Abstract
Myc proteins are often deregulated in human brain tumors, especially in embryonal tumors that affect children. Many observations have shown how alterations of these pleiotropic Myc transcription factors provide initiation, maintenance, or progression of tumors. This review will focus on the role of Myc family members (particularly c-myc and Mycn) in tumors like medulloblastoma and glioma and will further discuss how to target stabilization of these proteins for future brain tumor therapies.
Introduction
The Myc family comprises the transcription factors c-myc, Mycn, and Mycl (Citation1). Myc proteins are important for normal development, especially c-myc and Mycn which are embryonically lethal when knocked out (Citation2-4) in transgenic mice. Transgenic mice present a severely diminished forebrain and hindbrain when Mycn is specifically abrogated in nestin-positive neural stem and precursor cells (NSCs) of the developing brain (Citation5), while a similar conditional c-myc knock-out moderately impairs brain growth (Citation6). Nestin-driven transgenes for double c-myc and Mycn depletion have a nervous system phenotype that is much more severe than either Myc gene knock-out alone (Citation7,8). These findings collectively suggest that Myc proteins are essential for brain development. Overexpression of c-myc in nestin-positive NSCs promotes cell proliferation (Citation9), and many human tumors including brain tumors express high levels of or show gene amplifications of Myc family members. This is true for medulloblastoma (Citation10–12) and glioma (Citation13–15), the most common malignant types of brain tumors in children and adults, respectively. It is also true for other brain tumor types, like primitive neuroectodermal tumors (PNETs) (Citation16). Similarly, overexpressed c-myc or Mycn have been found to initiate different types of brain tumors like medulloblastoma (Citation17–20), PNETs (Citation21), and glioma (Citation22,23) in mice. In most of these cases Myc genes generate tumors after a rather long latency or in combination with other oncogenes (like Ras, Akt, Shh, or beta-catenin) or tumor suppressor genes (e.g. p53, Ptc, Ink4c). This suggests a necessary involvement of one or more additional transforming events before a full-blown cancer can develop. It is not known in detail how Myc proteins use other cancer genes to induce these tumors and if there is a difference in how the different Myc proteins are involved in brain tumor initiation. This is also difficult to study as Myc genes target a large number of other genes and regulate important cellular mechanisms like proliferation, apoptosis, DNA repair, metabolism, ribosome biogenesis, and protein synthesis (Citation24). Clearly, Myc proteins can use different strategies in their role as master regulators of cell proliferation and in tumor maintenance.
Medulloblastoma biology
Brain tumors represent the most common solid tumor of childhood. The embryonal tumor medulloblastoma (MB) is next to leukemia the most common malignancy in children (Citation25). Treatment includes surgery, radiation, and chemotherapy, which cures about 70% (Citation26), although survivors can have severe long-term side-effects following this treatment. MB can be categorized into different subtypes (Citation27) presenting a desmoplastic or nodular, classic, and large cell/anaplastic (LCA) pathology, where the LCA subtype shows monomorphic cells with large nuclei and correlates with the poorest outcome. Advanced genomic and transcriptional profiling has recently offered reliable molecular classification of human MB (Citation28–31). This provides a better classification of these childhood tumors, which will facilitate better strategies for improved patient treatment. Abnormal activation of molecular pathways like Sonic Hedgehog (SHH) and Wingless (WNT) signaling contributes to tumorigenesis in some patients; however, the majority show no abnormalities in these pathways and are referred to as Group 3 or Group 4 MB (Citation32). Wnt signaling abnormalities involving beta-catenin nuclear staining occur in 10%–15% of patients and have been described as a marker for favorable outcome (Citation33,34). Such tumors have recently been found to originate from BLBP-positive cells of the lower rhombic lip structure in the developing dorsal brain stem (Citation35). Another developmental pathway believed to have a critical role in MB maintenance is Notch. Inhibiting Notch suppresses MB in vivo by apoptosis and may prove effective in MB therapy (Citation36,37), even if Notch is not required in Shh-dependent MB subtypes (Citation38,39). Shh-dependent MB only represents about 25% of MB cases. Nevertheless there are numerous reports of Shh-dependent models that recapitulate human Shh-dependent MB in mice (see recent review (Citation40)); the first were models that activated the pathway through loss of Patched (Citation41). Amplification of MYC or MYCN occurs in about 10% of human MB and correlates with a 5-year overall survival of only 13% (Citation11). Both MYC and MYCN amplifications further associate with an aggressive LCA medulloblastoma pathology (Citation10,42,43). MYCN is expressed at high levels in SHH-associated MB (Citation10) and is actually essential for Shh-dependent tumors in mouse models of this disease (Citation17). However, most Shh-independent human MB also express MYCN (Citation20,44,45). Indeed, in a model where human MYCN drives MB formation from the glutamate transporter 1 (Glt1) promoter in cerebellar cells, the majority of the developed brain tumors are actually Shh-independent (Citation20). This MB model thus correlates with recent findings (Citation34,46) that suggest a majority of MYCN-amplified human MB are categorized as non-SHH Group 4 MB (Citation46).
Glioma biology
Glioma, the most common primary brain tumors in adults, is classified as grade I to IV according to the World Health Organization (WHO) (Citation47). Of high-grade gliomas (grade III and IV), glioblastoma (GBM) is the most common and most malignant (grade IV) tumor with dismal outcome. GBMs account for 60%–70% of malignant glioma (Citation48). While grade III gliomas (anaplastic astrocytoma, anaplastic oligodendroglioma, and anaplastic oligoastrocytoma) are characterized by increased cellularity, nuclear atypia, and proliferative activity, GBMs also contain areas of microvascular proliferation and necrosis. GBMs have a 5-year overall survival of less than 10% despite an increased survival effect from the use of the alkylating agent temozolomide following tumor resection and radiation (Citation49). An effort to stratify GBMs further and to enable a more individualized therapy is further to classify these malignant brain tumors into different subtypes characterized by molecular abnormalities (Citation50–52). The most recent efforts of genomic profiling from The Cancer Genome Atlas (TCGA) Research Network have defined four subgroups of GBM: proneural, neural, classical, and mesenchymal. These subgroups show signature aberrations where the gene expression of EGFR, NF1, and PDGFRA/IDH1 can help define a classical, mesenchymal, and proneural subtype, respectively (Citation52). There are many reports in which Myc proteins have been amplified or overexpressed in glioma (Citation53–56). Interestingly, MYCN showed high-level focal amplifications in a subset of GBM samples (Citation51), and MYC or MYCN is found to be amplified in almost half of brain cancers that have combined features of malignant glioma and primitive neuroectodermal tumors (MG-PNET) (Citation15). Interestingly, p53, which is directly or indirectly inactivated in 87% of GBMs (TCGA), represses MYC transcription by directly binding to the MYC promoter (Citation57). Most important, c-myc plays a critical role in the regulation and especially the proliferation of glioma stem cells, which are the putative cells of origin for these brain tumors (Citation58-60). This suggests that Myc proteins are master regulators also for these types of brain tumors.
Myc signaling in normal brain
Myc proteins are members of a large family of basic helix-loop-helix (bHLH) transcription factors. These protein members can form homo- or heterodimeric complexes with themselves or with other members of this family. One important prerequisite for Myc activity is its interaction with a dimerization partner, the bHLH protein Max () (Citation61). Such generated Myc-Max complexes bind to E-box sequences which are found in promoters of many genes that act to promote transcription and cell proliferation (Citation62,63). Through Miz1-complexes Myc and Max have been shown to have repressive functions (Citation64) inhibiting tumor suppressor genes/cell cycle regulators like p15Ink4b and also p21Cip1 (Citation65), where Myc represses p21Cip1 expression via a Miz1-dependent interaction with the p21 promoter (Citation66). Moreover, the partners Mad or Mnt (another bHLH protein) act in Max-dimeric complexes transcriptionally to repress genes associated with E-box sequences (Citation67).
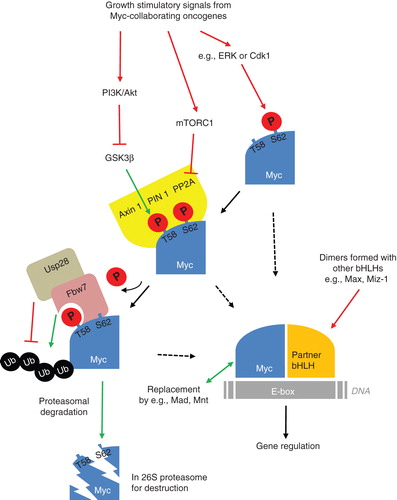
Figure 1. Schematic model of the regulatory pathways for Myc proteins from activating signals of collaborating oncogenes to protein destabilization and proteasomal degradation. Suggested activating pathways (in red) and inhibiting pathways (in green) with putative involvement in brain tumor development and maintenance.
Myc members are differently expressed in normal brain. Mycl has no reported phenotype in knock-out mice but has particularly high expression in the ventricular zone of the embryonic midbrain (Citation8,68). Mycn is highly expressed in developing forebrain and hindbrain (Citation5), while c-myc is expressed at lower levels (Citation6,8). In normal cerebellar granule cells, Mycn is important for cell replication, whereas Mycn protein degradation is essential for cell cycle exit (Citation69–71). Importantly, Mycn depletion in nestin-driven NSCs reduces both germinal zones of the developing cerebellum, including the neuroepithelial cells lining the fourth ventricle as well as the rhombic lip structure with the granule precursor cells forming the external germinal layer (Citation5). The depletion also leads to disrupted neuronal differentiation which correlates with the findings that Myc genes (including c-myc, Mycn, and Mycl) promote (but are not required for (Citation72)) the dedifferentiation processes for producing induced pluripotent stem (iPS) cells (Citation73,74). High expression of Myc proteins, at least when using v-myc (the retroviral homolog of c-myc) transduction, is important for immortalization and self-renewal of NSCs (Citation75,76). Interestingly, Mad proteins also appear to have a role in neuronal differentiation (Citation77). Mxi1, another member of the Mad family of proteins, can antagonize Myc oncoproteins in vivo (Citation78). Overexpression of Mxi1 in human glioma cell lines reduces their mitotic activity (Citation79), possibly through down-regulation of cyclin B1 expression (Citation80). The data suggest that Mxi1 that interacts with Max can antagonize Myc and thereby suppress glioma progression.
Myc protein stability
Myc proteins are extremely unstable with half-lives of only 20–30 min (Citation81). There are two phosphorylation sites on Myc proteins which are primarily responsible for stabilization (). First, serine (S62) is phosphorylated which leads to Myc stabilization (Citation82). The extracellular-regulated kinase 1, 2 (ERK) is known to mediate S62 phosphorylation (Citation83). However, in cerebellar neural precursors, the mitotic cyclin-dependent kinase 1 (Cdk1) is the priming kinase for Mycn at S62 (Citation70). Following S62 phosphorylation of Myc, threonine 58 (T58) is phosphorylated by glycogen synthase kinase 3 beta (GSK3β) which will target Myc for degradation (Citation84). Under normal conditions T58 is always phosphorylated after S62.
The turnover of Myc is mostly controlled by ubiquitin-mediated proteolysis (Citation85,86), which targets the specific Myc proteins with great precision. Specific ubiquitin-protein ligases (of type E3) like Fbw7 can recognize and bind to Myc, but only when T58 is phosphorylated alone. This requires dephosphorylation of S62 with the help of a prolyl isomerase, Pin1, that binds the phosphorylated sites and isomerizes Myc on proline 59 (Citation87). This provides a conformation change of Myc which enables protein phosphatase 2A (PP2A) to remove the phosphate group. Fbw7 can now recognize Myc with a single phosphorylation on T58 and send Myc for proteolytic degradation (Citation88) by ubiquitylating Myc on its amino-terminus. It is suggested that at least four ubiquitin groups need to be linked to a substrate in order for it to be recognized by a proteasome that will later dissect it into small peptide fragments (Citation89).
Increased stability of Myc proteins can explain the reported inconsistency between gene amplifications and protein levels of Myc found in solid tumors, like in breast cancer (Citation90,91). There are numerous reports of enhanced expression levels of Myc oncoproteins in many different types of tumors, that suggesting this is an important event for tumor initiation or an apparent advantage for tumor progression (reviewed in (Citation92)). Myc protein levels are elevated in subtypes of glioma (Citation14), but there are few reports in which a correlation between expression/amplification and protein levels of Myc proteins is assessed. Such a relationship would be required in order to understand fully the extent of putative Myc protein stabilization in these brain tumors.
Reports of mutations or alterations in genes that can regulate Myc protein stability are increasing (for a recent review, see (Citation93)). For example, Fbw7 can keep Myc levels low and therefore act as a tumor suppressor. In the absence of Fbw7, the levels of Myc and the activity of the protein will increase (Citation94), and mutations in Fbw7 prevent Myc degradation in T-cell acute lymphoblastic leukemia (T-ALL) (Citation95). With regard to the discussions of how brain tumor develops it is interesting to find that Fbw7 is highly expressed during normal brain development (Citation96,97), where it can possibly regulate Myc protein stability. Other ubiquitin ligases recognize Myc on other sites than Fbw7 does, like S phase kinase-associated protein 2 (Skp2). Skp2 is responsible for Myc protein turnover (Citation98) but is paradoxically also promoting Myc activity by functionally amplifying the Myc response (Citation99). How Skp2 is working like an oncogene and how it correlates with Myc is not fully known (Citation93), but Skp2 is actually associating with Myc target genes when Myc is active (Citation100). Other proteins involved in regulating stability of Myc proteins are also mutated in cancer, like Usp28, which has been found frequently up-regulated in colon adenocarcinomas (Citation101). This soluble deubiquitylating enzyme (DUB) stabilizes Myc by removing linked ubiquitin chains that have been conjugated by Fbw7 (). TRIM32 is another ubiquitin ligase that is asymmetrically distributed during NSC division and is inhibiting cell proliferation by promoting Myc degradation and neuronal differentiation (Citation102). However, whether TRIM32 can also regulate proliferation by differentiating brain tumor cells is currently not known.
Myc mutants
A mutation causing a switch from threonine to alanine on residue 58 was discovered in the transforming v-myc viral gene and in Myc or Mycn in Burkitt's lymphoma patients (Citation103,104). This mutation prevents the phosphorylation from GSK3β that is so important for Fbw7 recognition which can lead to proteolytic degradation of Myc proteins. When searching the Catalogue of Somatic Mutations In Cancer (COSMIC) (Citation105), very few mutations for Myc genes are found in the category of central nervous system (CNS) tumors (). Here, only one mutation in MYC and two (with one being a silent mutation) in MYCN were reported (from The Cancer Genome Atlas (TCGA) genotyping screen (Citation51)). Whether any of the non-silent mutations () could affect Myc protein function or stability has not been reported.
Table I. Few mutations of Myc genes are found in brain tumor samplesa.
Myc and Ras collaborate to transform cells
Expression of c-myc together with a co-operative oncogene like Ras is necessary for the stable transformation of primary or early-passage fibroblasts (Citation106). Ras can stabilize Myc proteins in tumors. One example is through high ERK-mediated Myc S62 phosphorylation in Ras-transformed cells (Citation83). Different activating mutations of Ras promote ERK-mediated phosphorylation and are implicated in brain tumors, but it is important to distinguish if and how the different isoforms collaborate with Myc proteins in brain tumor formation and maintenance. For example, H-rasG12V can collaborate with v-myc to generate brain tumors in human fetal neural stem cells (Citation23), but H-rasQ61L cannot help v-myc to generate brain tumors in human adult neural stem cells in another report (Citation107). It is possible that fetal neural stem cells are more prone to transformation than adult neural stem cells or that the different activating mutations have different effects on brain tumor development. Myc proteins might require different isoforms of Ras proteins as collaborators in brain tumorigenesis. For example, when searching COSMIC (Citation105) there are reports of mutations in CNS tumors (like glioma and PNETs) only for N-ras (8/1017) and K-ras (8/1054), but not for H-ras (Citation108). Mutations found in K-ras and N-ras were at residues 12 or 61. For N-ras it is evident that all mutations at residue 12 (4/1017) were found only in glioma samples, while mutations at residue 61 (4/1017) were found only in PNET or medulloblastoma. Interestingly, in the Myc-Ras collaboration, Myc represses the cellular senescence induced by Ras. This repression of senescence by Myc requires phosphorylation of Myc at S62 by the cell cycle kinase Cdk2 that is needed for inhibiting cellular senescence induced by c-myc (Citation109,110). Many oncogenes like Ras (Citation22,23), Akt (Citation22), Shh (Citation17), or beta-catenin (Citation21) can be involved in collaborating with Myc proteins to induce brain tumors. This property of Myc proteins to launch collaborations during tumor formation warrants more high-throughput screens in order to identify critical Myc collaborating genes using previously successful strategies with retroviral tagging (Citation111,112) or Sleeping Beauty techniques (Citation113–116) in mice.
How can we inhibit Myc proteins in brain tumors?
Many tumors show addiction to Myc oncoproteins (Citation117). This is also true in Mycn-driven medulloblastoma models in which inhibition of Mycn will result in total tumor regression and cellular senescence (Citation20). In glioma where suppressor genes like p53 and Pten are inactivated, c-myc is essential for tumor maintenance, and c-myc inhibition will suppress tumors by promoting differentiation of the glioma cells (Citation60). It is evident that Myc proteins are validated targets for cancer therapies (as reviewed in (Citation118,119)). Targeting these transcription factors that lack clear binding domains have always proved difficult, and using short interfering RNAs (like specific short hairpin RNAs) that target Myc directly is not yet a treatment option for patients. There are, however, small molecular drugs that inhibit Myc-Max interactions that are effective in cancers like human acute myeloid leukemia (Citation120-122). Promoting Mad, another bHLH protein that antagonizes Myc, would be another promising approach in brain tumor therapy (Citation123).
There are other ways to induce growth arrest and senescence in childhood medulloblastoma and in atypical teratoid/rhabdoid tumor cells by using G-quadruplex interactive agents in order to disable c-myc at the promoter level (Citation124). It is also possible to target regulatory components in which Myc controls ribosome biogenesis (Citation125). Myc can regulate transcription of ribosomal proteins through RNA polymerase II (RNA pol II) (Citation126,127). MYC participates in release of paused RNA pol II, as c-myc can bind positive elongation factor b (P-TEFb) and stimulate transcriptional elongation in cancer cells. Combined targeting of c-myc and P-TEFb could prove effective for tumors maintained by Myc proteins (Citation128). Another strategy is to force changes in chromatin modification controlled by Myc genes (Citation129). For example, histone lysine side-chain acetylation increased by c-myc can be effectively suppressed by inhibition of acetyl-lysine recognition domains (bromodomains) in multiple myeloma, a Myc-dependent hematologic cancer (Citation130).
Pathways downstream of receptor tyrosine kinases like MAPK/ERK and PI3K/Akt/mTOR that indirectly control Myc protein stability () are often overexpressed or altered in brain tumors. MAPK/ERK kinase inhibitors can dephosphorylate c-myc and reduce cell proliferation and anchorage-independent growth of rhabdomyosarcoma (Citation131), a soft tissue sarcoma in children. However, downstream of Shh signaling, Cdk1 rather than ERK is associated with S62 activity, at least in granule neuron precursors (Citation69,70) that could serve as medulloblastoma cells of origin (Citation13,132). The cyclin-dependent kinase sibling Cdk2 can also phosphorylate S62. Yet, while Cdk2 inhibitors will promote senescence in Myc-induced cells (Citation133), Cdk1 inhibitors (Citation134) can be used to promote the apoptotic effects induced by Myc proteins (Citation135). This is also the suggested cell death that temozolomide treatment promotes from c-myc via Akt signaling in O6-methylguanine-DNA methyl transferase (MGMT) expressing glioblastoma (Citation136). Akt activity also determines the sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating c-myc expression (Citation137). Moreover, mTOR exists in a complex, mTORC1, that directly phosphorylates and inhibits PP2A (Citation138) which (as described above and visualized in ) will lead to sustained Myc protein activity. Consequently, clinical inhibitors of PI3K/mTOR prove efficacy when used to degrade Mycn in neuroblastoma (Citation139,140), a childhood tumor thought to originate from the peripheral neural crest. Such PI3K/mTOR inhibitors are indeed also effective in suppressing glioma (Citation141) and medulloblastoma (Citation142). Other examples to target Myc protein stability include treatment with clinically available synthetic steroid drugs, like dexamethasone, that can be used to destabilize Mycn, leading to inhibited growth of Shh-dependent medulloblastoma (Citation143). Ultimately, drugs that can target regulators of the ubiquitin-proteasome system (reviewed in (Citation144)) can promote final degradation of long-lived and harmful Myc proteins also in brain tumor cells. To summarize, there are numerous observations about how Myc proteins co-ordinate cell transformation and many promising ideas on how to target these proteins in brain tumors, so let's keep on hunting.
Acknowledgements
Research in the author's laboratory has been supported by grants from The Swedish Childhood Cancer Foundation, The Swedish Cancer Society, The Pediatric Brain Tumor Foundation, The Swedish Research Council, Åke Wibergs stiftelse, Lions Cancerforskningsfond, and Stiftelsen Lars Hiertas Minne.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- Eilers M, Eisenman RN. Myc's broad reach. Genes Dev. 2008;22:2755–66.
- Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992;6:2235–47.
- Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6:2248–57.
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–82.
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–712.
- Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alboran IM, Olson JM, N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006;66:8655–61.
- Zindy F, Knoepfler PS, Xie S, Sherr CJ, Eisenman RN, Roussel MF. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc Natl Acad Sci USA. 2006;103:11579–83.
- Wey A, Knoepfler PS. c-myc and N-myc promote active stem cell metabolism and cycling as architects of the developing brain. Oncotarget. 2010;1:120–30.
- Fults D, Pedone C, Dai C, Holland EC. MYC expression promotes the proliferation of neural progenitor cells in culture and in vivo. Neoplasia. 2002;4:32–9.
- Eberhart CG, Kratz J, Wang Y, Summers K, Stearns D, Cohen K, Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol. 2004;63:441–9.
- Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009;27:1627–36.
- Aldosari N, Bigner SH, Burger PC, Becker L, Kepner JL, Friedman HS, MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children's Oncology Group. Arch Pathol Lab Med. 2002;126:540–4.
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–45.
- Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4:e7752.
- Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC, Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol. 2009;19:81–90.
- Rouah E, Wilson DR, Armstrong DL, Darlington GJ. N-myc amplification and neuronal differentiation in human primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1989;49:1797–801.
- Browd SR, Kenney AM, Gottfried ON, Yoon JW, Walterhouse D, Pedone CA, N-myc can substitute for insulin-like growth factor signaling in a mouse model of sonic hedgehog-induced medulloblastoma. Cancer Res. 2006;66:2666–72.
- Kessler JD, Hasegawa H, Brun SN, Yang ZJ, Dutton JW, Wang F, N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–70.
- Zindy F, Uziel T, Ayrault O, Calabrese C, Valentine M, Rehg JE, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67:2676–84.
- Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–72.
- Momota H, Shih AH, Edgar MA, Holland EC. C-Myc and beta-catenin cooperate with loss of p53 to generate multiple members of the primitive neuroectodermal tumor family in mice. Oncogene. 2008;27:4392–401.
- Lassman AB, Dai C, Fuller GN, Vickers AJ, Holland EC. Overexpression of c-MYC promotes an undifferentiated phenotype in cultured astrocytes and allows elevated Ras and Akt signaling to induce gliomas from GFAP-expressing cells in mice. Neuron Glia Biol. 2004;1:157–63.
- Lee JS, Lee HJ, Moon BH, Song SH, Lee MO, Shim SH, Generation of Cancerous neural stem cells forming glial tumor by oncogenic stimulation. Stem Cell Rev. 2011;Jul14 Epub ahead of print.
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–90.
- Davis FG, McCarthy BJ, Berger MS. Centralized databases available for describing primary brain tumor incidence, survival, and treatment: Central Brain Tumor Registry of the United States; Surveillance, Epidemiology, and End Results; and National Cancer Data Base. Neuro Oncol. 1999;1:205–11.
- Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5:209–18.
- Eberhart CG, Burger PC. Anaplasia and grading in medulloblastomas. Brain Pathol. 2003;13:376–85.
- Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–31.
- Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088.
- Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29:1424–30.
- Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–14.
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2011; [Epub ahead of print].
- Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666–70.
- Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–96.
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–9.
- Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–52.
- Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Phase I trial of MK-0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–34.
- Hatton BA, Villavicencio EH, Pritchard J, LeBlanc M, Hansen S, Ulrich M, Notch signaling is not essential in sonic hedgehog-activated medulloblastoma. Oncogene. 2010;29:3865–72.
- Julian E, Dave RK, Robson JP, Hallahan AR, Wainwright BJ. Canonical Notch signaling is not required for the growth of Hedgehog pathway-induced medulloblastoma. Oncogene. 2010;29:3465–76.
- Wu X, Northcott PA, Croul S, Taylor MD. Mouse models of medulloblastoma. Chin J Cancer. 2011;30:442–9.
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13.
- Eberhart CG, Kratz JE, Schuster A, Goldthwaite P, Cohen KJ, Perlman EJ, Comparative genomic hybridization detects an increased number of chromosomal alterations in large cell/anaplastic medulloblastomas. Brain Pathol. 2002;12:36–44.
- Stearns D, Chaudhry A, Abel TW, Burger PC, Dang CV, Eberhart CG. C-myc overexpression causes anaplasia in medulloblastoma. Cancer Res. 2006;66:673–81.
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42.
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28.
- Korshunov A, Remke M, Kool M, Hielscher T, Northcott PA, Williamson D, Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. 2011; [Epub ahead of print].
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507.
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66.
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73.
- TCGA research network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
- Herms JW, von Loewenich FD, Behnke J, Markakis E, Kretzschmar HA. C-myc oncogene family expression in glioblastoma and survival. Surg Neurol. 1999;51:536–42.
- Hui AB, Lo KW, Yin XL, Poon WS, Ng HK. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Lab Invest. 2001;81:717–23.
- Faria MH, Khayat AS, Burbano RR, Rabenhorst SH. C--MYC amplification and expression in astrocytic tumors. Acta Neuropathol. 2008;116:87–95.
- Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro Oncol. 2009;11:477–87.
- Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol. 2005;25:7423–31.
- Broaddus WC, Chen ZJ, Prabhu SS, Loudon WG, Gillies GT, Phillips LL, Antiproliferative effect of c-myc antisense phosphorothioate oligodeoxynucleotides in malignant glioma cells. Neurosurgery. 1997;41:908–15.
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769.
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33.
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72:233–45.
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:1149–51.
- Papoulas O, Williams NG, Kingston RE. DNA binding activities of c-Myc purified from eukaryotic cells. J Biol Chem. 1992;267:10470–80.
- Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3:392–9.
- Herold S, Herkert B, Eilers M. Facilitating replication under stress: an oncogenic function of MYC? Nat Rev Cancer. 2009;9:441–4.
- Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22:351–60.
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer. 2004;4:562–8.
- Hatton KS, Mahon K, Chin L, Chiu FC, Lee HW, Peng D, Expression and activity of L-Myc in normal mouse development. Mol Cell Biol. 1996;16:1794–804.
- Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–28.
- Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell. 2005;9:327–38.
- Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell. 2006;21:509–19.
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–6.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6.
- Kim SU, Park IH, Kim TH, Kim KS, Choi HB, Hong SH, Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease. Neuropathology. 2006;26:129–40.
- Villa A, Snyder EY, Vescovi A, Martinez-Serrano A. Establishment and properties of a growth factor-dependent, perpetual neural stem cell line from the human CNS. Exp Neurol. 2000;161:67–84.
- Queva C, Hurlin PJ, Foley KP, Eisenman RN. Sequential expression of the MAD family of transcriptional repressors during differentiation and development. Oncogene. 1998;16:967–77.
- Schreiber-Agus N, DePinho RA. Repression by the Mad(Mxi1)-Sin3 complex. Bioessays. 1998;20:808–18.
- Wechsler DS, Shelly CA, Petroff CA, Dang CV. MXI1, a putative tumor suppressor gene, suppresses growth of human glioblastoma cells. Cancer Res. 1997;57:4905–12.
- Manni I, Tunici P, Cirenei N, Albarosa R, Colombo BM, Roz L, Mxi1 inhibits the proliferation of U87 glioma cells through down-regulation of cyclin B1 gene expression. Br J Cancer. 2002;86:477–84.
- Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol. 1984;4:2486–97.
- Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene. 1994;9:59–70.
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14.
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12.
- Flinn EM, Busch CM, Wright AP. Myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Mol Cell Biol. 1998;18:5961–9.
- Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–26.
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–18.
- Schrader EK, Harstad KG, Matouschek A. Targeting proteins for degradation. Nat Chem Biol. 2009;5:815–22.
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102.
- Blancato J, Singh B, Liu A, Liao DJ, Dickson RB. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br J Cancer. 2004;90:1612–19.
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–8.
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–30.
- Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2011;110:77–106.
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–25.
- O'Neil J, Look AT. Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene. 2007;26:6838–49.
- Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, Fbw7 controls neural stem cell differentiation and progenitor apoptosis via Notch and c-Jun. Nat Neurosci. 2010;13:1365–72.
- Jandke A, Da Costa C, Sancho R, Nye E, Spencer-Dene B, Behrens A. The F-box protein Fbw7 is required for cerebellar development. Dev Biol. 2011;358:201–12.
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–88.
- Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci USA. 2001;98:5043–8.
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–200.
- Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. 2007;9:765–74.
- Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–25.
- Papas TS, Lautenberger JA. Sequence curiosity in v-myc oncogene. Nature. 1985;318:237.
- Hoang AT, Lutterbach B, Lewis BC, Yano T, Chou TY, Barrett JF, A link between increased transforming activity of lymphoma-derived MYC mutant alleles, their defective regulation by p107, and altered phosphorylation of the c-Myc transactivation domain. Mol Cell Biol. 1995;15:4031–42.
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8.
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602.
- Foroni C, Galli R, Cipelletti B, Caumo A, Alberti S, Fiocco R, Resilience to transformation and inherent genetic and functional stability of adult neural stem cells ex vivo. Cancer Res. 2007;67:3725–33.
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008;Chapter 10:Unit 10.11.
- Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der Lehr N, Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci USA. 2010;107:58–63.
- Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol. 2010;12:54–9; sup pp 1–14.
- Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA. 2004;101:11334–7.
- Johansson Swartling F. Identifying candidate genes involved in brain tumor formation. Ups J Med Sci. 2008;113:1–38.
- Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–6.
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–6.
- Bender AM, Collier LS, Rodriguez FJ, Tieu C, Larson JD, Halder C, Sleeping beauty-mediated somatic mutagenesis implicates CSF1 in the formation of high-grade astrocytomas. Cancer Res. 2010;70:3557–65.
- Collier LS, Adams DJ, Hackett CS, Bendzick LE, Akagi K, Davies MN, Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer Res. 2009;69:8429–37.
- Felsher DW. MYC inactivation elicits oncogene addiction through both tumor cell-intrinsic and host-dependent mechanisms. Genes Cancer. 2010;1:597–604.
- Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29:1249–59.
- Westermark UK, Wilhelm M, Frenzel A, Henriksson MA. The MYCN oncogene and differentiation in neuroblastoma. Semin Cancer Biol. 2011;21:256–66.
- Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–9.
- Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34:1480–9.
- Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–408.
- Chen J, Willingham T, Margraf LR, Schreiber-Agus N, DePinho RA, Nisen PD. Effects of the MYC oncogene antagonist, MAD, on proliferation, cell cycling and the malignant phenotype of human brain tumour cells. Nat Med. 1995;1:638–43.
- Shalaby T, von Bueren AO, Hurlimann ML, Fiaschetti G, Castelletti D, Masayuki T, Disabling c-Myc in childhood medulloblastoma and atypical teratoid/rhabdoid tumor cells by the potent G-quadruplex interactive agent S2T1-6OTD. Mol Cancer Ther. 2010;9:167–79.
- van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–9.
- Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31:6148–56.
- Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20:1383–93.
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, c-Myc regulates transcriptional pause release. Cell. 2010;141:432–45.
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723–34.
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17.
- Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer. 2006;5:31.
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–34.
- Hydbring P, Larsson LG. Tipping the balance: Cdk2 enables Myc to suppress senescence. Cancer Res. 2010;70:6687–91.
- Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13:820–7.
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28.
- De Salvo M, Maresca G, D'Agnano I, Marchese R, Stigliano A, Gagliassi R, Temozolomide induced c-Myc-mediated apoptosis via Akt signalling in MGMT expressing glioblastoma cells. Int J Radiat Biol. 2011;87:518–33.
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–46.
- Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci USA. 1999;96:4438–42.
- Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006;66:8139–46.
- Johnsen JI, Segerstrom L, Orrego A, Elfman L, Henriksson M, Kagedal B, Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27:2910–22.
- Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–9.
- Baryawno N, Sveinbjornsson B, Eksborg S, Chen CS, Kogner P, Johnsen JI. Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res. 2010;70:266–76.
- Heine VM, Priller M, Ling J, Rowitch DH, Schuller U. Dexamethasone destabilizes Nmyc to inhibit the growth of hedgehog-associated medulloblastoma. Cancer Res. 2010;70:5220–5.
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–44.