rosner A, Krause M, Hessel F, et al. Does heterogeneity of pimonidazole labelling correspond to the heterogeneity of radiation-response of FaDu human squamous cell carcinoma? Radiother Oncol 2005;76:206–12.
- Jordan BF, Grégoire V, Demeure RJ, Sonveaux P, Feron O, O’Hara J, et al. Insulin increases the sensitivity of tumors to irradiation: Involvement of an increase in tumor oxygenation mediated by a nitric oxide-dependent decrease of the tumor cells oxygen consumption. Cancer Res 2002;62:3555–61.
- Crokart N, Jordan BF, Baudelet C, Cron GO, Hotton J, Radermacher K, et al. Glucocorticoids modulate tumor radiation response through a decrease in tumor oxygen consumption. Clin Cancer Res 2007;13:630–5.
- Crokart N, Radermacher K, Jordan BF, Baudelet C, Cron GO, Grégoire V, et al. Tumor radiosensitization by antiinflammatory drugs: Evidence for a new mechanism involving the oxygen effect. Cancer Res 2005;65: 7911–6.
- Diepart C, Karroum O, Magat J, Feron O, Verrax J, Calderon PB, et al. Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res 2012;72:482–90.
- Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res 2013;19:6741–50.
- Durand RE, Biaglow JE. Radiosensitization of hypoxic cells of an in vitro tumor model by respiratory inhibitors. Radiat Res 1977;69:359–66.
- Nieri D, Fioramonti M, Berardinelli F, Leone S, Cherubini R, De Nadal V, et al. Radiation response of chemically derived mitochondrial DNA-deficient AG01522 human primary fibroblasts. Mutat Res 2013;756:86–94.