
Abstract
Antibiotics are the medical wonder of our age, but an increasing frequency of resistance among key pathogens is rendering them less effective. If this trend continues the consequences for cancer patients, organ transplant patients, and indeed the general community could be disastrous. The problem is complex, involving abuse and overuse of antibiotics (selecting for an increasing frequency of resistant bacteria), together with a lack of investment in discovery and development (resulting in an almost dry drug development pipeline). Remedial approaches to the problem should include taking measures to reduce the selective pressures for resistance development, and taking measures to incentivize renewed investment in antibiotic discovery and development. Bringing new antibiotics to the clinic is critical because this is currently the only realistic therapy that can ensure the level of infection control required for many medical procedures. Here we outline the complex process involved in taking a potential novel antibiotic from the initial discovery of a hit molecule, through lead and candidate drug development, up to its entry into phase I clinical trials. The stringent criteria that a successful drug must meet, balancing high efficacy in vivo against a broad spectrum of pathogens, with minimal liabilities against human targets, explain why even with sufficient investment this process is prone to a high failure rate. This emphasizes the need to create a well-funded antibiotic discovery and development pipeline that can sustain the continuous delivery of novel candidate drugs into clinical trials, to ensure the maintenance of the advanced medical procedures we currently take for granted.
Introduction
There is an urgent and growing need for new classes of antibiotics to maintain the advanced medical procedures we now take for granted. Antibiotics fulfil a critical infection control function in many areas of medicine including during invasive surgery, in cancer chemotherapy, and in the treatment of elderly or immune-compromised patients. The success of antibiotics as a drug class is due to several factors: they are in general very effective in helping the body to clear bacterial infections rapidly, they are relatively safe to use and many can be taken orally, and they are relatively cheap drugs and thus very widely used. The great success of antibiotics has also been their downfall: widespread and inappropriate overuse (including outside human medicine, in agriculture and aquaculture) has driven the rapid evolution and spread of antibiotic-resistant bacterial pathogens that now threaten our ability to control bacterial infections. In many parts of the developing world, where controls are lax, the situation is already out of control, but even in Europe and North America the problem is serious, and for many bacterial pathogens the options for antibiotic therapy are extremely limited (Citation1). The problem therefore is that we are possibly facing into a future where advanced medical procedures may entail extremely high risks of being associated with untreatable hospital-acquired bacterial infections, and where common community-acquired bacterial infections, such as pneumonia, will again carry a high risk of death as they did a century ago. How to tackle this problem has been widely discussed in recent years, including recently in a comprehensive discussion paper co-authored by Otto Cars (Citation2). The issue is complex and probably requires several simultaneous actions (including greater investment in antibiotic development and more effective controls to reduce the misuse of antibiotics), and possibly the development of some novel infection control approaches (developing alternatives to antibiotics, exploring the possibilities of using antibiotics in combination to treat resistant pathogens, and developing antibiotic dosing regimens that reduce the selection of resistance). However, the best solution to the current resistance problem would be to discover and develop several novel classes of antibiotics for which there is no pre-existing resistance among human bacterial pathogens. Here we outline how to discover novel molecules with antibiotic activity and the steps required to develop them into clinically useful antibiotics.
Defining a ‘good' antibiotic
Before launching into an antibiotic drug discovery programme one should discuss what makes a good antibiotic class (good in terms of medical and commercial success). Firstly, we make no distinction between antibiotic (a natural product) and antimicrobial (a synthetic product) and will refer to all antibacterial drugs as antibiotics. The antibiotic class that has been most successful in terms of medical usefulness is the β-lactam class (including the penicillins, cephalosporins, and carbapenems), followed by the macrolides and fluoroquinolones, with tetracyclines, aminoglycosides, and rifampicins taking an important but minor position (). What all of these drug classes have in common is that they are relatively small molecules (facilitating synthesis and chemical modification where desired), but they differ in many other respects. Most were originally natural products (although the marketed versions are typically synthetic or semi-synthetic), but the fluoroquinolones are completely synthetic molecules. The most successful drugs medically and commercially are bacteriocidal (β-lactams and fluoroquinolones), while others are conditionally bacteriocidal (rifampicins and aminoglycosides) or bacteriostatic (tetracyclines and macrolides). Their primary targets are enzymes for critical cellular functions: peptidoglycan synthesis in the cell wall (β-lactams); chromosome supercoiling dynamics in replication and transcription (fluoroquinolones); transcription of DNA into RNA (rifampicin); translation of mRNA into protein (aminoglycosides, macrolides, and tetracyclines). While each antibiotic class inhibits one major cell activity, the actual molecular targets of some drugs can be multiple: for example, fluoroquinolones interact with two different type II topoisomerases (DNA gyrase and DNA topoisomerase IV), each of which is essential. From this brief survey two important features emerge: 1) that successful antibiotics inhibit critical pathways in the bacterial cell, and the very best are bacteriocidal; 2) that successful antibiotics are usually molecules that can be manipulated and modified for the purpose of identifying analogues with more desirable antibacterial activities against the desired range of target pathogens, and to achieve good pharmacokinetic/pharmacodynamic properties with acceptably low levels of toxicity against mammalian cells. The most valuable antibiotics are soluble drugs, chemically stable, that can be taken orally and act systemically. This range of desirable properties places a premium on smaller molecules that in principle should be easier to modify chemically, achieve wide distribution and tissue penetration within the body, and be amenable to production on an industrial scale.
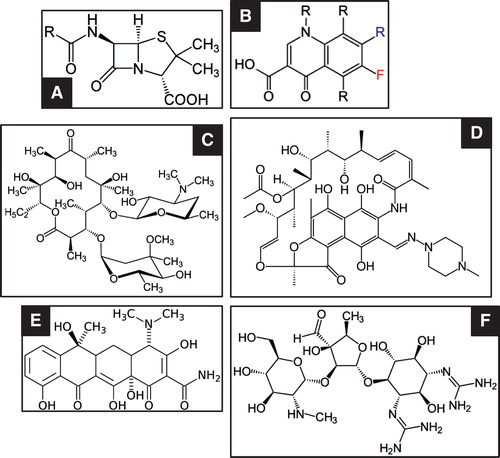
Figure 1. Examples of the small molecule structure of six major antibiotic classes. A: Penicillin (core); B: Fluoroquinolone (core); C: Macrolide (erythromycin); D: Rifampicin; E: Tetracycline; F: Aminoglycoside (streptomycin).
The principles of choosing and validating a good antibiotic target
Based on what we know of the most successful antibiotic classes (previous section) the ideal drug target should be an enzyme with a critical function in a biochemical pathway of central importance to the bacterial cell. In the search for novel antibiotics for which there is no pre-existing resistance, an obvious starting-point is to identify novel targets. Several complementary approaches have been taken to identify genes coding for potential essential target functions in relevant pathogen species. First is the use of genetics to test the essentiality of individual genes, usually by random transposon mutagenesis, or by making systematic gene-by-gene knock-outs. Complementing gene essentiality information is the use of pathogen genome sequence analysis to compile lists of genes that are conserved among important pathogens (for commercial and diagnostic reasons there is a preference for broad-spectrum antibiotics). These genes should preferably be absent or significantly different in the human genome. The aim is to identify novel and essential targets (not targeted by currently used antibiotics) present in most interesting pathogens, whose inhibition will not risk collateral damage to the human host. However, there are several caveats and fine points to consider before limiting the search for novel drug hits to what might be a very short target list. First, consider that the definition of a ‘novel target' is not always obvious. Take for example macrolides, tetracyclines, and aminoglycosides, each of which interacts directly with the ribosome to inhibit protein synthesis. However, resistance to any one of these antibiotic classes, at the level of alterations to the structure of the ribosome, does not confer resistance against either of the others, because the targets at the molecular level are different. The ribosome is a large and complex machine, and the actual targets for each antibiotic class are structurally distinct. For these antibiotics the specific drug target is not protein synthesis as a process, nor the ribosome as a machine. Instead, it is in each individual case a very specific molecular binding site on a large and structurally complex machine. Indeed, several other antibiotics also target the ribosome at structurally distinct targets. The message is that when compiling a list of potential targets one should not necessarily discard those for which successful antibiotics already exist, because there may be several distinct ways of inhibiting the functional entity. We argue that one should probably keep such targets on the list of interest precisely because they have been validated as useful antibiotic targets. Thus, the essential features we require in a novel antibiotic–target interaction are that it should inhibit bacterial growth (and preferably be bacteriocidal), that it should not be affected by existing resistance mechanisms, and that it should be available in a wide range of key bacterial pathogens. The prejudice to avoid targets present in human cells also comes with an important caveat. Take again for example the ribosome. The ribosome is present in all cells and is in large part very highly conserved at both the structural and functional levels. If we did not already know that many important and safe antibiotic classes target the ribosome we would probably, based on a simple bioinformatics analysis, have rejected the ribosome as a potential target out of fear for significant toxicity against human cells. In reality, the ribosome is the single most targeted molecular machine that we know of for natural antibiotics. Another significant target choice issue is the probability that resistance due to a target alteration should come with a significant fitness cost, but this is currently difficult to predict in advance.
First- and second-generation hit discovery approaches
The early days of antibiotic discovery were based on fortuitous discoveries of antimicrobial activity (penicillin) or systematic screens of soil and fungal extracts for bacterial growth inhibition (the Waksman screening platform). This first-generation approach was very successful and resulted in the discovery of most of the major antibiotic classes, discovered through the 1940s and 1950s (Citation3). However, this approach eventually ran into the law of diminishing returns when screens for growth inhibition began returning antibiotics that had already been discovered (Citation4). With the development of technologies that allowed rapid bacterial genome sequencing, improvements in protein structure determination, and advanced genetic engineering, many pharmaceutical companies initiated a second-generation approach to antibiotic drug discovery. They used genomics and genetics to identify novel essential targets present in a set of interesting pathogens. The structure of the target protein from one or more of the pathogens would then be solved. With this information in hand either or both of two approaches would be used to find hit molecules: 1) a high-throughput biochemical assay for target inhibition, or 2) structure-based design of small molecules that bound to and inhibited the function of the target molecule. The high-throughput screening was based either on available chemical libraries at each company (typically 10,000s to 100,000s of molecules), or on partially purified extracts of natural products. This rational approach to novel hit discovery was initially regarded as very productive. Molecules that inhibited the chosen target were usually identified and could be structurally optimized based on the 3-dimensional structure of the target protein. However, this approach was eventually abandoned by all of the major pharmaceutical companies because in every case it ran into serious problems later in the development pipeline (Citation5). The single most important problem was that in vitro inhibition of a target in a high-throughput assay turned out to be a very poor predictor of whole-cell antimicrobial activity. In essence, the primary problem was that most molecules identified failed to get into bacterial cells. A serious consequence of this large-scale failure in drug discovery was that most large pharmaceutical companies abandoned their antibiotic drug discovery programmes. In the meantime the problem of antibiotic resistance has significantly worsened worldwide. This has motivated both the European Union and the government of the United States of America to provide funds to stimulate a third-generation effort to discover and develop new antibiotics.
Third-generation hit discovery approaches
The failure of high-throughput biochemical assays to deliver useful hit molecules that could be developed into drugs has refocused the hit discovery process back onto whole-cell activity screening, but with the significant difference that the screen is coupled in some way to target specificity. The aim is to create and exploit biosensor strains. This approach combines the advantages of target choice (to reduce the possibility of rediscovering old antibiotics) with timely information on the ability of a molecule to inhibit the growth of whole bacterial cells (Citation6). A drug discovery team will consist of microbiologists, preparative chemists, and molecular modellers that are able to react to different target scenarios. The role of the microbiologist is to identify and validate potential drug targets, and to set up an assay system for chemical screening. One way to focus the screen on the chosen target is to use genetics to attenuate the target gene and its product, making bacterial growth hypersensitive to even a very modest interference with the product of interest. This can be coupled with a parallel screen against a strain in which the target is overproduced, resulting in a strain that is refractory to inhibition by the test molecule. The strains being screened might also be made generally hypersensitive to potential hit molecules by using genetic mutants that lack some permeability barriers or efflux pumps. The use of hyperpermeable cells with an attenuated target will allow even weakly active molecules to be detected, providing potentially important information to direct the optimization of structures by modelling and chemical modification. The role of the chemist is to prepare novel and potent compounds that can ultimately be developed into new antibacterial drugs. For this a chemical starting-point, a hit, is needed to work on and to progress further. Several strategies have been used to identify these early hits, including for example literature surveys, identification of endogenous ligands or active natural products, high-throughput screening of chemical libraries (including whole-cell phenotypic screens), and virtual screening. The molecular modeller plays an important role in the hit identification process as well as in the later hit and lead optimization stages. For example, if the 3D structure of the target is known, a structure-based virtual screen can be performed where large numbers of commercially available compounds are docked to the active site and prioritized based on different criteria. Alternatively, if the 3D structure is not known, compounds can be screened according to the similarity of each compound to any of the known actives (ligand-based virtual screening). The prioritized hits, from either approach, are then acquired and evaluated in the activity assay. Once a confirmed hit has been identified and the chemistry is determined to be tractable, the chemist will start to modify the compounds and build in lead-like properties. These include, but are not limited to, improved potency, aqueous solubility, stability, and selectivity versus relevant related targets. In all cases it is important to monitor that the compound class is active in terms of minimal inhibitory concentrations (MICs). Lead optimization is a complicated, non-linear process in which drug-like characteristics are refined. The medicinal chemists aim to optimize the lead series and generate a pharmacological, safety, and pharmacokinetic profile to select one or more candidates for early trial development.
Validating and declaring a hit
In an initial screening programme several different potential hits might be discovered, but typically not all of them will have equally desirable qualities. It is essential to make an initial evaluation of their potential and liabilities before settling on a single hit as suitable for expansion and development into a lead compound. An antibiotic hit is usually defined as a molecule that binds to a target that is important to the pathogen of interest and inhibits its growth. Accordingly, the first issue is to determine that growth inhibition is actually associated with target binding. Depending on the particular target, a suitable in vitro binding assay should be set up to test hit–target interaction. The structure of the hit molecule should be determined (if not already known) to ensure that it is a novel chemical with the possibility for patent protection. Knowledge of the chemical structure will also indicate whether it is likely to be amenable for future modifications and for scaled-up synthesis as required.
The chemical and biological stability of the molecule should be determined, as well as its solubility in water. An advantage of molecules that are soluble in water is that they would support systemic administration, and if they also exhibit chemical and biological stability they might be suitable for oral administration if sufficient bioavailability could be achieved.
The structure of the target (if not already known) should also be determined so as to direct modelling and chemistry efforts to enhance target binding or other relevant features. The molecule (or family of related molecules) should be tested for antimicrobial activity against non-attenuated bacterial strains of the initial test species and also against other relevant Gram-negatives and Gram-positives (and other micro-organisms as appropriate to the particular project). The aim should be to identify molecules with a low MIC, preferably against more than one interesting pathogen. Preliminary tests of safety (against mammalian cell lines) should also be conducted. It is preferable that preliminary assays of spontaneous resistance be made to ensure that this is not an immediate and serious liability.
The purpose of these preliminary assays is to choose between several potential hit molecules or classes of molecules (that may have different chemical scaffolds) and ensure as far as possible that the one chosen for further development has at least some of the properties of a useful antibiotic, and that it does not suffer from critical liabilities that will with high probability cause failure at a later stage in development.
It is important at this stage, if not earlier, to draft a target product profile (TPP), which defines the key features of the drug to be developed bearing in mind probable clinical needs. The TPP is an organized list that prioritizes key features of the drug and lists both ideal and acceptable values for important features (for example, in vitro and in vivo efficacy, bacteriocidal or bacteriostatic, safety versus human targets, rate of resistance, bioavailability, stability, spectrum of antibacterial activity, treatment duration, route of administration, etc.). The TPP may be revised during the course of the development programme, but care must be taken not to compromise it by lowering the acceptable standards. This preliminary data defining a hit, together with the TPP, will be used to make an initial evaluation of the economic potential of the drug if it were to succeed all the way to the market. The most valuable antibiotics economically will be those that can be synthetized easily, modified readily, taken orally, have a wide bioavailability, low toxicity, and can be prescribed for a broad spectrum of bacterial pathogens. However, in an era when antibiotic resistance is becoming a very serious problem, molecules for intravenous use, or for a narrow spectrum of pathogens, will also be of interest.
Before declaring a hit and initiating hit-to-lead and lead-to-candidate development, a drug discovery manual should be drafted to guide the optimization process. This would define key decision points during the preclinical drug discovery process and provide objective measures for making stop/go decisions, with reference to potency, physicochemical properties, and in vitro and in vivo drug metabolism and pharmacokinetics (DMPK) and toxicology. These guidelines will be critical in making decisions on hit, lead, and candidate selections. In the context of being serious about bringing a novel antibiotic to the market the reality is that multiple independent hits (different chemical structures, possibly also different targets) will need to be discovered and validated, because during the subsequent stages of development most antibiotic development programmes will eventually suffer from serious liabilities and be discontinued.
The scenario outlined here and below represents the path of that rare species, a drug development programme that succeeds at every stage and eventually results in the successful entry of a novel antibiotic in clinical trials ().
Figure 2. Outline diagram of a drug discovery funnel.
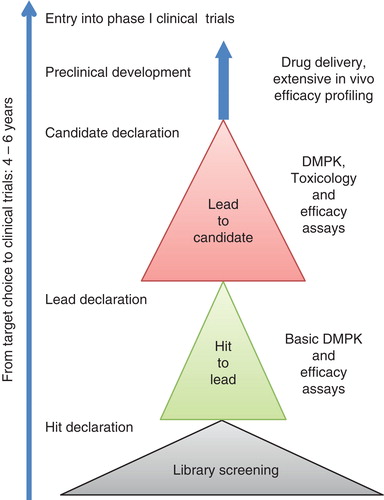
Hit to lead development
Once a hit has been declared it should be tested more extensively to ensure it does not suffer from a critical liability, and modified chemically to improve its activity and safety profile. Successive series of molecules will be processed through a funnel of hit-to-lead screening assays to identify liabilities and inform successive cycles of chemical modification. The funnel should be structured to ensure a rapid turnaround of the most critical activity and liability information to the chemists responsible for synthetizing new series of molecules. Initial assays should measure compound purity and solubility.
Screening assays should early on measure in vitro cytotoxicity (Citation7); measure MIC against a panel of relevant pathogens, including some hypersensitive strains; measure target activity; and if appropriate, selectivity versus human counter-targets. Evaluation of these data would be used to inform a new round of chemical synthesis (if necessary) and to direct the choice of which, if any, of the compounds to progress.
If the TPP requires an oral route of administration, it would be important to test the ability of the drug compounds to cross physiological membranes. An established method for predicting the in vivo absorption of drugs across the gut wall is by measuring the rate of transport of a compound across the Caco-2 cell line (Citation8). This cell line is derived from a human colon carcinoma, and the cells have characteristics that resemble intestinal epithelial cells. An added advantage is that transport can be assessed in both directions across the cell monolayer, enabling an efflux ratio to be determined which provides an indicator as to whether a compound undergoes active efflux.
A number of assays would focus on assessing potential liabilities (Citation9). The inhibitory potency and time-dependent inhibition of novel compounds against cytochrome P450s (CYPs) should be tested with liver microsomes. CYPs are a family of haem-containing enzymes that play a key role in the metabolism of pharmaceutical agents and that could potentially be inhibited by novel compounds. CYP inhibition can result in several undesirable consequences: 1) increased pharmacological effects or toxicity caused by decreased drug metabolism, 2) decreased pharmacological effects caused by decreased formation of reactive metabolites, and 3) drug–drug interactions caused by multiple medication. Knowledge of the potential for a drug to decrease CYP activity at an early stage of drug development therefore reduces the risk of failure during clinical trials.
Another important potential liability to assess in hit-to-lead work is drug interference with human ion channels. Inhibition of the hERG potassium channel by drug binding can cause an increase in the risk of cardiac problems such as QTc prolongation (Citation10). This makes these channels important anti-targets that must be avoided during drug development. Assays of inhibition should be made at appropriate concentrations and can be evaluated using patch-clamp electrophysiology (Citation11).
The in vitro metabolic stability of the compounds is another key property important for drug administration as well as toxicity, and should be tested in liver microsomes and hepatocytes (Citation12). This allows the evaluation of metabolic stability as a result of phase I oxidation. If there is a high intrinsic clearance, then metabolic profiling should also be performed to identify metabolic ‘soft spots' in the compound series. The ideal drug should be relatively stable, maintain an effective concentration in the blood for a reasonable period of time, be metabolized by multiple CYP enzymes, and not form pharmacologically active or chemically reactive metabolites.
The most promising compounds would be elaborated further in more extensive assays/models. Compounds would be screened against a secondary MIC panel of multi-drug-resistant strains (to identify liabilities such as inability to deal with existing resistance problems), and against strains to be used in in vivo animal assays. In addition, MIC90 values would be determined against panels of contemporary clinical isolates of each of the intended target pathogen species. Time-kill assays would be made against key target pathogens to distinguish between bacteriocidal and bacteriostatic modes of action, and provide data to support early PKPD modelling studies (Citation13). Physiochemical properties of compounds (pK a, logD, and thermodynamic stability) would need to be determined before formulation for in vivo studies. An Ames test would also be performed to test for mutagenicity potential (Citation14).
At this stage in late hit-to-lead phase, solution formulations for in vivo pharmacokinetic (PK) and efficacy studies would be prepared for testing of the stability of the compounds in formulation. Bioanalytical methods for the progressed compounds would be developed and PK measured after a single dose in a rodent (oral, subcutaneous, or intravenous as appropriate) with the preferred outcome being ‘sufficient' in vivo exposure. Tolerability of single doses in rodents would also be evaluated to estimate appropriate doses for further tests. In hit-to-lead screening a single animal model efficacy test would probably be sufficient. The mouse neutropenic thigh infection model (Citation15) could be used to assess efficacy in the complex in vivo environment (simulating an acute infection) after intramuscular injection with bacteria. Finally, resistance studies would be made to measure the frequency or rate of spontaneous resistance development to the progressed compounds as a function of drug exposure, and the relative fitness of resistant mutants, using strains of each of the key target profile pathogens (Citation16,17). The preferred outcome would be a low mutation rate, with resistant mutants associated with a high fitness cost and a low fold-shift in MIC relative to the parent strains. Resistance mechanism studies could also be made at this stage on selected strains of key pathogens, by whole-genome sequencing and proteomics, to expand knowledge of the drug targets and to identify issues related to drug uptake and drug efflux. At this stage a lead compound would be declared to enter a lead-to-candidate screening funnel.
Lead-to-candidate development
Lead-to-candidate development follows a similarly structured funnel of assays as described in the previous section for hit-to-lead development. The process begins with medicinal chemistry to expand the chosen lead molecule, with the primary aims being to improve on desired activities and reduce known liabilities. The initial suite of assays would include those described above. Most likely re-synthesis would have to be made to facilitate profiling of the remaining advanced lead compounds. A series of assays, similar to those performed during hit-to-lead development, but more extensive and with stricter criteria, would be made in the lead-to-candidate phase. Also new assays/models would be introduced, including for example PK and PKPD modelling based on time-kill data to facilitate design and interpretation of in vivo efficacy assays and in vitro–in vivo translation. In addition extended in vivo PK studies in rats would be required, to determine plasma concentrations and a range of important PK parameters. The preferred outcome of the PK assays is establishing that there is sufficient exposure to allow subsequent toxicology studies. After the PK studies the efficacy of the advanced lead compounds would be measured in comparison with suitable reference compounds in multiple rodent infection models, as appropriate (Citation18–20). The dosing and assay times would be based on the measured in vivo PK, and the preferred outcome would be that they should be at least comparable to the reference compounds. Finally, resistance rate, relative fitness of resistant mutants, and mechanisms of resistance would be determined for key pathogens as a function of compound exposure (Citation21,22). Resistance fitness parameters are expected strongly to influence the probability of resistance increasing to fixation in a pathogen population. The few remaining interesting compounds would then be re-synthesized on a large scale to fuel pre-candidate profiling, and to prepare future studies beyond candidate selection.
Pre-candidate profiling
Based on an evaluation of the totality of data from previous assays, and in the absence of serious liabilities, a candidate drug might be declared for entry into a preclinical development programme in preparation for phase I clinical studies. The specific activities conducted in this preparation would vary but would include assessment of toxicity in two species, assessment of genotoxicity, more extensive in vivo efficacy profiling, and assays to ensure appropriate delivery of drug.
Conclusion and future perspectives
The problems that antibiotic-resistant bacterial pathogens cause for effective medical therapies are continuing to increase, and solutions will need to be found soon if we are not to run the serious risk of losing the ability to carry out advanced medical procedures. The current shortage of effective antibiotics is not simply a biological problem (evolution of resistance), or even a social problem (misuse of antibiotics and selection of resistance), but is in large part the result of a long-term lack of investment in discovery and development programmes by the large pharmaceutical companies. There are currently very few antibiotics in late-phase clinical trials, and nearly all of these belong to existing classes (Citation23,24). The reasons for this investment gap in novel classes are complex, but they are associated with a lack of confidence on the part of the pharmaceutical industry in the relative profitability of antimicrobials as an area for investment. This may be about to change with the help of public–private partnership programmes like the recently launched EU Innovative Medicines Initiative (New Drugs 4 Bad Bugs, ND4BB) wherein academics, small companies and major pharmaceutical companies are working together, to kick-start a new generation of antimicrobial drug discovery and development.
In the sections above we have outlined a non-exhaustive list of efficacy and liability assays that a potential novel antibiotic drug must satisfy at each stage of early development before it will be approved for entry into the lengthy and expensive process of clinical trials. The number and breadth of these preclinical assays, and the exacting criteria that must be met by a potential novel antibiotic, make it unsurprising that very few hits make it all the way through to clinical trials. Because clinical trials represent the vast bulk of the expense of drug development it is preferable to discard potentially problematic molecules in the early hit-to-lead or lead-to-candidate stages, rather than have an expensive failure late during clinical trials. The high rate of attrition in antibiotic drug development makes it essential, therefore, that investment in early-stage discovery and development is maintained at a high enough level to generate a sustainable pipeline of novel hits moving into later-stage development, and feeding into clinical trials.
Declaration of interest: D.H. acknowledges support from Vetenskapsrådet (Swedish Science Council), SSF (Swedish Strategic Science Foundation), Vinnova (Swedish Innovation Science), and the Knut and Alice Wallenberg Foundation (RiboCore Project). A.K. acknowledges support from Vinnova (Swedish Innovation Science). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- CDC. Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR Morb Mortal Wkly Rep. 2013;62:165–70.
- Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, et al. Antibiotic resistance - the need for global solutions. Lancet Infect Dis. 2013;13:1057–98.
- Schatz A, Bugie E, Waksman SA. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc Soc Exp Biol Med. 1944;55:66–9.
- Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109.
- Livermore DM; British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug Discovery and Development. Discovery research: the scientific challenge of finding new antibiotics. J Antimicrob Chemother. 2011;66:1941–4.
- Hughes D. Exploiting genomics, genetics and chemistry to combat antibiotic resistance. Nat Rev Genet. 2003;4:432–41.
- Seeland S, Torok M, Kettiger H, Treiber A, Hafner M, Huwyler J. A cell-based, multiparametric sensor approach characterises drug-induced cytotoxicity in human liver HepG2 cells. Toxicol In Vitro. 2013;27:1109–20.
- Volpe DA. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med Chem. 2011;3:2063–77.
- Nettleton DO, Einolf HJ. Assessment of cytochrome p450 enzyme inhibition and inactivation in drug discovery and development. Curr Top Med Chem. 2011;11:382–403.
- Staudacher I, Schweizer PA, Katus HA, Thomas D. hERG: protein trafficking and potential for therapy and drug side effects. Curr Opin Drug Discov Devel. 2010;13:23–30.
- Farre C, Haythornthwaite A, Haarmann C, Stoelzle S, Kreir M, George M, et al. Port-a-patch and patchliner: high fidelity electrophysiology for secondary screening and safety pharmacology. Comb Chem High Throughput Screen. 2009;12:24–37.
- Chiba M, Ishii Y, Sugiyama Y. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009;11:262–76.
- Nielsen EI, Friberg LE. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol Rev. 2013;65:1053–90.
- Biran A, Yagur-Kroll S, Pedahzur R, Buchinger S, Reifferscheid G, Ben-Yoav H, et al. Bacterial genotoxicity bioreporters. Microb Biotechnol. 2010;3:412–27.
- Craig WA. Proof of concept: performance testing in models. Clin Microbiol Infect. 2004;10:12–17.
- Hughes D, Andersson DI. Selection of resistance at lethal and non-lethal antibiotic concentrations. Curr Opin Microbiol. 2012;15:555–60.
- Andersson DI, Hughes D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev. 2011;35:901–11.
- Hvidberg H, Struve C, Krogfelt KA, Christensen N, Rasmussen SN, Frimodt-Moller N. Development of a long-term ascending urinary tract infection mouse model for antibiotic treatment studies. Antimicrob Agents Chemother. 2000;44:156–63.
- Sandberg A, Lemaire S, Van Bambeke F, Tulkens PM, Hughes D, von Eiff C, et al. Intra- and extracellular activities of dicloxacillin and linezolid against a clinical Staphylococcus aureus strain with a small-colony-variant phenotype in an in vitro model of THP-1 macrophages and an in vivo mouse peritonitis model. Antimicrob Agents Chemother. 2011;55:1443–52.
- Sandberg A, Hessler JH, Skov RL, Blom J, Frimodt-Moller N. Intracellular activity of antibiotics against Staphylococcus aureus in a mouse peritonitis model. Antimicrob Agents Chemother. 2009;53:1874–83.
- Andersson DI, Hughes D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist Updat. 2012;15:162–72.
- Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8:260–71.
- Coates AR, Halls G. Antibiotics in phase II and III clinical trials. Handb Exp Pharmacol. 2012;211:167–83.
- Hernandez V, Crepin T, Palencia A, Cusack S, Akama T, Baker SJ, et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother. 2013;57:1394–403.