
Abstract
Tuberculosis (TB) represents a major public health problem. The growing number of (extensively) multi-drug resistance cases indicates that there is an urgent need for discovery of new anti-TB entities, addressed towards new and specific targets, and continuous development of fast and efficient synthetic strategies to access them easily. Microwave-assisted chemistry is well suited for small-scale laboratory synthetic work, allowing full control of reaction conditions, such as temperature, pressure, and time. Microwave-assisted high-speed organic synthesis is especially useful in the lead optimization phase of drug discovery. To illustrate the advantages of modern microwave heating technology, we herein describe applications and approaches that have been useful for the synthesis of new drug-like anti-TB compounds.
Introduction
Tuberculosis (TB) is a common and often lethal infectious disease caused primarily by the bacillus Mycobacterium tuberculosis (Mtb) and occasionally by M. bovis or M. africanum. It typically affects the lungs (pulmonary TB) but can affect other organs or tissues as well (extrapulmonary TB). The disease is transmitted through the air when people with pulmonary TB expel bacteria, for example by coughing. The origin of the disease was unknown until 1882, when Robert Koch showed that TB was caused by Mtb, for which he was later awarded the Nobel Prize (in 1905) (Citation1,2). The only treatment available at that time consisted of rest at sanatoria supplemented with pulmonary collapse procedures, aimed to rest infected parts of lungs and to close cavities. Although this was widely applied, it could not be extended to the entire population affected by TB (Citation3). In 1921, the Bacille Calmette–Guérin (BCG) vaccine was developed, but widespread vaccination campaigns were not initiated until 1943 after the introduction of the freeze-drying technique, which enabled vaccine mass production (Citation4). The vaccine is still used mainly in countries with endemic TB, but its efficacy has been questioned in several studies, most notably in India, where very limited or no protection has been reported (Citation5). Effective drug treatments were first developed in the 1940s–1950s. Streptomycin was the first drug proven to have a clinical effect, but it was not until the introduction of isoniazid, the first highly potent and orally available bactericidal entity, in 1952, that TB could be effectively treated. In 1965, with the advent of combination chemotherapy with isoniazid and rifampicin, the duration of the treatment could be shortened to 6 months. This allowed widespread TB control programmes to be implemented, which rapidly reduced the incidence of TB disease in industrialized countries. Unfortunately, the same success could not be observed in developing countries. The emergence of drug-resistant Mtb strains along with the human acquired immunodeficiency syndrome (AIDS) epidemic in the early 1980s, resulted in a crucial and sharp increase in TB incidence in developed countries. These issues, along with socio-economic problems and ineffective health care systems in many countries, enabled the disease to re-establish itself (Citation4). The World Health Organization (WHO) in 1990 estimated that one-third of the world's population was infected with Mtb, with 8 million new infections and millions of death annually (Citation6). Accordingly, in 1993 WHO officially recognized TB as a global emergency. Since then, significant effort has been put into overcoming the disease, and this has resulted in the recent launch of bedaquiline, the first effective drug to reach the market since the introduction of rifampicin almost 50 years ago (in 1965) (Citation7).
Mycobacterium tuberculosis life cycle
Infection with Mtb follows a pattern of events that has been established through animal models, as well as observations from human TB (Citation8,9). The infectious bacilli are inhaled as droplet nuclei that have been exhaled into the atmosphere. Initially, these droplets are small enough to remain airborne for several hours. The bacteria are phagocytosed by alveolar (Citation10) macrophages, which then invade the epithelial layer. At this stage, a localized inflammatory response occurs and leads to recruitment of mononuclear cells from neighbouring blood vessels, providing fresh host cells for the expanding bacterial population. These cells are the building blocks for the generation of the granuloma, which initially appears as an amorphous mass of macrophages. Subsequently, it differentiates into several specialized cell types, such as multinucleated giant cells, foamy macrophages, and epithelioid macrophages. With the development of an acquired immune response and the arrival of lymphocytes, the granuloma becomes more organized and adopts a stratified structure, where the macrophage centre is surrounded by a mantle of lymphocytes. The appearance of Mtb lymphocytes, about 2 to 3 weeks post-infection, marks the end of the phase of rapid bacterial replication. At this time the granuloma is extensively vascularized, and cells are actively recruited to the site of the infection. In the late stage, the granuloma becomes hypoxic (Citation11), a condition that can induce a state of non-replicative persistence of Mtb in culture. An active granuloma is highly infective, and ultimately its rupture spills thousands of infectious bacilli into the airways, which results in the development of a productive cough that facilitates aerosol spread of infectious bacilli.
Tuberculosis treatment
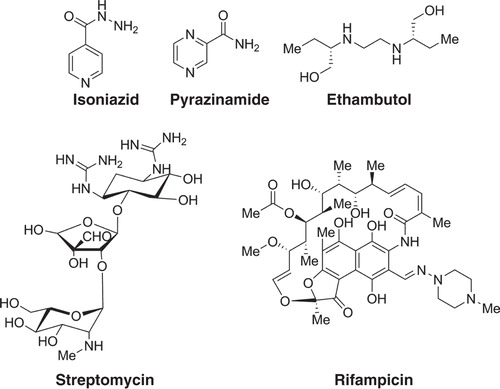
TB is not a disease of the past. Rather, TB remains a major public health problem. According to the latest TB global report released by the WHO (Citation12), in 2012 an estimated 8.6 million people developed TB and 1.3 million died from the disease (including 320,000 deaths among HIV-positive people). The African region has the highest TB/HIV burden. The majority of cases worldwide were in the Southeast Asian (29%), African (27%), and Western Pacific (19%) regions. India and China alone accounted for 26% and 12% of total cases, respectively. The currently recommended treatment for new cases of drug-susceptible TB is a 2-month regimen with four first-line drugs (isoniazid, rifampicin, ethambutol, and pyrazinamide), followed by a continuous treatment with rifampicin and isoniazid for an additional 4 months () (Citation12).
Figure 1. First line anti-TB drugs used in combination treatment.
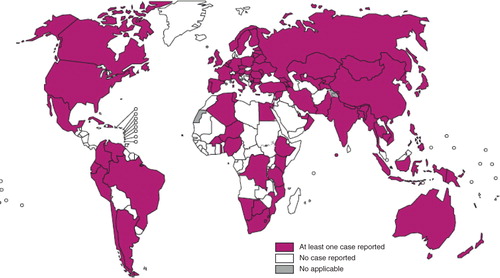
Treatment for multi-drug resistant TB (MDR-TB), defined as resistance to rifampicin and isoniazid (the two most powerful anti-TB drugs) is longer, and requires second-line drugs, which are more expensive and more toxic. In 2013, an estimated 3.6% of new TB cases and 20.2% of previously treated cases have MDR-TB (Citation12). Eastern European countries and especially Asia continue to have the highest numbers of MDR-TB. Globally in 2012, there were an estimated 450,000 new cases of MDR-TB. Extensively drug-resistant Mtb (XDR-TB) strains have also been reported in at least 92 countries, and these make up around 10% of all MDR-TB cases (). This is a form of multi-drug resistance with additional resistance to at least one fluoroquinolone (DNA gyrase inhibitor) and at least one second-line injectable aminoglycoside antibiotic (ribosome inhibitor). Thirteen of these countries reported more than 10 XDR-TB cases last year. Among these cases, the highest proportion was found in Azerbaijan (12.8%), Belarus (11.9%), Latvia (16.0%), Lithuania (24.8%), and Tajikistan (21%) (Citation12).
Figure 2. Countries that had notified at least one case of XDR-TB by the end of 2012. Figure reproduced with kind permission from the World health Organization (Citation12).
Currently available drugs in the market target mainly Mtb enzymes, such as DNA gyrase, topoisomerase IV, and ATP-phosphoribosyl transferase (His-G) (Citation13). Isoniazid and ethambutol act as inhibitors for the synthesis of mycolic acids that are essential for building the cell wall of Mtb. They can generate electrophilic intermediates during their oxidation, capable of reacting with a nucleophilic group of the enoyl-reductase (InhA), a specific enzyme of Mtb, leading to its inactivation (Citation14). Aminoglycosides such as amikacin and streptomycin have several potential antibiotic mechanisms, such as protein synthesis inhibition, inhibition of ribosomal translocation, and disruption of the integrity of the bacterial cell membrane (Citation15). Rifamycins (rifampicin, rifabutin, and rifalazil) act directly on messenger RNA synthesis.
The growing number of MDR-TB and XDR-TB cases highlights the urgent need for development of new anti-TB drugs, addressed towards new and specific targets. This has resulted in the need for the continuous development of new TB entities. Moreover there exists an unmet need for the development of new enabling technologies to accelerate the anti-TB drug discovery and the development process. In this context, this review will focus on the use of controlled microwave irradiation for the synthesis of small molecules designed and exploited as potential anti-TB drugs with novel Mtb targets.
Importance of MW in the synthesis of new anti-TB entities
Microwave (MW) irradiation has proven to be a highly effective heating source for driving chemical reactions in sealed vessels. Microwaves can accelerate the reaction rate, provide uniform and selective heating, achieve greater reproducibility of reaction outcome, and help in developing cleaner and greener synthetic routes (Citation16,17). Dedicated microwave equipment is suitable for small-scale laboratory synthetic work, allowing full control of the reaction conditions, such as temperature, pressure (depending on the heating properties of the solvent used and on the selected temperature), and time (generally short) (Citation18). Microwaves heat reaction system directly (in situ heating); therefore, usage of solvents in the chemical reaction can be reduced or eliminated. Microwave-assisted organic synthesis is being widely applied in the pharmaceuticals industry, particularly for developing compounds in the lead optimization phase of drug development (Citation19,20). To illustrate the advantages of using modern microwave heating technology, we herein describe applications and approaches used in the synthesis of anti-TB entities (Citation21).
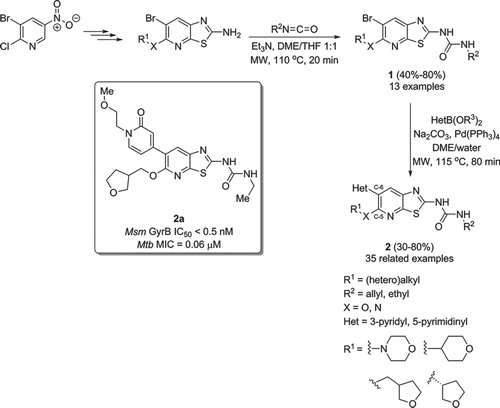
The Mtb genome is unique in encoding only two topoisomerases, topoisomerase I and II, also known as DNA gyrase (Citation22) (Escherichia coli genome, for example, encodes four topoisomerases (Citation22)). Unlike other bacteria, Mtb does not possess a topoisomerase type IV. The presence of a single type I topoisomerase and a single type II topoisomerase in Mtb makes the enzyme attractive from a drug discovery perspective, because it can be more vulnerable to inhibition. Very recently, Ghorpade et al. have disclosed the structure-activity relationship (SAR) studies of a library of thiazolopyridine ureas as novel anti-TB compounds active towards Mtb DNA gyrase B (GyrB) (Citation23). Potent compounds with GyrB half maximal inhibitory concentration (IC50) ≤ 1 nM and Mtb minimum inhibitory concentration (MIC) ≤0.1 µM were obtained (), with a combination of side chains at the C-5 position and introduction of heterocyclic moieties at the C-6 position of the benzothiazole core. Urea formation was performed by microwave heating of benzothiazol-2-amine at 110°C for 20 min in the presence of the appropriate isocyanate. Additionally, heterocyclic groups were introduced at the C-6 position via a microwave-heated Suzuki cross-coupling reaction (Citation24,25). By heating at 110°C the reaction time could be shortened to 30 min compared with a longer time of 4–15 h when cross-coupling was performed under standard heating conditions at 90°C. The best compound in this series, , showed a Mtb MIC of 0.06 µM and was very active against GyrB in M. smegmatis (Msm GyrB IC50 < 0.05 nM).
Figure 3. Preparation of anti-TB thiazolopyridine ureas by microwave irradiation.
Fluoroquinolones are known to be Mtb DNA GyrB inhibitors (see Introduction). In 2013, De Rosa et al. reported the synthesis of a set of new fluoroquinolones bearing a (hetero)aromatic moiety at the C-7 position and an alkyl group at the N-1 position () that were very active against the Mtb H37Rv strain (Citation26). Compound was found to be the most active (MIC50 = 0.25 µM), showing a similar MIC to the reference compound ciprofloxacin. MICs were also determined for Mtb fluoroquinolone-sensitive and -resistant clinical isolate strains; compound was the most active compound, and it was found to be 4-fold more potent than ciprofloxacin. Diversity at the C-7 position was introduced via an efficient and rapid microwave-heated Suzuki cross-coupling reaction. 3-Quinolone-carboxylic acids were reacted with the appropriate arylboronic acids or esters at 150°C, to afford the desired final inhibitors in moderate to very good yields after only 5 min of irradiation.
Figure 4. Preparation of new fluoroquinolones via microwave assisted Suzuki cross-coupling reaction.
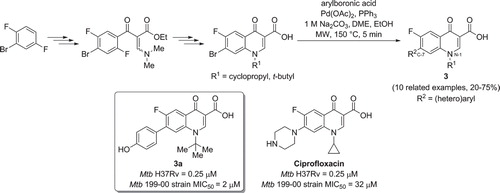
Figure 5. a) Microwave assisted preparation of a Mtb inhibitor via Fischer indolization. b) Synthesis of Mtb inhibitors via a microwave assisted ammonolysis.
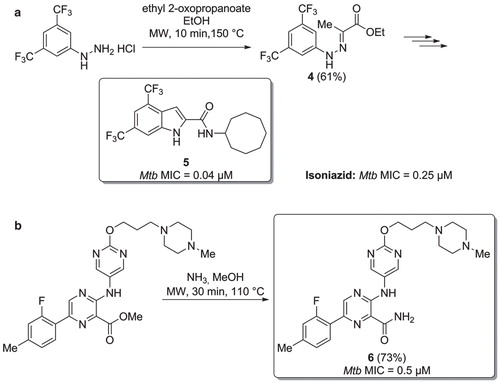
In the same year, Kozikowski and co-workers designed and synthesized a series of indole-2-carboxamide structure-based molecules active against resistant and multi-drug resistant Mtb strains (Citation27). Compound showed very good activity against Mtb (MIC = 0.04 µM). Its heterocyclic scaffold was prepared via a microwave-assisted hydrazone formation (Citation4), followed by a Fischer indole cyclization (). Compound was significantly more active than isoniazid against the Mtb H37Rv strain. However, a close analogue to , with the two trifluoromethyl groups both replaced by a methyl substituent, showed lower cytotoxicity, good pharmacokinetic profile in mice, and excellent anti-TB activity against a panel of five drug-resistant and multi-drug resistant Mtb strains.
Aminopyrazinamides have been reported as a novel class of Mtb GryB inhibitors, able to kill both replicating and non-replicating mycobacteria (Citation28). The development of compounds active against non-replicating Mtb is one of the most promising strategies to achieve new and shorter TB treatment regimes. Inhibitor () was the most active compound of this class of new molecules (MIC = 0.5 µM), and the primary amide moiety was obtained via a microwave-assisted ammonolysis of the precursor methyl ester.
Mtb glutamine synthetase (Mtb-GS) is an essential enzyme involved in bacterial cell wall biosynthesis (Citation29), the ability of pathogens to inhibit phagosome-lysosome fusion, and in phagosome acidification (Citation30,31). Mtb-GS is believed to be a promising TB target since it was shown that L-methionine-(SR)-sulfoximine (MSO, ), a well-known inhibitor of this enzyme, was able to inhibit bacterial growth both in vitro and in vivo (Citation32,33). Recently, Gising et al. reported a new class of imidazole-based Mtb-GS inhibitors. Microwave-assisted synthesis played an important role for the preparation of the most potent compound of this series (, ) (Citation34). Intermediate was obtained in excellent yield of 93% via a palladium-catalysed alkoxycarbonylation of the starting-material 2-bromo-6-methoxynaphthalene in ethanol, using Mo(CO)6 as the carbon monoxide source (Citation35), in the presence of palladium(II) acetate as the precatalyst and Xantphos as the ligand. Fluorine was replaced by a primary amine, through a sluggish SNAr substitution using diphenylmethanamine, followed by a deprotection reaction, affording the final inhibitor . In both reactions, the superheated microwave conditions were necessary for product formation. When compared with MSO, compound showed 100-times higher activity towards Mtb-GS, with an IC50 of 0.049 μM. Interestingly, compound was found to inhibit bacterial growth and exhibited a MIC of 2 μM against Mtb.
Figure 6. a) L-methionine-(SR)-sulfoximine (MSO). b) Synthesis of Mtb-GS inhibitors via a multi-step microwave assisted synthetic route. c) Microwave assisted synthesis of Mtb-GS inhibitors via multi-component heterocyclization.
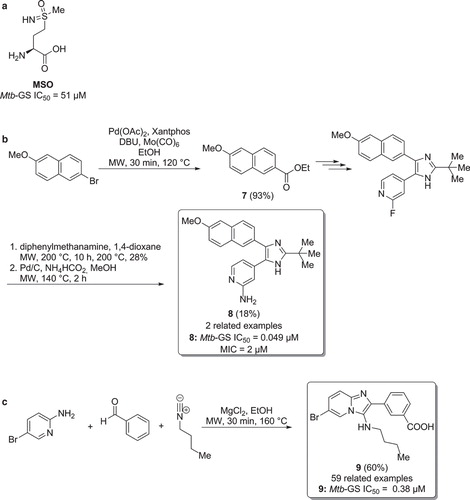
Odell et al. and Nordqvist et al. have reported that functionalized 3-amino-imidazo[1,2 a]pyridines, identified via a high-throughput screening (HTS) of AstraZeneca's corporate library, are potent inhibitors of Mtb-GS (Citation36,37). The synthetic approach was based on a microwave-assisted Groebke–Blackburn–Bienaymé heterocyclization, using various substituted 2-aminopyridines, benzaldehydes, and isonitriles (). Compound , with an IC50 of 0.38 µM, was the most active compound in this study, being considerably more potent than MSO (51 µM) against Mtb-GS.
Mtb 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Mtb-DXR, also known as IspC) is an important Mtb enzyme involved in the non-mevalonate pathway and responsible for the production of isopentyl diphosphate, the precursor of isoprenoid units, essential for the TB bacilli. In eukaryotes, isopentyl diphosphate is instead produced via the mevalonate pathway, making IspC an attractive anti-TB drug target. Fosmidomycin () is a known inhibitor of this enzyme and is currently in clinical trials for the treatment of malaria, confirming that it is a promising target for drug development (Citation38). In 2013, Dowd et al. used the Mtb-DXR-fosmidomycin co-crystal structure complex to design bi-substrate ligands able to bind to both the natural substrate of IspC, 1-deoxy-D-xylulose 5-phosphate (DXP), and nicotinamide adenine dinucleotide phosphate hydrogenated (NADPH), with the aim of improving the whole-cell activity due to increased lipophilicity () (Citation38). The most potent compound produced in this study (compound ) exhibited an IC50 of 17.8 µM, and, importantly, the diethyl ester of was found to inhibit Mtb growth (H37Rv Mtb MIC = 200 µM). The synthesis of commenced with a microwave-assisted Arbuzov reaction between triethylphosphite and 1,3-dibromopropane to generate the key intermediate (Citation38).
Figure 7. a) Structure of fosmidomycin. b) Synthesis of fosmidomycin analogues. c) Synthesis of Mtb-IspC inhibitors via a microwave promoted oxidative Heck reaction. d) Synthesis of Mtb inhibitors via a heterocyclization.
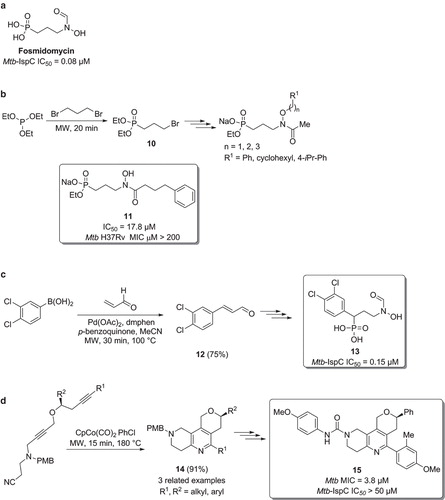
Andaloussi et al. prepared a series of fosmidomycin analogues bearing various aromatic groups in the phosphonic acid α position () (Citation39). The most potent inhibitor was prepared using a key microwave-assisted oxidative Heck reaction between 3,4-dichloroboronic acid and acrylaldehyde, using palladium(II) acetate and 2,9-dimethyl-1,10-phenanthroline (dmphen) as the catalytic system (Citation40). The corresponding cinnamaldehyde was then easily converted into the final compound . This displayed Mtb-IspC inhibition similar to fosmidomycin.
Zhou et al. have reported the synthesis of pyrano-annulated 5,6,7,8-tetrahydro-1,6-naphthyridines (), with the aim of discovering novel anti-TB scaffolds (Citation41). The tricyclic core could be easily prepared via a microwave-assisted intramolecular [2+2+2] cyclization, affording compound in very good yield. Three additional analogues, with different alkyl and aryl groups (R1 and R2) were also prepared, using the same synthetic approach. Removal of the protecting para-methoxybenzyl group (PMB) furnished the corresponding secondary amines, which were subsequently reacted with a diverse set of eight isocyanates, acyl chlorides, and sulfonyl chlorides, yielding the corresponding ureas, amides, and sulphonamides, respectively. Most of the synthesized entities showed potency in the micromolar range, and structure was identified as the most promising anti-TB compound.
In 2009, Lin et al. described the synthesis of oxathiazol-2-ones derived from a HTS targeting the Mtb proteasome (Citation42). Proteasomes represent an important class of complex enzymes involved in protein degradation and have been investigated as potential targets for the treatment of several human diseases. Interestingly, the active compounds were found to react covalently but reversibly with the enzyme. Structure () was one of the most active compounds in this class and exhibited a 13-fold selectivity for the rate of inactivation (kobs) over the human proteasome. The heterocyclic core was prepared by reacting a (hetero)aryl-amide with chlorocarbonylsulfenyl chloride, at room temperature for 16 h or by microwave irradiation at 100°C for only 15 min, both reactions affording similar yields.
Figure 8. Synthesis of a Mtb proteasome inhibitor under batch or continuous flow MW conditions.
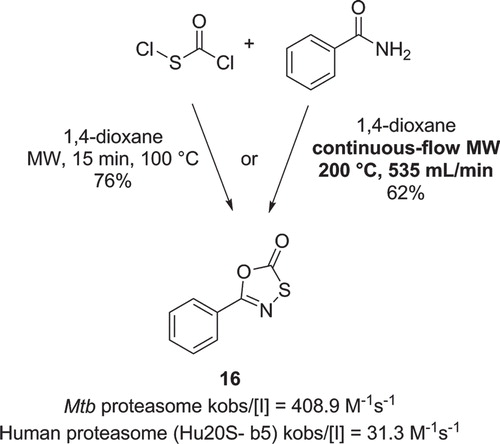
Öhrngren et al. have described a non-resonant microwave reactor designed specifically for continuous-flow chemistry applications (Citation43). One of the reported applications was the synthesis of compound (). The reaction was performed by pumping two stock solutions of benzamide and chlorocarbonylsulfenyl chloride through a mixer before reaching the microwave applicator. Under optimized conditions (353 µL/min of flow, residence time of 1 min and a temperature of 200°C) product was isolated in 62% yield, with a throughput of 3.3 mol/h.
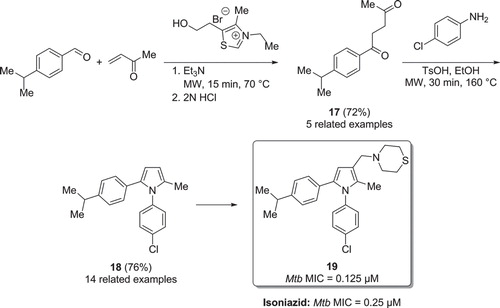
In 2008, the group of Biava reported on the activity of 1,5-diarylpyrrole derivatives towards Mtb (Citation44). Under microwave conditions, a Stetter reaction was carried out reacting cuminaldehyde and methyl vinyl ketone (). The reaction was driven by the addition of 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazolium bromide which gave the 1,4-diketone in good yield. Intermediate was then cyclized according to Paal–Knorr conditions with 4-chloroaniline, yielding the tri-substituted pyrrole . The reaction temperature could be safely raised to 160°C (double the boiling-point of the solvent EtOH) by using an appropriate microwave reactor and sealed vials, and full conversion of starting-material was observed within 30 min. One of the most active compounds, , was twice as effective as isoniazid on wild-type Mtb. It was also active towards isoniazid- and rifampicin-resistant Mtb (MIC = 8.0 and 0.125 µM, respectively). The same research group later on presented further exploration of the 1,5-diarylpyrrole derivatives (Citation45).
Figure 9. Microwave assisted synthesis of Mtb inhibitors via a condensation reaction.
In a report from 2010, Manna et al. described the synthesis of anti-TB 1,3,5-trisubstituted-4,5-dihydro-1H-pyrazoles anti-TB molecules (Citation46). The heterocyclization reaction was carried out using a single-mode microwave reactor, affording high yield and decreased reaction time, when compared with a previous protocol using standard conditions (12–22 min instead of reflux for 6–10 h). The final products were prepared by reacting a set of 14 α,β-unsaturated ketones with two hydrazides (). Out of 28 substituted pyrazoles synthesized, inhibitor 20 was the most active compound and was twice as active as isoniazid against Mtb. Furthermore, compound 20 was studied in a mouse model and was shown to lower the Mtb counts in lung and spleen two log units further than isoniazid.
Figure 10. a) Synthesis of Mtb inhibitors via a microwave heated 5-membered heterocyclisation. b) Synthesis of Mtb inhibitors via a microwave assisted Wittig reaction. c) Synthesis of Mtb inhibitors via a microwave promoted intramolecular SNAr reaction.
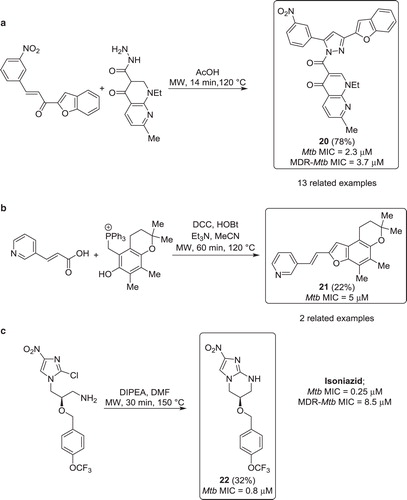
In an effort to develop new anti-TB entities, Alvey et al. investigated a Wittig-type cyclization to prepare benzofurans (Citation47). Using microwave irradiation, the reaction times could be shortened from 88 h to 1 h, compared with traditional reflux conditions. The inhibitors were assembled in a one-pot two-step synthesis, involving an initial coupling between acids or acyl chlorides and the hydroxyl group of the phosphonium species. The esters were subsequently cyclized via Wittig reaction, yielding the desired benzofurans in moderate yields (). Compound 21 was found to be active, showing moderate activity in the whole bacterial assay.
In 2009, Kim and co-workers exploited a structure–activity relationship study of nitroimidazoles active towards Mtb (Citation48). One of the synthetic pathways, an intramolecular SNAr reaction, was performed under single-mode microwave irradiation (). Compound 22 was obtained in modest yield, by heating the chloroimidazole starting-material at 150°C for 30 min in the presence of DIPEA. Importantly, inhibitor 22 exhibited a low MIC of 0.8 µM.
Conclusions
Herein, we have depicted a number of examples of microwave-promoted synthetic approaches used in discovery projects for the development of new anti-TB molecules. In many of these applications, the use of microwave technology enabled the fast, smooth, and reliable preparation of a diverse range of biologically active molecules. In a number of cases, microwave superheating allowed a dramatic reduction in reaction times and the preparation of compounds that are difficult to synthesize using traditional synthetic methods. Microwave-assisted synthesis is not restricted to anti-TB research but is today widely used in many medicinal chemistry projects to accelerate lead optimization. It is believed that microwave chemistry will continue to play an important role in accelerating the development process of all kinds of new drug-like molecules, including novel antibiotics.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Koch R. Die Ätiologie der Tuberkulose. Berliner Klinischen Wochenschrift. 1882;19:221–30.
- Koch R. The etiology of tuberculosis. Rev Infect Dis. 1982;4:1270–4.
- Daniel TM. The history of tuberculosis. Respir Med. 2006;100:1862–70.
- Harries AD, Dye C. Tuberculosis. Ann Trop Med Parasit. 2006;100:415–31.
- Verma I, Grover A. Antituberculous vaccine development: a perspective for the endemic world. Expert Rev Vaccines. 2009;8:1547–53.
- Tuberculosis control and research strategy for the 1990s. World Health Organization: Geneva, 26–27 October, 1991.
- Palomino JC, Martin A. TMC207 becomes bedaquiline, a new anti-TB drug. Future Microbiol. 2013;8:1071–80.
- World Health Organization. Global tuberculosis control: a short update to the 2009 report. Geneva: World Health Organization; 2009.
- Dye C, Lonnroth K, Jaramillo E, Williams BG, Raviglione M. Trends in tuberculosis incidence and their determinants in 134 countries. Bull World Health Organ. 2009;87:683–91.
- Ax A, Schaal W, Vrang L, Samuelsson B, Hallberg A, Karlén A. Cyclic sulfamide HIV-1 protease inhibitors, with sidechains spanning from P2/P2′ to P1/P1′. Bioorg Med Chem. 2005;13:755–64.
- Bacaer N, Ouifki R, Pretorius C, Wood R, Williams B. Modeling the joint epidemics of TB and HIV in a South African township. J Math Biol. 2008;57:557–93.
- Global tuberculosis report. Geneva: World Health Organization: 2013.
- Aubry A, Fisher LM, Jarlier V, Cambau E. First functional characterization of a singly expressed bacterial type II topoisomerase: the enzyme from Mycobacterium tuberculosis. Biochem Biophys Res Commun. 2006;348:158–65.
- Johnsson K, King DS, Schultz PG. Studies on the mechanism of action of isoniazid and ethionamide in the chemotherapy of tuberculosis. J Am Chem Soc. 1995;117:5009–10.
- Shakil S, Khan R, Zarrilli R, Khan AU. Aminoglycosides versus bacteria: a description of the action, resistance mechanism, and nosocomial battleground. J Biomed Sci. 2008;15:5–14.
- Kappe CO. Microwave dielectric heating in synthetic organic chemistry. Chem Soc Rev. 2008;37:1127–39.
- Nilsson P, Gold H, Larhed M, Hallberg A. Microwave-assisted enantioselective Heck reactions: expediting high reaction speed and preparative convenience. Synthesis. 2002;11:1611–14.
- Noteberg D, Schaal W, Hamelink E, Vrang L, Larhed M. High-speed optimization of inhibitors of the malarial proteases plasmepsin I and II. J Comb Chem. 2003;5:456–64.
- Wannberg J, Ersmark K, Larhed M. Microwave-accelerated synthesis of protease inhibitors. Top Curr Chem. 2006;266:167–98.
- Gising J, Odell LR, Larhed M. Microwave-assisted synthesis of small molecules targeting the infectious diseases tuberculosis, HIV/AIDS, malaria and hepatitis C. Org Biomol Chem. 2012;10:2713–29.
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44.
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–62.
- Kale GM, Raichurkar A, Hameed SP, Waterson D, McKinney D, Manjunatha MR, et al. Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA gyrase B. J Med Chem. 2013;56:8834–48.
- Larhed M, Hallberg A. Microwave-promoted palladium-catalyzed coupling reactions. J Org Chem. 1996;61:9582–4.
- Wu X, Ohrngren P, Joshi AA, Trejos A, Persson M, Arvela RK, et al. Synthesis, X-ray analysis, and biological evaluation of a new class of stereopure lactam-based HIV-1 protease inhibitors. J Med Chem. 2012;55:2724–36.
- Guerrini V, De Rosa M, Pasquini S, Mugnaini C, Brizzi A, Cuppone AM, et al. New fluoroquinolones active against fluoroquinolones-resistant Mycobacterium tuberculosis strains. Tuberculosis. 2013;93:405–11.
- Onajole OK, Pieroni M, Tipparaju SK, Lun S, Stec J, Chen G, et al. Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. J Med Chem. 2013;56:4093–103.
- Shirude PS, Madhavapeddi P, Tucker JA, Murugan K, Patil V, Basavarajappa H, et al. Aminopyrazinamides: novel and specific gyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem Biol. 2013;8:519–23.
- Harth G, Clemens DL, Horwitz MA. Glutamine-synthetase of Mycobacterium tuberculosis: extracellular release and characterization of its enzymatic activity. Proc Natl Acad Sci USA. 1994;91:9342–6.
- Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257–70.
- Gordon AH, Darcyhart P, Young MR. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature. 1980;286:79–80.
- Harth G, Horwitz MA. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J Exp Med. 1999;189:1425–35.
- Harth G, Horwitz MA. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: demonstration of efficacy in vivo. Infect Immun. 2003;71:2297–8.
- Gising J, Nilsson MT, Odell LR, Yahiaoui S, Lindh M, Iyer H, et al. Trisubstituted imidazoles as Mycobacterium tuberculosis glutamine synthetase inhibitors. J Med Chem. 2012;55:2894–8.
- Odell LR, Russo F, Larhed M. Molybdenum hexacarbonyl mediated CO gas-free carbonylative reactions. Synlett. 2012;23:685–98.
- Nordqvist A, Nilsson MT, Lagerlunda O, Muthas D, Gising J, Yahiaoui S, et al. Synthesis, biological evaluation and X-ray crystallographic studies of imidazo[1,2-a]pyridine-based Mycobacterium tuberculosis glutamine synthetase inhibitors. Medchemcomm. 2012;3:620–6.
- Odell LR, Nilsson MT, Gising J, Lagerlund O, Muthas D, Nordqvist A, et al. Functionalized 3-amino-imidazo[1,2-a]pyridines: a novel class of drug-like Mycobacterium tuberculosis glutamine synthetase inhibitors. Bioorg Med Chem Lett. 2009;19:4790–3.
- Jose GS, Jackson ER, Uh E, Johny C, Haymond A, Lundberg L, et al. Design of potential bisubstrate inhibitors against Mycobacterium tuberculosis (Mtb) 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr)-evidence of a novel binding mode. Medchemcomm. 2013;4:1099–104.
- Andaloussi M, Henriksson LM, Wieckowska A, Lindh M, Björkelid C, Larsson AM, et al. Design, synthesis, and X-ray crystallographic studies of α-aryl substituted fosmidomycin analogues as inhibitors of Mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate reductoisomerase. J Med Chem. 2011;54:4964–76.
- Nordqvist A, Bjorkelid C, Andaloussi M, Jansson AM, Mowbray SL, Karlen A, et al. Synthesis of functionalized cinnamaldehyde derivatives by an oxidative Heck reaction and their use as starting materials for preparation of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase inhibitors. J Org Chem. 2011;76:8986–98.
- Zhou Y, Beeler AB, Cho SY, Wang YH, Franzblau SG, Snyder JK. Library synthesis using 5,6,7,8-tetrahydro-1,6-naphthyridines as scaffolds. J Comb Chem. 2008;10:534–40.
- Lin G, Li DY, de Carvalho LPS, Deng HT, Tao H, Vogt G, et al. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–63.
- Ohrngren P, Fardost A, Russo F, Schanche JS, Fagrell M, Larhed M. Evaluation of a nonresonant microwave applicator for continuous-flow chemistry applications. Org Process Res Dev. 2012;16:1053–63.
- Biava M, Porretta GC, Poce G, De Logu A, Saddi M, Meleddu R, et al. 1,5-Diphenylpyrrole derivatives as antimycobacterial agents. Probing the influence on antimycobacterial activity of lipophilic substituents at the phenyl rings. J Med Chem. 2008;51:3644–8.
- Biava M, Porretta GC, Poce G, De Logu A, Meleddu R, De Rossi E, et al. 1,5-Diaryl-2-ethyl pyrrole derivatives as antimycobacterial agents: design, synthesis, and microbiological evaluation. Eur J Med Chem. 2009;44:4734–8.
- Manna K, Agrawal YK. Design, synthesis, and antitubercular evaluation of novel series of 3-benzofuran-5-aryl-1-pyrazolyl-pyridylmethanone and 3-benzofuran-5-aryl-1-pyrazolylcarbony1-4-oxo-naphthyridin analogs. Eur J Med Chem. 2010;45:3831–9.
- Alvey L, Prado S, Saint-Joanis B, Michel S, Koch M, Cole SI, et al. Diversity-oriented synthesis of furo[3,2-f]chromanes with antimycobacterial activity. Eur J Med Chem. 2009;44:2497–505.
- Kim P, Kang S, Boshoff HI, Jiricek J, Collins M, Singh R, et al. Structure-activity relationships of antitubercular nitroimidazoles. 2. Determinants of aerobic activity and quantitative structure-activity relationships. J Med Chem. 2009;52:1329–44.