Abstract
Platelet-derived growth factor (PDGF) isoforms are important mitogens for different types of mesenchymal cells, which have important functions during the embryonal development and in the adult during wound healing and tissue homeostasis. In tumors, PDGF isoforms are often over-expressed and contribute to the growth of both normal and malignant cells. This review focuses on tumors expressing PDGF isoforms together with their tyrosine kinase receptors, thus resulting in autocrine stimulation of growth and survival. Patients with such tumors could benefit from treatment with inhibitors of either PDGF or PDGF receptors.
Key words::
Introduction
Members of the platelet-derived growth factor (PDGF) family are major mitogens for connective tissue cells, glial cells, and certain other cell types. Structurally they are homodimers of related A-, B-, C-, and D-polypeptide chains, and an AB heterodimer (Citation1).
PDGF isoforms exert their cellular effects by binding to α- and β-tyrosine kinase receptors. Ligand binding induces dimerization of the receptors; the α-receptor binds all PDGF chains except the D-chain, whereas the β-receptor binds the B- and D-chains. Thus, the different PDGF isoforms induce different homo- and heterodimeric complexes of α- and β-receptors. In the dimeric receptor complexes, receptors phosphorylate each other in trans positions on specific tyrosine residues, which allows binding and activation of SH2-domain-containing signaling molecules (Citation1). Some of these have enzymatic activities, such as phospholipase Cγ (PLCγ), the tyrosine kinase Src, the protein tyrosine phosphatase SHP-2, and the GTPase activating protein for Ras (RasGAP). Alternatively, they are adaptor molecules forming complexes with enzymes, such as Grb2, which forms a complex with SOS1, a nucleotide exchange molecule that activates Ras, and the p85 regulatory subunit of phosphatidylinositol 3'-kinase (PI3-kinase), which forms a complex with the p110 catalytic subunit. In addition, members of the STAT family of transcription factors bind to activated PDGF receptors, as do the adaptor molecules Grb7, Shc, Nck, and Crk.
PDGF isoforms stimulate proliferation, survival, chemotaxis, and differentiation of cells. They have important functions during embryonic development (Citation2), and in the adult during wound healing (Citation3) and in the control of interstitial fluid pressure (Citation4). Over-activity of PDGF has been linked to several pathological conditions, including malignancies and other conditions involving an excess cell proliferation, such as fibrotic conditions and atherosclerosis (Citation1).
PDGF isoforms are often over-expressed in malignancies and contribute to the growth of certain tumor types as well as to non-transformed cells in solid tumors, such as pericytes and smooth muscle cells of vessels and of stromal fibroblasts. Certain PDGF receptor-bearing tumor cells produce PDGF isoforms, which stimulate cell growth and survival in an autocrine manner. The aim of the present communication is to review the involvement of autocrine PDGF stimulation in malignancies.
Homology between PDGF and the Sis oncogene product
PDGF was originally purified from human platelets (Citation5–8). When the purified PDGF was subjected to amino acid sequencing, a homology to the product of the oncogene sis was noticed (Citation9,10). In fact, the gene for the B-chain of PDGF has been transduced by the simian sarcoma virus (SSV), and infected cells were shown to produce large amounts of a PDGF-BB-like growth factor (Citation11,12). Evidence that the autocrine stimulation is crucial for cell transformation was rapidly obtained, e.g. it was shown that the transformed phenotype of SSV-transformed fibroblasts can be normalized by inhibitory PDGF antibodies (Citation13).
The discovery of the homology between PDGF and Sis was rapidly followed by additional findings of homologies between products of retroviral oncogenes and growth factor receptors, as well as with components of their intracellular pathways. Together, these observations provided strong support for the hypothesis that oncogenes transform cells by subverting the mitogenic pathways of growth factors (Citation14). Furthermore, the findings triggered intensive efforts to investigate if autocrine mechanisms occur also in human malignancies.
Autocrine PDGF stimulation in human glioma, osteosarcoma, and other tumor types
During the 1970s, a hypothesis was formulated that tumor cells may make their own growth factors and thereby be self-sufficient with regard to growth stimulatory signals (Citation15). To explore this hypothesis, a growth factor produced by the human osteosarcoma cell line U-2OS was purified (Citation16,17). Initial characterization revealed that this factor was similar but not identical to PDGF purified from platelets; sequencing showed that it was in fact PDGF-AA, whereas platelets contain mainly PDGF-AB (Citation18). Autocrine PDGF receptor activation was demonstrated in U-2OS cells, but effects on growth stimulation were more difficult to show, probably because of the numerous other mutations these cells have acquired during many years of in-vitro culturing (Citation19). Similar analyses of glioma cell lines revealed that co-expression of PDGF isoforms and PDGF receptors is common, suggesting autocrine mechanisms (Citation20–24). Furthermore, analysis of expression of PDGF isoforms and PDGF receptors in sections of human glioblastomas provided evidence that both types of PDGF receptors are involved in autocrine and paracrine growth stimulation of gliomas, affecting different cellular compartments, however. Thus, the α-receptor is expressed mainly in the tumor cells, whereas the β-receptor is expressed in cells of the supporting stroma (Citation25–29). The levels of expression of PDGF ligands as well as receptors are higher in more malignant tumors, suggesting that autocrine and paracrine effects of PDGF increase with degree of malignancy. Gliomas are probably the tumor type in which PDGF autocrine mechanisms are most important, and nearly 30% of human gliomas show over-activity of PDGF receptor signaling (Citation30). Gliomas are discussed further by Lindberg and Holland (Citation31) in this series.
PDGF has also been implicated in autocrine mechanisms of other tumor types. Thus, malignancy-dependent expressions of PDGF and PDGF receptors were observed in sarcomas (Citation32,33). Co-expression of PDGF and PDGF receptors has also been reported in an AIDS-related Kaposi's sarcoma (Citation34) and in meningeomas (Citation35,36). Moreover, an autocrine PDGF-BB/PDGF β-receptor loop was found to mediate survival of large granular lymphocyte leukemia of both T- and NK-cell origin (Citation37). In addition, co-expression of PDGF-AA and PDGF α-receptor in the epithelial part of Wilms' tumor of the kidney is common; in contrast to other tumors with autocrine PDGF stimulation, the expression of PDGF-A and PDGF α-receptor in Wilms' tumor correlates to favorable prognosis (Citation38).
Screening of 637 human tumor-derived cell lines revealed that only 2 were sensitive to sunitinib, an inhibitor which targets the PDGF receptor kinases as well as other kinases, i.e. a non-small-cell lung cancer and a rhabdomyosarcoma (Citation39). Both these cell lines co-express the PDGF α-receptor and PDGF-C. Moreover, investigation of a large number of human and mouse rhabdomyosarcomas revealed that the PDGF α-receptor is a target of the Pax3/Fkhr chimeric transcription factor, which is found in a majority of this tumor type (Citation40). This results in over-expression of the PDGF α-receptor, which is correlated to poor prognosis (Citation41), and often occurs together with expression of PDGF-A or -C, thus creating autocrine loops.
In the rare skin tumor dermatofibrosarcoma protuberans (DFSP), a specific genetic perturbation is responsible for the establishment of autocrine PDGF stimulation. Thus, in this disease the PDGF-B gene is fused to the collagen 1A1 gene, leading to the production of a collagen 1A1/PDGF-B fusion protein, which is processed to mature PDGF-BB that activates PDGF receptors on fibroblasts in an autocrine manner (Citation42–45).
Taken together, there are thus now several examples of autocrine mechanisms involving PDGF and PDGF receptors in different forms of malignancies.
Intracrine versus extracrine PDGF stimulation
While all PDGF isoforms are produced as inactive precursor molecules, the N-terminal parts of PDGF-A and -B are removed already intracellularly by furin-like proteases. In contrast, PDGF-C and -D are secreted as latent precursor molecules containing N-terminal CUB domains, which need to be cleaved off by proteases before these PDGF isoforms can bind to receptors. Thus, tissue plasminogen activator (tPA) has been shown to cleave and activate PDGF-CC (Citation46) and urokinase plasminogen activator (uPA) PDGF-DD (Citation47), but other proteases may also be involved. Thus, in cells which express PDGF-AA, -AB, or -BB together with PDGF receptors, the active ligands will be present together with the extracellular, ligand-binding parts of the receptors in the endoplasmic reticulum, Golgi apparatus, and secretory vesicles (). In such cells, there is evidence that receptors are activated intracellularly before they have obtained their mature glycosylation and have reached the cell surface (Citation48-50). However, several observations support the notion that only a subset of the intracellular pathways can be activated in intracellular vesicles, and that the ligand–receptor complex needs to reach the cell surface before an efficient mitogenic signal is initiated () (Citation13,51–53). One mechanistic explanation could be that certain signal transduction components, critical for the mitogenic response, are located at the plasma membrane. In support of this possibility, activated PDGF β-receptors in sis-transformed cells were found to interact with certain signaling molecules intracellularly, e.g. PLCγ, RasGAP, and PI3-kinase, whereas efficient interaction with SHP-2, Grb2, and Src occurred only after the receptor had reached the plasma membrane (Citation54).
Figure 1. A: A PDGF receptor-bearing cell that does not produce PDGF itself responds to PDGF via activation of receptors at the cell surface. This initiates activation of intracellular signaling pathways leading to cell growth, proliferation, and survival. B: In a PDGF receptor-bearing cell that produces PDGF-A or -B, the ligands will meet and activate the receptors already in the ER, Golgi, and secretary vesicles. Whereas some intracellular signaling pathways are activated intracellularly, other pathways important for mitogenesis are not activated until the ligand–receptor complex reaches the cell membrane.
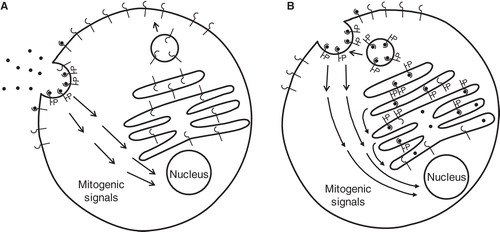
Interestingly, the C-terminals of PDGF-B and a long splice variant of PDGF-A contain basic amino acid sequences, which mediate binding to extracellular matrix molecules (Citation55,56). Moreover, proteolytic activation of PDGF-DD was found to reveal a retention motif mediating interactions with pericellular components (Citation57). The presence of these retention motifs restricts the action of the PDGF isoforms and thus enhances autocrine and paracrine stimulation of cells in the local environment, at the expense of stimulation of cells at a distance.
Mutations of genes for PDGF receptors in human malignancies
In addition to classical autocrine stimulation, there are examples of mutations in the genes for PDGF receptors, which cause their activation and promote tumorigenesis. Thus, in chronic myelomonocytic leukemia (CMML), the kinase domain of the PDGF β-receptor is fused to different partners, e.g. the transcription factor Tel or rabaptin 5, which have in common that they can dimerize or oligomerize (Citation58,59). Analogously, in patients with idiopathic hypereosinophilia, the kinase domain of the PDGF α-receptor is fused to FIP1L1 (Citation60,61). A similar FIP1L1-PDGF α-receptor fusion has been observed in systemic mastocytosis (Citation62). In addition to the juxtaposition of the kinase domains of the receptors, the loss of regulatory sequences in the juxtamembrane (Citation63) and transmembrane (Citation64) domains is important for the increased autophosphorylation and initiation of signaling pathways promoting cell growth and survival.
A majority of gastrointestinal stromal tumors (GISTs) have activating point mutations in Kit, a tyrosine kinase receptor for stem cell factor, which is structurally similar to PDGF receptors. However, some of these tumors are instead driven by point mutations in the PDGF α-receptor gene affecting the control mechanisms of the kinase; such mutations make the receptor kinase constitutively active (Citation65).
Finally, the PDGF α-receptor has been found to be amplified in a subset of glioblastoma tumors (Citation66–68), in anaplastic oligodendrogliomas (Citation69), in esophageal squamous cell carcinoma (Citation70), and in pulmonary artery intimal sarcoma (Citation71). The large amounts of receptors expressed on such cells may cause constitutive activation of the receptors, since the high receptor density may promote ligand-independent receptor–receptor interactions. Alternatively, the cells at least become very sensitive to PDGF stimulation. In addition, a transforming deletion mutant of the PDGF α-receptor has been described in gliomas (Citation72).
Treatment of PDGF-dependent tumors with PDGF antagonists
Several types of PDGF antagonists have been developed, including antibodies and aptamers against PDGF or PDGF receptors, and low-molecular-weight inhibitors of the receptor kinases as reviewed by Östman and Heldin (Citation73). A few kinase inhibitors have been approved for clinical use, including imatinib, which is fairly selective for PDGF receptors, Kit and Abl tyrosine kinases, and sorafinib and sunitinib which have broader specificities and inhibit also other kinases.
In the rather rare tumor types in which mutations of PDGF or PDGF receptor genes drive tumorigenesis, e.g. DFSP (Citation74–76), CMML (Citation77,78), hypereosinophilic syndrome (Citation79), and GIST (Citation80), treatment with imatinib has been shown to have beneficial effects. Whereas inhibition of glioma cell growth by imatinib or other PDGF receptor kinase inhibitors has been observed in animal tumor models (Citation28,81,82), no clear benefit has been noticed when glioma patients have been treated with PDGF receptor kinase inhibitors, suggesting that in human glioma perturbations of PDGF signaling pathways are not of unique importance. Other genetic alterations have, however, occurred, which also drive tumor cell growth and survival.
In addition to a direct effect on tumor cells with over-active PDGF signaling, PDGF antagonists have also been shown be useful to target cells of the stroma of solid tumors (Citation83).
Multistep induction of malignancies
It is well established that the development of a fully malignant tumor requires several genetic or epigenetic alterations. It is therefore likely that autocrine PDGF stimulation is an initial event in tumor progression, which leads to an expansion of cells that are targets for neoplastic transformation. Alternatively, the aberrant production of PDGF may stimulate the growth of cells that are already genetically altered. In the case of SSV-induced transformation, there is also the possibility that the sis oncogene is inserted in regions of the genome where it affects the expression of oncogenes or tumor suppressor genes.
The possible importance of insertional mutagenesis in PDGF-driven gliomagenesis has been explored in Bengt Westermark's laboratory using a recombinant Moloney leukemia virus encoding the PDGF B-chain (Citation84). Sixty-six common retroviral insertion sites were identified (Citation85), and retroviral insertion was found to affect the expression of a number of genes with a potential role in the regulation of glial cell growth and survival (Citation86). One of the targeted genes was the gene for cGMP-dependent protein kinase II; an anti-proliferative role of this kinase was demonstrated, which was lost during the loss-of-function retroviral insertion (Citation87). Another common integration site was a gain-of-function insertion in the gene for the transcription factor Sox10 (Citation88); over-expression of Sox10 was shown to enhance the tumorigenic activity of PDGF-B but did not alone induce gliomas. Integrations in the gene for Sox5 were also observed, but in this case suppression of Sox5 activity was correlated to gliomagenesis (Citation89). Loss-of-function retroviral insertion in the gene for p190RhoGAP was also shown to promote glioma development, most likely via loss of control of Rho signaling (Citation90).
In high-grade oligodendrogliomas, perturbation of PDGF, or epidermal growth factor, signaling is often accompanied by homozygous deletion of the INK4a-ARF locus (Citation91,92). This locus encodes the tumor suppressor proteins p16INK4a and p14ARF, which control the Rb and p53 pathways, respectively. In PDGF-induced oligodendroglioma development, loss of Ink4a was found to render astrocytes susceptible to PDGF-BB-induced tumorigenesis, whereas loss of Arf caused increased malignancy (Citation93). Taken together, these observations illustrate that over-activity of PDGF receptors need to be complemented by other cellular alterations to promote tumorigenesis.
Autocrine PDGF stimulation during epithelial-mesenchymal transition
Epithelial cells normally do not contain PDGF receptors. However, epithelial tumors can undergo epithelial-mesenchymal transition (EMT), a change in phenotype which makes the tumor cells more invasive and prone to make metastases (Citation94). In vitro, EMT is promoted e.g. by stimulation by transforming growth factor-β (TGFβ), certain tyrosine kinase receptor ligands, and Notch. In conjunction with EMT, PDGF and PDGF receptors are induced (Citation95). Interestingly, the metastatic potential of mammary epithelial tumors was shown to be dependent on an autocrine PDGF/PDGF receptor loop; inhibition of PDGF by a dominant negative receptor or by imatinib inhibited metastasis in mouse models (Citation96). The invasiveness of human mammary carcinomas correlates to the expression of PDGF α- and β-receptors (Citation97), and earlier studies had shown that the expression of PDGF correlates with unfavorable prognosis (Citation98). Expression of PDGF and PDGF receptors also correlates to poor prognosis of lung carcinoma (Citation99). Moreover, TGFβ-induced EMT of mouse hepatocellular carcinoma was found to involve expression of PDGF-AA and PDGF α-receptor and the establishment of an autocrine loop (Citation99). In this tumor type, hypoxia was shown to induce PDGF-BB production via induction of HIF-1α (Citation100).
PDGF-D and the PDGF β-receptor have been implicated in autocrine mechanisms in prostate cancer cell lines (Citation101). Prostate cancer cells secrete matriptase, which activates PDGF-D by proteolytic removal of the CUB domain, thus inducing an autocrine stimulation. Moreover, immunohistochemical stainings of sections of human prostate cancers revealed co-staining of PDGF-D and matriptase (Citation101). In PC-3 prostate carcinoma cells, PDGF-DD has furthermore been shown to drive the EMT process by repressing miR-200 which targets ZEB1, ZEB2, and Snail2, critical components of the EMT transcriptional program (Citation102). A role for PDGF α-receptors in promoting metastasis of prostate cancer cells to bone has also been reported (Citation103), and targeting the PDGF α-receptor with a monoclonal antibody dramatically inhibited the growth of skeletal prostate cancer metastases in an animal model (Citation104). Evidence has been presented that in the latter case the PDGF α-receptor is not activated by ligand binding in a conventional way, but rather transactivated by an intracellular mechanism (Citation105).
Does autocrine PDGF stimulation occur in normal cells?
PDGF isoforms have important functions during embryonic development. Often the ligand is produced by epithelial or endothelial cells and acts on nearby mesenchymal cells in a paracrine manner (Citation2). There are examples of non-transformed cell types which both can express PDGF receptors and produce PDGF, e.g. smooth muscle cells, endothelial cells, and macrophages (Citation1,2). However, it is not clear whether normal cells express PDGF receptors and synthesize PDGF at the same time. If there are such examples of autocrine PDGF loops also in normal cells it is likely that they are transient and well controlled.
Future perspectives
The discovery that the Sis oncogene product is similar to PDGF-B led to the first demonstration of an oncogenic autocrine mechanism. Subsequent studies have shown that autocrine PDGF loops occur in human tumors, both in e.g. gliomas and sarcomas where the corresponding normal cell type expresses PDGF receptors, in epithelial cells that have undergone EMT, and in some rare tumors which aberrantly express the PDGF receptors. Examples of autocrine mechanisms involving other growth factors and cytokines are also accumulating. Thus, it is likely that autocrine stimulation is common in tumors.
In addition to autocrine stimulation, PDGF is involved in paracrine stimulation of normal cells in solid tumors; PDGF made by tumor cells or other cells can thus act on pericytes, smooth muscle cells, and endothelial cells, thereby promoting angiogenesis, as well as on stromal fibroblasts and myofibroblasts, thereby controlling the interstitial fluid pressure of tumors (Citation83). Paracrine mechanisms involving a number of different growth factors and cytokines with trophic effects on tumor cells as well as non-tumor cells have important roles in the balanced growth of tumor tissue and in the recruitment of other cell types to the tumor, including macrophages. Particularly M2 macrophages are well known to secrete many different growth factors and cytokines, thus creating a vicious cycle. The availability of sensitive and affordable microarray and proteomic techniques will make it possible in the future to perform systematic analysis of autocrine and paracrine mechanisms in human tumors. Such information will be important for optimal design of treatment.
Recent work supports the notion that tumor development is driven by a subpopulation of cells with self-regenerating capacity, so-called cancer stem cells. Importantly, PDGF-BB has been shown to promote expansion of neural stem/progenitor cells (Citation106) and to sustain self-renewal and tumorigenicity of glioma cancer-initiating cells by preventing oligodendrocyte differentiation (Citation107). Additional insights into the effect of PDGF on cancer stem cells are highly warranted.
The extensive autocrine and paracrine stimulations that occur in tumors, which are of crucial importance for the growth and survival of tumor cells, offer opportunities for selective treatment of tumor patients by targeting growth factors and their receptors. A few selective signal transduction antagonists have been approved for clinical use, and many others are under testing in clinical trials. It seems likely that such inhibitors will be useful tools in future treatment of tumor patients.
Acknowledgements
I would like to express my sincere thanks to Bengt Westermark for a more than 35-year-long very enjoyable collaboration and friendship. Bengt's vision and sharp analysis have been of utmost importance for all our work. I would also like to thank Ingegärd Schiller for help in the preparation of this manuscript.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Heldin C-H, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–316.
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312.
- Robson MC, Phillips LG, Thomason A, Robson LE, Pierce GF. Platelet-derived growth factor BB for the treatment of chronic pressure ulcers. Lancet. 1992;339:23–5.
- Rodt SÅ, Åhlén K, Berg A, Rubin K, Reed RK. A novel physiological function for platelet-derived growth factor-BB in rat dermis. J Physiol. 1996;495:193–200.
- Antoniades HN, Scher CD, Stiles CD. Purification of human platelet-derived growth factor. Proc Natl Acad Sci USA. 1979;76:1809–12.
- Deuel TF, Huang JS, Proffitt RT, Baenziger JU, Chang D, Kennedy BB. Human platelet-derived growth factor: purification and resolution into two active protein fractions. J Biol Chem. 1981;256:8896–9.
- Heldin C-H, Westermark B, Wasteson Å. Platelet-derived growth factor: purification and partial characterization. Proc Natl Acad Sci USA. 1979;76:3722–6.
- Raines EW, Ross R. Platelet-derived growth factor. I. High yield purification and evidence for multiple forms. J Biol Chem. 1982;257:5154–60.
- Waterfield MD, Scrace GT, Whittle N, Stroobant P, Johnsson A, Wasteson Å, Platelet-derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature. 1983;304:35–9.
- Doolittle RF, Hunkapiller MW, Hood LE, Devare SD, Robbins KC, Aaronson SA, Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science. 1983;221:275–7.
- Deuel TF, Huang JS, Huang SS, Stroobant P, Waterfield MD. Expression of a platelet-derived growth factor-like protein in simian sarcoma virus transformed cells. Science. 1983;221:1348–50.
- Johnsson A, Betsholtz C, von der Helm K, Heldin C-H, Westermark B. Platelet-derived growth factor agonist activity of a secreted form of the v-sis oncogene product. Proc Natl Acad Sci USA. 1985;82:1721–5.
- Johnsson A, Betsholtz C, Heldin C-H, Westermark B. Antibodies against platelet-derived growth factor inhibit acute transformation by simian sarcoma virus. Nature. 1985;317:438–40.
- Heldin C-H, Westermark B. Growth factors: mechanism of action and relation to oncogenes. Cell. 1984;37:9–20.
- Temin HM, Pierson RW Jr, Dulak NC. The role of serum in the control of multiplication of avian and mammalian cells in culture. In Rothblatt GH, Cristofalo VJ, editors. Growth, nutrition and metabolism of cells in culture. New York: Academic Press; 1972. p 50–81.
- Heldin C-H, Westermark B, Wasteson Å. Chemical and biological properties of a growth factor from human-cultured osteosarcoma cells: resemblance with platelet-derived growth factor. J Cell Physiol. 1980;105:235–46.
- Westermark B, Wasteson Å. The response of cultured normal glial cells to growth factors. In Luft R, Hall K, editors. Advances in metabolic disorders. New York: Academic Press; 1975. p 85–100.
- Heldin C-H, Johnsson A, Wennergren S, Wernstedt C, Betsholtz C, Westermark B. A human osteosarcoma cell line secretes a growth factor structurally related to a homodimer of PDGF A-chains. Nature. 1986;319:511–14.
- Betsholtz C, Westermark B, Ek B, Heldin C-H. Coexpression of a PDGF-like growth factor and PDGF receptors in a human osteosarcoma cell line: implications for autocrine receptor activation. Cell. 1984;39:447–57.
- Nistér M, Libermann TA, Betsholtz C, Pettersson M, Claesson-Welsh L, Heldin C-H, Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-α and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48:3910–18.
- Nistér M, Claesson-Welsh L, Eriksson A, Heldin C-H, Westermark B. Differential expression of platelet-derived growth factor receptors in human malignant glioma cell lines. J Biol Chem. 1991;266:16755–63.
- Vassbotn FS, Östman A, Langeland N, Holmsen H, Westermark B, Heldin C-H, Activated platelet-derived growth factor autocrine pathway drives the transformed phenotype of a human glioblastoma cell line. J Cell Physiol. 1994;158:381–9.
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–25.
- Shamah SM, Stiles CD, Guha A. Dominant-negative mutants of platelet-derived growth factor revert the transformed phenotype of human astrocytoma cells. Mol Cell Biol. 1993;13:7203–12.
- Guha A, Dashner K, Black PM, Wagner JA, Stiles CD. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer. 1995;60:168–73.
- Hermanson M, Nistér M, Betsholtz C, Heldin C-H, Westermark B, Funa K. Endothelial cell hyperplasia in human glioblastoma: coexpression of mRNA for platelet-derived growh factor (PDGF) B chain and PDGF receptor suggests autocrine growth stimulation. Proc Natl Acad Sci USA. 1988;85:7748–52.
- Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin C-H, Westermark B, Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–19.
- Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–35.
- Maxwell M, Naber SP, Wolfe HJ, Galanopoulos T, Hedley-Whyte ET, Black PM, Coexpression of platelet-derived growth factor (PDGF) and PDGF-receptor genes by primary human astrocytomas may contribute to their development and maintenance. J Clin Invest. 1990;86:131–40.
- Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4:e7752.
- Lindberg N, Holland E. PDGF in gliomas: more than just a growth factor? Ups J Med Sci. 2012;117:92–8.
- Smits A, Funa K, Vassbotn FS, Beausang-Linder M, af Ekenstam F, Heldin C-H, Expression of platelet-derived growth factor and its receptors in proliferative disorders of fibroblastic origin. Am J Pathol. 1992;140:639–48.
- Wang J, Coltrera MD, Gown AM. Cell proliferation in human soft tissue tumors correlates with platelet-derived growth factor B chain expression: an immunohistochemical and in situ hybridization study. Cancer Res. 1994;54:560–4.
- Stürzl M, Roth WK, Brockmeyer NH, Zietz C, Speiser B, Hofschneider PH. Expression of platelet-derived growth factor and its receptor in AIDS-related Kaposi sarcoma in vivo suggests paracrine and autocrine mechanisms of tumor maintenance. Proc Natl Acad Sci USA. 1992;89:7046–50.
- Maxwell M, Galanopoulos T, Hedley-Whyte ET, Black PM, Antoniades HN. Human meningiomas co-express platelet-derived growth factor (PDGF) and PDGF-receptor genes and their protein products. Int J Cancer. 1990;46:16–21.
- Todo T, Adams EF, Fahlbusch R, Dingermann T, Werner H. Autocrine growth stimulation of human meningioma cells by platelet-derived growth factor. J Neurosurg. 1996;84:852–8.
- Yang J, Liu X, Nyland SB, Zhang R, Ryland LK, Broeg K, Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood. 2010;115:51–60.
- Ghanem M, Nijman R, Safan M, van der Kwast T, Vansteenbrugge G. Expression and prognostic value of platelet-derived growth factor-AA and its receptor α in nephroblastoma. BJU Int. 2010;106:1389–93.
- McDermott U, Ames RY, Iafrate AJ, Maheswaran S, Stubbs H, Greninger P, Ligand-dependent platelet-derived growth factor receptor (PDGFR)-α activation sensitizes rare lung cancer and sarcoma cells to PDGFR kinase inhibitors. Cancer Res. 2009;69:3937–46.
- Taniguchi E, Nishijo K, McCleish AT, Michalek JE, Grayson MH, Infante AJ, PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27:6550–60.
- Armistead PM, Salganick J, Roh JS, Steinert DM, Patel S, Munsell M, Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: correlation with overall survival in 105 patients. Cancer. 2007;110:2293–303.
- O'Brien KP, Seroussi E, Dal Cin P, Sciot R, Mandahl N, Fletcher JA, Various regions within the α-helical domain of the COL1A1 gene are fused to the second exon of the PDGFB gene in dermatofibrosarcomas and giant-cell fibroblastomas. Gene Chrom Cancer. 1998;23:187–93.
- Shimizu A, O'Brien KP, Sjöblom T, Pietras K, Buchdunger E, Collins VP, The dermatofibrosarcoma protuberans-associated collagen type Iα1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res. 1999;59:3719–23.
- Simon M-P, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre J-M, Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95–8.
- Sjöblom T, Shimizu A, O'Brien KP, Pietras K, Dal Cin P, Buchdunger E, Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001;61:5778–83.
- Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204.
- Ustach CV, Kim HR. Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol Cell Biol. 2005;25:6279–88.
- Keating MT, Williams LT. Autocrine stimulation of intracellular PDGF receptors in v-sis transformed cells. Science. 1988;239:914–16.
- Huang SS, Huang JS. Rapid turnover of the platelet-derived growth factor receptor in sis-transformed cells and reversal by suramin. Implications for the mechanism of autocrine transformation. J Biol Chem. 1988;263:12608–18.
- Bejcek BE, Li DY, Deuel TF. Transformation by v-sis occurs by an internal autoactivation mechanism. Science. 1989;245:1496–9.
- Betsholtz C, Johnsson A, Heldin C-H, Westermark B. Efficient reversion of simian sarcoma virus-transformation and inhibition of growth factor-induced mitogenesis by suramin. Proc Natl Acad Sci USA. 1986;83:6440–4.
- Hannink M, Donoghue DJ. Autocrine stimulation by the v-sis gene product requires a ligand-receptor interaction at the cell surface. J Cell Biol. 1988;107:287–98.
- Huang JS, Huang SS, Deuel TF. Transforming protein of simian sarcoma virus stimulates autocrine growth of SSV-transformed cells through PDGF cell-surface receptors. Cell. 1984;39:79–87.
- Valgeirsdóttir S, Eriksson A, Nistér M, Heldin C-H, Westermark B, Claesson-Welsh L. Compartmentalization of autocrine signal transduction pathways in Sis transformed NIH 3T3 cells. J Biol Chem. 1995;270:10161–70.
- Afrakhte M, Nistér M, Östman A, Westermark B, Paulsson Y. Production of cell-associated PDGF-AA by a human sarcoma cell line: evidence for a latent autocrine effect. Int J Cancer. 1996;68:802–9.
- Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellström M, Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–40.
- Ehnman M, Li H, Fredriksson L, Pietras K, Eriksson U. The uPA/uPAR system regulates the bioavailability of PDGF-DD: implications for tumour growth. Oncogene. 2009;28:534–44.
- Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor β to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–16.
- Magnusson MK, Meade KE, Brown KE, Arthur DC, Krueger LA, Barrett AJ, Rabaptin-5 is a novel fusion partner to platelet-derived growth factor β receptor in chronic myelomonocytic leukemia. Blood. 2001;98:2518–25.
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14.
- Griffin JH, Leung J, Bruner RJ, Caligiuri MA, Briesewitz R. Discovery of a fusion kinase in EOL-1 cells and idiopathic hypereosinophilic syndrome. Proc Natl Acad Sci USA. 2003;100:7830–5.
- Pardanani A, Ketterling RP, Brockman SR, Flynn HC, Paternoster SF, Shearer BM, CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood. 2003;102:3093–6.
- Stover EH, Chen J, Folens C, Lee BH, Mentens N, Marynen P, Activation of FIP1L1-PDGFRalpha requires disruption of the juxtamembrane domain of PDGFRalpha and is FIP1L1-independent. Proc Natl Acad Sci USA. 2006;103:8078–83.
- Toffalini F, Hellberg C, Demoulin J-B. Critical role of the platelet-derived growth factor receptor (PDGFR) β transmembrane domain in the TEL-PDGFRβ cytosolic oncoprotein. J Biol Chem. 2010;285:12268–78.
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10.
- Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550–3.
- Kumabe T, Sohma Y, Kayama T, Yoshimoto T, Yamamoto T. Amplification of platelet-derived growth factor receptor gene lacking an exon coding for a portion of the extracellular region in a primary brain tumor of glial origin. Oncogene. 1992;7:627–33.
- Puputti M, Tynninen O, Sihto H, Blom T, Maenpaa H, Isola J, Amplification of KIT, PDGFRA, VEGFR2, and EGFR in gliomas. Mol Cancer Res. 2006;4:927–34.
- Smith JS, Wang XY, Qian J, Hosek SM, Scheithauer BW, Jenkins RB, Amplification of the platelet-derived growth factor receptor-A (PDGFRA) gene occurs in oligodendrogliomas with grade IV anaplastic features. J Neuropathol Exp Neurol. 2000;59:495–503.
- Arai H, Ueno T, Tangoku A, Yoshino S, Abe T, Kawauchi S, Detection of amplified oncogenes by genome DNA microarrays in human primary esophageal squamous cell carcinoma: comparison with conventional comparative genomic hybridization analysis. Cancer Genet Cytogenet. 2003;146:16–21.
- Zhao J, Roth J, Bode-Lesniewska B, Pfaltz M, Heitz PU, Komminoth P. Combined comparative genomic hybridization and genomic microarray for detection of gene amplifications in pulmonary artery intimal sarcomas and adrenocortical tumors. Genes Chromosomes Cancer. 2002;34:48–57.
- Clarke ID, Dirks PB. A human brain tumor-derived PDGFR-alpha deletion mutant is transforming. Oncogene. 2003;22:722–33.
- Östman A, Heldin C-H. Involvement of platelet-derived growth factor in disease: development of specific antagonists. Adv Cancer Res. 2001;80:1–38.
- Maki RG, Awan RA, Dixon RH, Jhanwar S, Antonescu CR. Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer. 2002;100:623–6.
- McArthur GA, Demetri GD, van Oosterom A, Heinrich MC, Debiec-Rychter M, Corless CL, Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866–73.
- Rubin BP, Schuetze SM, Eary JF, Norwood TH, Mirza S, Conrad EU, Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002;20:3586–91.
- Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–7.
- Magnusson MK, Meade KE, Nakamura R, Barrett J, Dunbar CE. Activity of STI571 in chronic myelomonocytic leukemia with a platelet-derived growth factor receptor fusion oncogene. Blood. 2002;100:1088–91.
- Gleich GJ, Leiferman KM, Pardanani A, Tefferi A, Butterfield JH. Treatment of hypereosinophilic syndrome with imatinib mesilate. Lancet. 2002;359:1577–8.
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9.
- Kilic T, Alberta JA, Zdunek PR, Acar M, Iannarelli P, O'Reilly T, Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res. 2000;60:5143–50.
- Uhrbom L, Hesselager G, Östman A, Nistér M, Westermark B. Dependence of autocrine growth factor stimulation in platelet-derived growth factor-B-induced mouse brain tumor cells. Int J Cancer. 2000;85:398–406.
- Pietras K, Sjöblom T, Rubin K, Heldin C-H, Östman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43.
- Uhrbom L, Hesselager G, Nistér M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–9.
- Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA. 2004;101:11334–7.
- Johansson FK, Goransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene. 2005;24:3896–905.
- Swartling FJ, Ferletta M, Kastemar M, Weiss WA, Westermark B. Cyclic GMP dependent protein kinase II inhibits cell proliferation, Sox9 expression and Akt phosphorylation in human glioma cell lines. Oncogene. 2009;28:3121–31.
- Ferletta M, Uhrbom L, Olofsson T, Ponten F, Westermark B. Sox10 has a broad expression pattern in gliomas and enhances platelet-derived growth factor-B-induced gliomagenesis. Mol Cancer Res. 2007;5:891–7.
- Tchougounova E, Jiang Y, Brasater D, Lindberg N, Kastemar M, Asplund A, Sox5 can suppress platelet-derived growth factor B-induced glioma development in Ink4a-deficient mice through induction of acute cellular senescence. Oncogene. 2009;28:1537–48.
- Wolf RM, Draghi N, Liang X, Dai C, Uhrbom L, Eklof C, p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev. 2003;17:476–87.
- Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473–9.
- Bigner SH, Rasheed BK, Wiltshire R, McLendon RE. Morphologic and molecular genetic aspects of oligodendroglial neoplasms. Neuro Oncol. 1999;1:52–60.
- Tchougounova E, Kastemar M, Brasater D, Holland EC, Westermark B, Uhrbom L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene. 2007;26:6289–96.
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
- Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–69.
- Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest. 2006;116:1561–70.
- Seymour L, Dajee D, Bezwoda WR. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res Treat. 1993;26:247–52.
- Kawai T, Hiroi S, Torikata C. Expression in lung carcinomas of platelet-derived growth factor and its receptors. Lab Invest. 1997;77:431–6.
- Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25:3170–85.
- Lau CK, Yang ZF, Ho DW, Ng MN, Yeoh GC, Poon RT, An Akt/hypoxia-inducible factor-1α/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin Cancer Res. 2009;15:3462–71.
- Ustach CV, Huang W, Conley-LaComb MK, Lin CY, Che M, Abrams J, A novel signaling axis of matriptase/PDGF-D/ss-PDGFR in human prostate cancer. Cancer Res. 2010;70:9631–40.
- Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Mir-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27:1712–21.
- Dolloff NG, Shulby SS, Nelson AV, Stearns ME, Johannes GJ, Thomas JD, Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor. Oncogene. 2005;24:6848–54.
- Russell MR, Jamieson WL, Dolloff NG, Fatatis A. The receptor for platelet-derived growth factor as a target for antibody-mediated inhibition of skeletal metastases from prostate cancer cells. Oncogene. 2009;28:412–21.
- Russell MR, Liu Q, Lei H, Kazlauskas A, Fatatis A. The receptor for platelet-derived growth factor confers bone-metastatic potential to prostate cancer cells by ligand- and dimerization-independent mechanisms. Cancer Res. 2010;70:4195–203.
- Erlandsson A, Brannvall K, Gustafsdottir S, Westermark B, Forsberg-Nilsson K. Autocrine/paracrine platelet-derived growth factor regulates proliferation of neural progenitor cells. Cancer Res. 2006;66:8042–8.
- Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492–503.