Abstract
Although AL amyloidosis usually is a systemic disease, strictly localized AL deposits are not exceptionally rare. Such case reports form a considerable body of published articles. Although both AL amyloidosis types are formed from an N-terminal segment of a monoclonal immunoglobulin light chain, a typical localized AL amyloid differs from the systemic counterpart by the morphological appearance of the amyloid, and presence of clonal plasma cells and of giant cells. In this article it is pointed out that localized AL amyloidosis (‘amyloidoma') represents a true plasma cell neoplasm and not a pseudotumor. The pathogenesis of localized AL amyloidosis may differ from that of the systemic type, a suggestion underlined by the fact that localized AL amyloidosis of kappa type is as common as that of lambda origin, in contrast to the systemic form where lambda chains constitute the overwhelming majority of cases. It is suggested that oligomeric assemblies of the produced immunoglobulin light chain are toxic to plasma cells, which in this way commit suicide.
Introduction
The amyloidoses constitute a large and heterogeneous group of protein misfolding disorders in which proteins adopt a high degree of beta-sheet structure, allowing molecules to bind to each other in long, cross-beta-sheet fibrils. In this form the protein is comparatively resistant to degradation, explaining why the amyloid masses may increase as long as the protein substrate is present. The mechanisms behind the fibril formations are still insufficiently understood, but it is generally accepted that generation of amyloid from a protein includes three phases. The first phase is nucleation, at which misfolded molecules assemble to form a nucleus, resembling the process of crystallization (Citation1). This nucleus can catalyze an amyloid-prone misfolding of other molecules, which adopt β-structure and form rapidly growing fibrils. This second elongation phase proceeds until a steady state (third phase) has been obtained. A particularly favorable milieu is consequently formed where there is a high concentration of substrate.
This mini-review deals with the nature of one localized form of amyloidosis that clinically often presents as a malignant tumor but that is often regarded as a pseudotumor or tumor-like nodule (Citation2,3). One of the main messages in this communication is that it is a manifestation of a real neoplasm.
AL amyloidosis
Amyloidosis can either be systemic or localized. In systemic forms, the precursor of the fibril protein is expressed at one site, e.g. liver or bone-marrow, released to blood plasma in soluble form, distributed via the circulation, and finally deposited as amyloid fibrils in different organs by yet unknown mechanisms. In human, 15 different proteins have been shown to be able to cause systemic amyloidosis, although most forms are very rare (Citation4). One of the least uncommon is AL amyloidosis where the fibril protein is derived from monoclonal immunoglobulin (Ig) light chains. The primary cause of this form of amyloid is consequently a clonal expansion of one plasma cell expressing one light chain, usually in excess. The plasma cell clone is spread in the bone-marrow, either as a malignant tumor (myeloma) or as apparently benign cells. The Ig light chain circulates in blood plasma before it aggregates into amyloid fibrils in virtually all organs outside the brain by so far unknown mechanisms. Systemic AL amyloidosis is usually lethal.
Although many of the subclasses of Ig light chains are represented in amyloid only, not all light chains are amyloidogenic. There is strong evidence that this restriction depends on the variability in the amino acid sequences, and certain variable subgroups and amino acid substitutions are particularly important (Citation5,6). AL protein of lambda type is 2–3 times more common than of kappa type (Citation7,8). The major part of amyloid AL protein lacks a portion of the constant region (Citation9-12), although a minor component always is the complete Ig light chain (Citation13). Whether or not this truncation is pathogenically important or occurs after fibrillogenesis is not clear.
Localized AL amyloidosis
Amyloid localized to one tissue, where multiple deposits appear, is very common, particularly in association with aging. Examples are amyloid in the brain in Alzheimer's disease and in islets of Langerhans in type 2 diabetes (Citation14). In contrast localized AL amyloidosis usually appears as one, often tumor-like lesion, although multiple nodules may occur. The symptoms may vary depending on location of the lesion, but, initially, malignant tumors are often suspected, particularly when the nodules are present in the breast, lung, or urinary tract (Citation3,15,16). Calcifications are common (Citation17), and in the case of breast nodules mammographic appearance may include microcalcifications, increasing the suspicion of malignancy () (Citation18,19). A monoclonal component in plasma is usually absent (Citation20). Treatment of choice seems to be local resections endoscopically, although radical excision has been performed successfully, e.g. for respiratory lesions (Citation3,21). Local resections often have to be repeated (Citation22). The long-term prognosis is usually favorable, although deaths due to severe hemorrhages or other complications have been described (Citation20,21). While a monoclonal gammopathy of unknown significance (MGUS) may develop to systemic amyloidosis or myeloma (Citation23) this does not seem to happen with a localized plasma cell clone associated with localized amyloidosis, which almost never progresses to a systemic disease (Citation20,24,25).
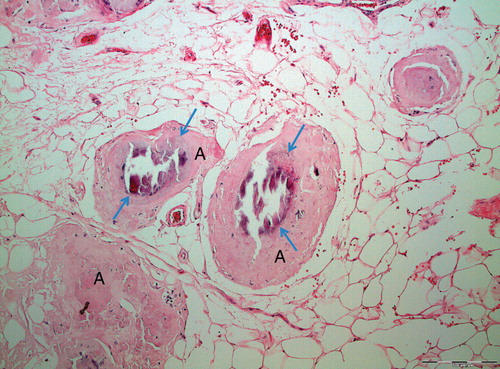
Figure 1. Localized AL amyloidosis in a breast. The localized nature and occurrence of microcalcifications (arrows) constitute a risk of misdiagnosis of mammary carcinoma.
Appearance of localized AL amyloid
Usually, amyloid deposits are observed in a strictly limited but not encapsulated area. Involvement of vessel walls is common but does only occur very close to the main deposit. Like systemic AL amyloidosis, affinity for Congo red is variable between patients but always present, as is the concomitant green birefringence in polarized light. Unlike deposits in most cases of systemic amyloidosis, crystal-like structures with Maltese cross appearance are as common as areas with concentric ring structures (). These formations usually have stronger affinity for Congo red and a brighter green birefringence. Typically, scattered and small groups of plasma cells occur in the amyloid deposits. Localized AL amyloid can occur at virtually any site of the body, including the brain (Citation26). Most common sites are eyelids, larynx, bronchi, skin, and urinary tract.
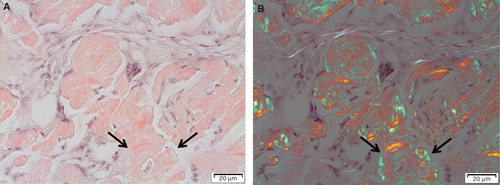
Figure 2. Typical appearance of localized AL amyloidosis with discrete amyloid formation and multiple giant cells. Note the concentric appearance of some amyloid particles (arrows).
Biochemical nature of localized AL amyloid
An immunoglobulin light chain nature of localized nodular pulmonary amyloid was indicated by peptide mapping and immunodiffusion soon after the monoclonal light chain origin of primary systemic amyloidosis had been demonstrated (Citation27). Later, amino acid sequence analyses proved the monoclonal immunoglobulin light chain nature in a number of cases (Citation28-35). AL kappa is as common as AL lambda (Solomon A., personal communication). On very rare occasions, a heavy chain fragment has been demonstrated (Citation36). As in systemic AL amyloidosis, most of the amyloid protein is not full-length light chain molecules, and a varying part of the constant region is missing (Citation27,29,34,35,37). Small amounts of full-length light chains are usually present as well as small C-terminal fragments (Citation13). The importance of fragmentation in the amyloidogenic process is still not quite settled, since it has been suspected to occur after fibril formation. On the other hand, the almost universal predominance of N-terminal fragments over fragments from the C-terminus has been suggested to indicate a major mechanism in fibrillogenesis (Citation37).
Is localized AL amyloid a pseudotumor or a real neoplasm?
According to Celsus a tumor originally meant swelling, but the designation is nowadays almost only used for neoplasm. Consequently, tumor-like lesions should be non-neoplastic swellings. It is obvious that localized AL amyloidosis appears as a tumor according to a classical definition. The descriptions ‘tumor-like' or ‘pseudotumor' are often used in order to discriminate from neoplasm. This may be correct for localized AL amyloidosis in the sense that the mass depends on the deposition of amyloid, but, as described below, the basic lesion is nevertheless a neoplasm in the form of a clonal expansion of one plasma cell.
A localized production of the amyloid immunoglobulin light chain was suspected at an early date but was difficult to prove. On most occasions, the plasma cell clone is very discrete and even difficult to recognize. Only very exceptionally is the background a clear-cut immunocytoma or plasmacytoma with atypia and mitoses (Citation38). Not only is the number of plasma cells in the amyloid lesion often fairly sparse, but trials to identify monoclonality of the plasma cells by immunohistochemistry have also generally failed (Citation22), although an abnormal overload of kappa or lambda plasma cells sometimes is evident (). By means of immunohistochemistry it is often evident that a mixture of plasma cell populations with both kappa and lambda-expressing cells is present. A predominance of one plasma cell type (kappa or lambda) has sometimes been taken as evidence for monoclonality (Citation39). However, gene rearrangement analysis of a few typical localized AL amyloidoses has shown presence of plasma cell clones (Citation40-44). A similar result was obtained by amino acid sequence analysis of the deposited amyloid combined with mRNA extraction and DNA sequencing (Citation33). Therefore it has been suggested that the process can be classified as a ‘low-grade B cell lymphoproliferative disease' (Citation44), a somewhat diffuse designation but one which in hematological terminology probably means a neoplasm.
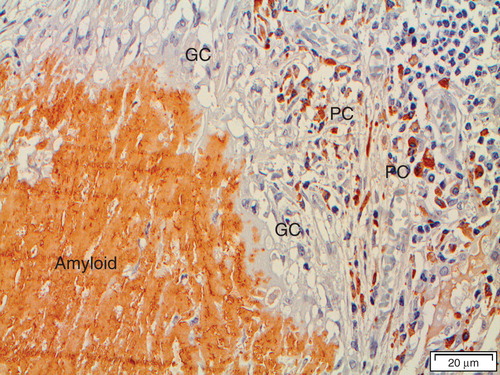
Figure 3. ‘Amyloidoma' immunolabeled with the immunoglobulin light chain lambda-specific monoclonal antibody pwlam (Citation73). Amyloid and a majority of plasma cells (PC) are strongly labeled. Note the giant cells (GC) between plasma cells and amyloid. The hypothesis is that plasma cells are synthesizing an amyloidogenic Ig light chain, which is modified by giant cells to amyloid fibrils.
Pathogenesis of the amyloidogenic plasma cell clone
Given that the most common localization of localized AL amyloidosis is at mucous membranes in contact with the environment, it has been suggested there is an antigenic induction of amyloidogenic plasma cell clones (Citation17). A chronic inflammation has often been encountered (Citation15). It seems likely that similar plasma cell clones without amyloid deposits may appear as commonly when the cells do not produce an amyloidogenic Ig light chain. It is noteworthy that a number of cases with localized AL amyloidosis have been associated with Sjögren's syndrome (Citation24,45).
Giant cells in localized AL amyloidosis
Foreign giant cells are very often mentioned in plentiful case reports on localized amyloidosis (e.g. (Citation17,18,22,36,39,44,46-55)). Their occurrence is usually explained as a reaction to the amyloid deposits (Citation54). However, amyloid in general does not induce a foreign body reaction, and there are almost never any giant cells present in the amyloid masses in systemic AL amyloidosis. Furthermore, amyloid fibrils close to giant cells are organized in the same way as seen at sites where cells participate in amyloid production, e.g. in the spleen in AA amyloidosis (Citation56) or in the islets of Langerhans in IAPP amyloidosis (Citation14). This is often suggested already when using polarization microscopy where a clear-cut orientation of amyloid fibril bundles against giant cells is evident (). Therefore, we proposed that giant cells in localized AL amyloidosis directly participate in the transformation of the soluble full-length light chains into insoluble fibrils formed from N-terminal light chain fragments (Citation34).
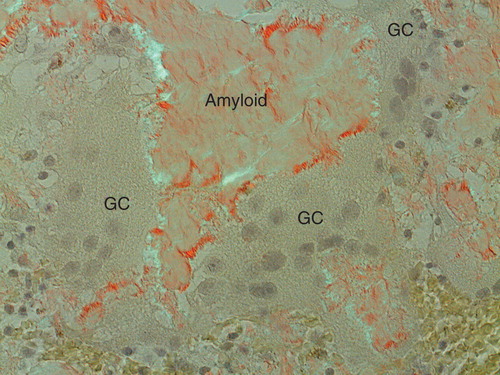
Figure 4. Amyloid adjacent to giant cells (GC). An organization of amyloid fibrils is apparent when in contact with giant cells. This organization is interpreted as assemblage of protein, probably after some modification, into fibrils on or at the surface of the cells. Congo red stained section in polarized light with crossed polars.
The genesis and function of multinuclear giant cells, typical of certain infections and inflammations, are still poorly understood. They develop by fusion of macrophages and are generally thought of as cellular elements that try to perform phagocytosis of particular structures, e.g. larger foreign particles. However, this simplistic explanation has been questioned, and giant cells may exhibit novel functions that are not yet understood (Citation57). Experimentally, giant cells have been reported to be induced by cultivation of monocytes in the presence of interleukin 3 (IL-3) and interferon-gamma (Citation58,59), IL-4 (Citation60-64), or IL-13 (Citation65). Plasma cells can express IL-4 (Citation66). Although T cells may be a more important source of IL-4, there is a normal foreign body response in T cell-deficient mice (Citation67). The inducer in localized AL amyloid is unknown, and giant cells do not appear in the bone-marrow in systemic AL amyloidosis in spite of presence of neoplastic plasma cells. It should also be pointed out that the bone-marrow is usually not particularly affected in the systemic disease, although the fibril protein is synthesized there.
Hypothesis and conclusion
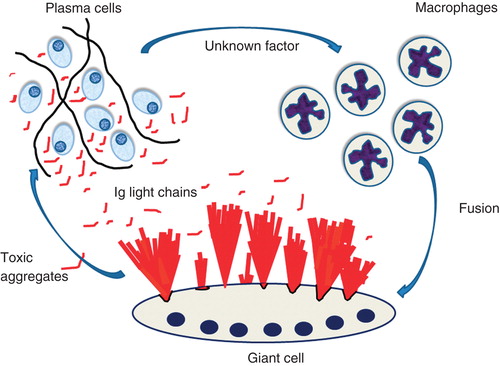
From the current knowledge, it is only possible to create a hypothesis for the pathogenesis of localized AL amyloidosis. A benign small plasma cell clone is formed at one site, perhaps after prolonged antigenic stimulation. The clonal plasma cells produce an immunoglobulin light chain which is amyloidogenic but needs to be modified. Only clones that can recruit macrophages, which fuse to giant cells by mechanisms yet to be elucidated, can form amyloid. The modification of the light chains may take place within giant cells which take up the soluble light chain molecules by endocytosis and process them so that the N-terminal parts can aggregate into fibrils. Alternatively, protein modification may occur on the cell surface. Most likely, the assembly take place on the cell surface, e.g. after lysosomal secretion, which is a regulated secretory pathway (Citation68). Such a scenario would explain the evident organization of amyloid fibrils close to giant cells, very different from what is seen in deposits in systemic amyloidosis. Another fact which points to a pathogenesis different from systemic amyloidosis is that localized AL amyloid of kappa type is common. Although it is only possible to speculate, this may depend on a lower propensity of kappa chains to form beta-sheet fibrils, overcome in the localized situation where the protein concentration close to plasma cells likely is high. The hypothetical formation of localized AL amyloidosis has been outlined in .
Figure 5. Suggested events in localized AL amyloidosis. A plasma cell clone develops at one site. The cells express amyloidogenic Ig light chains and, in addition, an unknown factor that attracts macrophages and cause them to fuse into giant cells. Ig light chains are modified by giant cells and aggregate into bundles of amyloid fibrils at the surface of these cells. In addition, smaller aggregates of Ig light chains are toxic to plasma cells, an event controlling the plasma cell clone.
Another interesting aspect of localized AL amyloid is the sparse occurrence of plasma cells in the lesions. In fact, the number of plasma cells is sometimes so low that the clonality can be difficult to demonstrate. The lesion has therefore been suggested to represent a ‘burnt-out' plasmacytoma (Citation9,69). In many other amyloid diseases, e.g. Alzheimer's disease or type 2 diabetes, there is increasing evidence that amyloid protein aggregation involves genesis of smaller aggregates (oligomers, protofibrils) that exert a toxic effect on nearby cells and generate apoptosis (reviewed in (Citation70,71)). In addition, such a mechanism is believed to explain the rapid effect on myocardial dysfunction after successful treatment of a plasma cell clone in systemic AL amyloidosis (Citation72). If generation of toxic oligomers is true also with the formation of localized AL amyloid, this may lead to apoptosis of plasma cells in an ‘amyloidoma'. This event may then be regarded as a suicide of tumor plasma cells creating a self-limiting neoplasm.
Acknowledgements
Thanks to Gunilla T. Westermark for help with figures. Supported by the Swedish Research Council.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Jarrett JT, Lansbury PT. Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8.
- Bhagwandeen BS, Taylor S. Primary localized amyloidosis of the bladder with a monoclonal plasma cell infiltrate. Pathology. 1988;20:67–9.
- Monge M, Chauveau D, Cordonnier C, Noël LH, Presne C, Makdassi R, Localized amyloidosis of the genitourinary tract: report of 5 new cases and review of the literature. Medicine (Baltimore). 2011;90:212–22.
- Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Amyloid fibril protein nomenclature: 2010 recommendations of the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17:101–4.
- Solomon A, Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V–VI subgroup of human light chains with amyloidosis AL(l). J Clin Invest. 1982;70:453–60.
- Stevens FJ. Protein structure, stability, and conformational disease: human antibody light chains 1999. Springfield, VA: National Technical Information Service, U.S. Department of Commerce; 1999.
- Isobe T, Osserman EF. Patterns of amyloidosis and their association with plasma-cell dyscrasia, monoclonal immunoglobulins and Bence-Jones proteins. N Engl J Med. 1974;290:473–7.
- Bellotti V, Mangione P, Merlini G. Review: immunoglobulin light chain amyloidosis—the archetype of structural and pathogenic variability. J Struct Biol. 2000;130:280–9.
- Glenner GG. Amyloid deposits and amyloidosis. The β-fibrilloses. N Engl J Med. 1980;302:1283–92; 1333–43.
- Tveteraas T, Sletten K, Westermark P. The amino acid sequence of a carbohydrate-containing immunoglobulin-light-chain-type amyloid-fibril protein. Biochem J. 1985;232:183–90.
- Ramstad HM, Sletten K, Husby G. The amino acid sequence and carbohydrate composition of an immunoglobulin kappa light chain amyloid fibril protein (AL) of variable subgroup I. Amyloid. 1995;2:223–8.
- Omtvedt LA, Bailey D, Renouf DV, Davies MJ, Paramonov NA, Haavik S, Glycosylation of immunoglobulin light chains associated with amyloidosis. Amyloid. 2000;7:227–44.
- Enqvist S, Sletten K, Westermark P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J Pathol. 2009;219:473–80.
- Westermark P. Amyloid in the islets of Langerhans: thoughts and some historical aspects. Ups J Med Sci. 2011;116:81–9.
- Toyoda M, Ebihara Y, Kato H, Kita S. Tracheobronchial AL amyloidosis: histologic, immunohistochemical, ultrastructural, and immunoelectron microscopic observations. Hum Pathol. 1993;24:970–6.
- Merrimen JL, Alkhudair WK, Gupta R. Localized amyloidosis of the urinary tract: case series of nine patients. Urology. 2006;67:904–9.
- Krishnan J, Chu W, Elrod JP, Frizzera G. Tumoral presentation of amyloidosis (amyloidomas) in sof tissues. A report of 14 cases. Am J Clin Pathol. 1993;100:135–44.
- Fernandez BB, Hernandez FJ. Amyloid tumor of the breast. Arch Pathol. 1973;95:102–5.
- Charlot M, Seldin DC, O'hara C, Skinner M, Sanchorawala V. Localized amyloidosis of the breast: a case series. Amyloid. 2011;18:72–5.
- Paccalin M, Hachulla E, Cazalet C, Tricot L, Carreiro M, Rubi M, Localized amyloidosis: a survey of 35 French cases. Amyloid. 2005;12:239–45.
- Piazza C, Cavaliere S, Foccoli P, Toninelli C, Bolzoni A, Peretti G. Endoscopic management of laryngo-tracheobronchial amyloidosis: a series of 32 patients. Eur Arch Otorhinolaryngol. 2003;260:349–54.
- Godbersen GS, Leh JF, Hansmann ML, Rudert H, Linke RP. Organ-limited laryngeal amyloid deposits: clinical, morphological, and immunohistochemical results of five cases. Ann Otol Rhinol Laryngol. 1992;101:770–5.
- Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med. 1978;64:814–26.
- Moon AO, Calamia KT, Walsh JS. Nodular amyloidosis. Arch Dermatol. 2003;139:1157–9.
- Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006;13:135–42.
- Cohen M, Lanska D, Roessmann U, Karaman B, Ganz E, Whitehouse P, Amyloidoma of the CNS. I. Clinical and pathologic study. Neurology. 1992;42:2019–23.
- Page DL, Isersky C, Harada M, Glenner GG. Immunoglobulin origin of localized nodular pulmonary amyloidosis. Res Exp Med. 1972;159:75–86.
- Westermark P, Sletten K, Pitkänen P, Natvig JB, Lindholm CE. Localized laryngeal amyloidosis: partial characterization of an amyloid fibril protein AL. Mol Immunol. 1982;19:447–50.
- Sletten K, Westermark P, Pitkänen P, Natvig JB, Lindholm C-E. Amino acid sequences in amyloid proteins of κIII immunoglobulin light-chain origin. Scand J Immunol. 1983;18:557–60.
- Berg AM, Troxler RF, Grillone G, Kasznica J, Kane K, Cohen AS, Localized amyloidosis of the larynx: evidence for light chain composition. Ann Otol Rhinol Laryngol. 1993;102:884–9.
- Eriksson L, Sletten K, Benson L, Westermark P. Tumour-like localized amyloid of the brain is derived from immunoglobulin light chain. Scand J Immunol. 1993;37:623–6.
- Olsen KE, Sandgren O, Sletten K, Westermark P. Primary localized amyloidosis of the eyelid: two cases of immunoglobulin light chain-derived amyloid proteins, subtype lV respectevely lVI. Clin Exp Immunol. 1996;106:362–6.
- Hamidi Asl K, Liepnieks JJ, Nakamura M, Benson MD. Organ-specific (localized) synthesis of Ig light chain amyloid. J Immunol. 1999;162:5556–60.
- Olsen KE, Sletten K, Sandgren O, Olsson H, Myrvold K, Westermark P. What is the role of giant cells in localized AL amyloidosis? Amyloid. 1999;6:89–97.
- Zaky ZS, Liepnieks JJ, Rex DK, Cummings OV, Benson MD. Lambda II immunoglobulin light chain protein in primary localized rectal amyloidosis. Amyloid. 2007;14:299–304.
- Kaplan B, Martin BM, Boykov O, Gal R, Pras M, Shechtman I, Co-deposition of amyloidogenic immunoglobulin light and heavy chains in localized pulmonary amyloidosis. Virchows Arch. 2005;447:756–61.
- Buxbaum J. Mechanisms of disease: monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol Oncol Clin North Am. 1992;6:323–46.
- Ohmori T, Tabel R, Arita N, Kondo M, Kozawa T, Chikamori Y, Solitary plasmacytoma of the submandibular lymph node with stromal amyloid deposits. Acta Pathol Jpn. 1983;33:999–1007.
- Lucas DR, Knox F, Davies S. Apparent monoclonal origin of lymphocytes and plasma cells infiltrating ocular adnexal amyloid deposits: report of 2 cases. Br J Ophthalmol. 1982;66:600–6.
- Hagari Y, Mihara M, Hagari S. Nodular localized cutaneous amyloidosis: detection of monoclonality of infiltrating plasma cells by polymerase chain reaction. Br J Dermatol. 1996;135:630–3.
- Laeng RH, Altermatt HJ, Scheithauer BW, Zimmermann DR. Amyloidomas of the nervous system. A monoclonal B-cell disorder with monotypic amyloid light chain l amyloid production. Cancer. 1997;82:362–74.
- Miyamoto T, Kobayashi T, Makiyama M, Kitada S, Fujishima M, Hagari Y, Monoclonality of infiltrating plasma cells in primary pulmonary nodular amyloidosis: detection with polymerase chain reaction. J Clin Pathol. 1999;52:464–70.
- Setoguchi M, Hoshii Y, Kawano H, Ishihara T. Analysis of plasma cell clonality in localized AL amyloidosis. Amyloid. 2000;7:41–5.
- Ross P, Magro CM. Clonal light chain restricted primary intrapulmonary nodular amyloidosis. Ann Thorac Surg. 2005;80:344–7.
- Meijer JM, Schonland SO, Palladini G, Merlini G, Hegenbart U, Ciocca O, Sjögren's syndrome and localized nodular cutaneous amyloidosis. Arthritis Rheum. 2008;58:1992–9.
- Dyke PC, Demaray MJ, Delavan JW, Rasmussen RA. Pulmonary amyloidoma. Am J Clin Pathol. 1974;61:301–5.
- Barnes EL, Zafar T. Laryngeal amyloidosis. Clinicopathologic study of seven cases. Ann Otol Rhinol Laryngol. 1977;86:856–63.
- Michaels L, Hyams VJ. Amyloid in localized deposits and plasmacytomas of the respiratory tract. J Pathol. 1979;128:29–38.
- Schoen FJ, Alexander RW, Hood I, Dunn LJ. Nodular pulmonary amyloidosis. Arch Pathol Lab Med. 1980;104:66–9.
- Willén R, Willén H, Lindstedt E, Ekelund L. Localized primary amyloidosis of the ureter. Scand J Urol Nephrol. 1983;17:385–9.
- Da Costa P, Corrin B. Amyloidosis localized to the lower respiratory tract: probably immunoamyloid nature of the tracheobronchial and nodular pulmonary forms. Histopathology. 1985;9:703–10.
- Cordier JF, Loire R, Brune J. Amyloidosis of the lower respiratory tract. Clinical and pathologic features in a series of 21 patients. Chest. 1986;90:827–31.
- Grünewald K, Sepp N, Weyrer K, Lhotta K, Feichtinger H, Konwalinka G, Gene rearrangement studies in the diagnosis of primary systemic and nodular primary localized cutaneous amyloidosis. J Invest Dermatol. 1991;97:693–6.
- Mukhopadhyay S, Damron TA, Valente AL. Recurrent amyloidoma of soft tissue with exuberant giant cell reaction. Arch Pathol Lab Med. 2003;127:1609–11.
- Makheshwari AV, Muro-Cacho CA, Kransdorf MJ, Temple HT. Soft-tissue amyloidoma of the extremities: a case report and review of literature. Skeletal Radiol. 2009;38:287–92.
- Shirahama T, Cohen AS. Intralysosomal formation of amyloid fibrils. Am J Pathol. 1975;81:101–16.
- Helming L, Gordon S. The molecular basis of macrophage fusion. Immunobiology. 2008;212:785–93.
- Enelow RI, Sullivan GW, Carper HT, Mandell GL. Induction of multinucleated giant cell formation from in vitro culture of human monocytes with interleukin-3 and interferon-gamma: comparison with other stimulating factors. Am J Respir Cell Mol Biol. 1992;6:57–62.
- Fais S, Burgio VL, Silvestri M, Capobianchi MR, Pacchiarotti A, Pallone F. Multinuclear giant cell generation induced by interferon-gamma. Changes in the expression and distribution of the intercellular adhesion molecule-1 during macrophage fusion and multinucleated giant cell formation. Lab Invest. 1994;71:737–44.
- McNally AK, Anderson JM. Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol. 1995;147:1487–99.
- Dugast C, Gaudin A, Toujas L. Generation of multinucleated giant cells by culture of monocyte-derived macrophages with IL-4. J Leukoc Biol. 1997;61:517–21.
- Jenney CR, DeFife KM, Colton E, Anderson JM. Human monocyte/macrophage adhesion, macrophage motility, and IL-4-induced foreign body giant cell formation on silane-modified surfaces in vitro. J Biomed Mater Res. 1998;41:171–84.
- Helming L, Winter J, Gordon S. The scavenger receptor CD36 plays a role in cytokine-induced macrophage fusion. J Cell Sci. 2009;122:453–9.
- McNally AK, Anderson JM. Foreign body-type multinucleated giant cells induced by interleukin-4 express select lymphocyte co-stimulatory molecules and are phenotypically distinct from osteoclasts and dendritic cells. Exp Mol Pathol. 2011;91:673–81.
- Ikeda T, Ikeda K, Sasaki K, Kawakami K, Hatake K, Kaji Y, IL-13 as well as IL-4 induces monocytes/macrophages and a monoblastic cell line (UG3) to differentiate into multinucleated giant cells in the presence of M-CSF. Biochem Biophys Res Commun. 1998;253:265–72.
- Zhu J, Qiu Y, Valobra M, Qiu S, Majumdar S, Matin D, Plasma cells and IL-4 in chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:1125–33.
- Rodriguez A, MacEwan SR, Meyerson H, Kirk JT, Anderson JM. The foreign body reaction in T-cell-deficient mice. J Biomed Mater Res. 2009;90A:106–13.
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–30.
- Kanoh T, Suzuki K, Kawaguchi S. Multifocal nodular AL amyloidosis in primary Sjögren's syndrome. Rinsho Ketsueki. 1998;39:1157–62; Japanese.
- Götz J, Eckert A, Matamales M, Ittner LM, Liu X. Modes of Aβ toxicity in Alzheimer's disease. Cell Mol Life Sci. 2011;68:3359–75.
- Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid and diabetes mellitus. Physiol Rev. 2011;91:795–826.
- Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, Serum N-terminal and pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107:2440–5.
- Westermark GT, Sletten K, Westermark P. Alkali-degradation of amyloid: an ancient method useful for making monoclonal antibodies against amyloid fibril proteins. Scand J Immunol. 2009;70:535–40.