
Abstract
Background. It has been proposed that the histamine 1-receptor (H1-receptor) not only promotes allergic reactions, but also modulates innate immunity and autoimmune reactions. In line with this, we have recently reported that the H1-receptor antagonist cetirizine partially counteracts cytokine-induced beta-cell signaling and destruction. Therefore, the aim of this study was to determine whether cetirizine affects diabetes in NOD mice, a model for human type 1 diabetes, and glucose intolerance in high-fat diet C57BL/6 mice, a model for human glucose intolerance.
Methods. Female NOD mice were treated with cetirizine in the drinking water (25 mg/kg body weight) from 9 until 30 weeks of age during which precipitation of diabetes was followed. Male C57BL/6 mice were given a high-fat diet from 5 weeks of age. When the mice were 12 weeks of age cetirizine was given for 2 weeks in the drinking water. The effects of cetirizine were analyzed by blood glucose determinations, glucose tolerance tests, and insulin sensitivity tests.
Results. Cetirizine did not affect diabetes development in NOD mice. On the other hand, cetirizine treatment for 1 week protected against high-fat diet-induced hyperglycemia. The glucose tolerance after 2 weeks of cetirizine treatment was improved in high-fat diet mice. We observed no effect of cetirizine on the insulin sensitivity of high-fat diet mice.
Conclusion. Our results suggest a protective effect of cetirizine against high-fat diet-induced beta-cell dysfunction, but not against autoimmune beta-cell destruction.
Introduction
Histamine 1 (H1)-receptor antagonists are commonly used in the treatment of allergic disorders, but histamine may also influence non-allergic inflammatory and autoimmune events (Citation1). For example, in the absence of histamine, achieved by using histidine decarboxylase-knockout mice and a histamine-deficient diet, inflammatory processes in arthritis (Citation2) and colitis (Citation3) are attenuated. In addition, using H1-receptor knockout mice reduced arteriosclerosis, and steatohepatitis-associated inflammation was observed (Citation4,5). Also adaptive immunity is influenced by histamine-induced signaling events. Indeed, results from H1-receptor knockout mice indicate expansion of Th2 cells (Citation6,7), decreases in cytotoxic T-cells (Citation8), and reduced autoimmune encephalomyelitis in an experimental mouse model lacking the H1-receptor (Citation9,10). As type 1 diabetes is an autoimmune disease in which pancreatic beta-cells are destroyed via inflammatory and T-cell-mediated events (Citation11), and as type 2 diabetes is associated with macrophage activation and increased release of pro-inflammatory cytokines (Citation12), it is possible that histamine via activation of the H1-receptor modulates events pertinent to the pathogenesis of both type 1 and type 2 diabetes. This notion is supported by our recent observation that cetirizine, a second-generation H1-receptor antagonist (Citation13), partially counteracted pro-inflammatory-induced beta-cell death (Citation14). In line with this, it has also been reported that H1-receptor antagonists can inhibit NF-κB, a transcription factor that plays an important role in expression of adhesion molecules and inflammatory mediators (Citation15-17). In addition, cetirizine suppressed the activation and chemotaxis of T-cells, monocytes, and granulocytes at sites of inflammation, and inhibited production of reactive oxygen radicals and lipid mediators from eosinophils, neutrophils, and mast cells (Citation18-22). Therefore, the aim of the present study was to investigate the putative effects of cetirizine on diabetes development in non-obese diabetes (NOD) mice, a model for human type 1 diabetes, and glucose intolerance in high-fat diet mice, a model for human glucose intolerance. We observed that cetirizine improved the glucose tolerance of high-fat diet mice, but failed to protect against diabetes in NOD mice.
Methods
Development of diabetes in NOD mice
A local colony of non-obese diabetic (NOD) mice was kept under standard pathogen-free conditions, with free access to tap water and pelleted food. To study the spontaneous development of diabetes, 20 female NOD mice were given cetirizine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA; 25 mg/kg body weight) in the drinking water between 9 and 30 weeks of age. The dose was chosen to be high but far below the sub-lethal dose, which is 55 mg/kg, and the lethal dose, which is 750 mg/kg, in mice (Citation23). Twenty control mice were given only drinking water with no extra supplement. The amount of cetirizine in the drinking water was continuously adjusted according to water intake and body weight of the mice. Blood glucose levels were measured every week until the first sign of hyperglycemia, then daily. Animals were considered diabetic and killed upon two consecutive days of blood glucose >12 mM. The NOD mice experiments were approved by the local animal ethics committee.
High-fat diet treatment of C57BL/6 mice
Four-week-old male C57BL/6 mice were purchased from Scanbur AB (Sollentuna, Sweden). When 5 weeks of age, the mice were divided into two groups with 20 mice in each: one given a normal diet (ND) and one given a high-fat diet (HFD). The high-fat diet (HFD) (D12492, Research Diets, New Brunswick, NJ, USA) contained 60 kcal% fat, whereas the normal diet (ND) (D12450B, Research Diets) contained only 10% kcal fat (Citation24). After 7 weeks of diet, both groups were randomly divided into two subgroups, one receiving cetirizine (25 mg/kg body weight) in the drinking water and one group receiving no supplementation. The amount of cetirizine in the drinking water was continuously adjusted according to water intake and body weight of the mice. Water intake was not affected by the addition of cetirizine (results not shown). Directly before start of cetirizine treatment and after 1 week of cetirizine treatment non-fasting blood glucose levels were determined. For this purpose, blood was withdrawn from the tip of the tail and analyzed using the Freestyle Mini System (Abbot, TheraSense Inc., Alameda, CA, USA). The same person collected all blood samples for blood glucose determinations. The high-fat diet experiments were approved by the local animal ethics committee.
Blood glucose tolerance test
After 2 weeks of cetirizine treatment the mice were fasted for approximately 8 h. The mice were then given a single dose of 2.5 g/kg body weight of 30% w/v D-glucose intravenously by injection in the tail vein. Blood glucose was determined prior to injection and then at 10, 30, 60, and 120 min after injection. A small drop of blood was withdrawn from the tail and analyzed with the Freestyle Mini System.
Insulin sensitivity test
Mice were given an intraperitoneal injection (1.6 U/kg body weight) of the insulin analog Actrapid (Novo Nordisk, Bagsværd, Denmark). Blood glucose was determined on blood samples from the tail, before injection and 15, 60, 120, and 180 min later using the Freestyle Mini System.
Statistics
The chi-square test was used for analysis of cumulative diabetes incidence. Student’s t test was used for comparison of control mice with experimental group when the sample data were normally distributed. Non-normally distributed sample data were analyzed using the Mann–Whitney U test.
Results
Cetirizine did not protect against development of diabetes in NOD mice
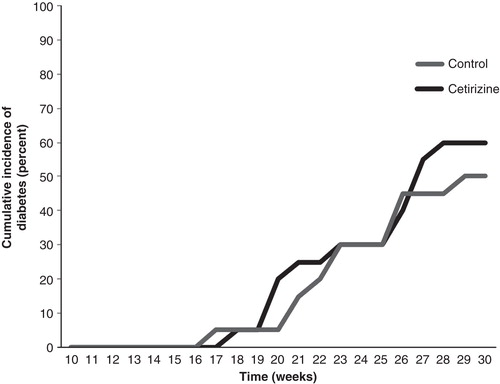
The precipitation of overt diabetes in female NOD mice usually occurs from approximately 15 weeks of age. To study whether the activity of the H1-receptor affects the onset of disease, we treated female NOD mice from 9 to 30 weeks of age with 25 mg/kg body weight per day of cetirizine administered via the drinking water. At 30 weeks of age 10 out of 20 non-cetirizine-treated mice had developed diabetes (). Among the 20 cetirizine-treated mice 12 developed diabetes (P > 0.05) (). Cetirizine did not affect the age at which diabetes precipitated ().
Figure 1. Cetirizine does not affect diabetes development in the NOD mouse. Female NOD mice were treated from 9 to 30 weeks of age with 25 mg/kg × day with cetirizine in the drinking water. Blood samples were taken at indicated time points for the assessment of hyperglycemia. Mice were considered diabetic when the blood glucose values were >12 mM on two consecutive days. At 30 weeks of age, 10 out of 20 control mice and 12 out of 20 cetirizine-treated mice had developed diabetes (P > 0.05 using the chi-square test).
Cetirizine ameliorated high-fat diet-induced glucose intolerance in C57BL/6 mice
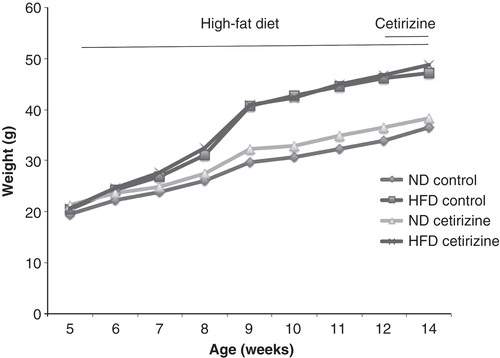
The H1-receptor antagonist cetirizine was given to C57BL/6 mice in the drinking water at 25 mg/kg/day, to determine whether high-fat diet (HFD)-induced glucose intolerance is affected. We found that cetirizine supplemented via the drinking water during the last 2 weeks of a 9-week long HFD period did not significantly affect the weight increase of the normal diet (ND) and HFD mice (). We also assessed food intake by weighing the cages without mice, before and after addition of new pellets, and observed similar food intake in cetirizine-treated mice as compared to non-cetirizine-treated mice (results not shown).
Figure 2. Weight curve of C57BL/6 mice given a normal diet or a high-fat diet from 5 to 14 weeks of age. After 7 weeks of normal diet (ND, 20 mice) and high-fat diet (HFD, 20 mice), the mice (aged 12 weeks) were divided into four groups with 10 mice in each: ND or HFD, with or without cetirizine (25 mg/kg/day) and followed for another 2 weeks.
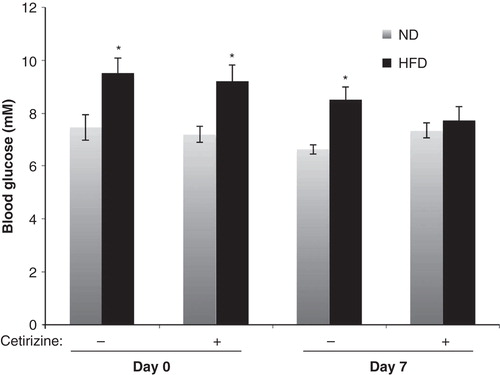
The non-fasting blood glucose levels of HFD mice were higher than those of ND mice after 7 weeks of diet (). A 1-week treatment with cetirizine resulted in a normalization of the blood glucose levels of HFD mice (). Cetirizine did not affect the blood glucose concentrations of ND mice ().
Figure 3. Non-fasting blood glucose of ND or HFD mice treated with cetirizine for 1 week. The non-fasting blood glucose concentrations were analyzed after 7 and 8 weeks of HFD, directly before start of cetirizine treatment (Day 0) and after 1 week of cetirizine treatment (Day 7). Results are means ± SEM; n = 10 for the two ND groups and 8–9 for the two HFD groups. *P < 0.05 versus corresponding ND mice using Student’s unpaired t test.
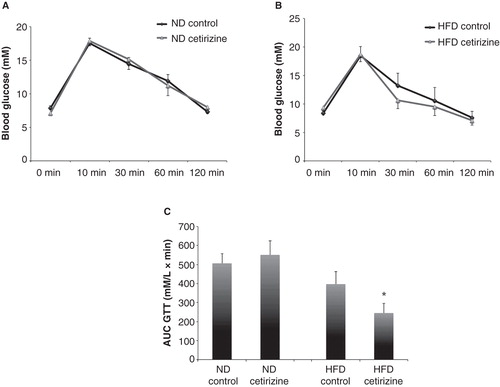
The glucose tolerance at fasting conditions of the different study groups was assessed by an intravenous glucose injection followed by determinations of blood glucose levels at 0, 10, 30, 60, and 120 min. We observed that cetirizine did not affect the glucose tolerance of ND mice (). The glucose concentrations of HFD mice, however, tended to be lower in the cetirizine-treated mice at all time points after the glucose injection (). To test whether there was a significant difference in the glucose tolerance, we calculated the area under the curve (AUC) and observed a significantly lower AUC in the cetirizine-treated mice (). Cetirizine did not exert any effect on the AUC of ND mice ().
Figure 4. Glucose tolerance test of ND and HFD mice treated with cetirizine for 2 weeks. After 2 weeks of cetirizine treatment ND (A) and HFD (B) mice were fasted for 8 h and injected intravenously with glucose (2.5 g/kg). Blood glucose levels were analyzed at the time points given in the Figure. Results are means ± SEM for 8–10 mice in each group. C: Data from were recalculated to area under the curve (AUC). *P < 0.05 versus HFD mice without cetirizine treatment using the Mann–Whitney U test for non-parametric populations.
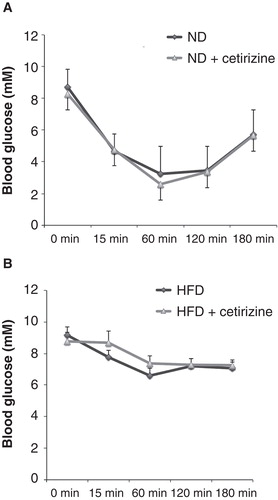
An insulin sensitivity test was also performed after the 9 weeks of HFD and 2 weeks of cetirizine treatment, and we observed that cetirizine did not affect the insulin sensitivity of ND mice (). HFD mice displayed a marked reduction in insulin sensitivity as compared to ND mice (). The insulin sensitivity of HFD mice was not affected by the cetirizine treatment ().
Figure 5. Insulin sensitivity test of ND (A) and HFD (B) mice treated for 2 weeks with cetirizine. Insulin (1.6 U/kg, Actrapid, Novo Nordisk, Bagsværd, Denmark) was injected intraperitoneally, and blood glucose was analyzed at the time points given. Results are means ± SEM for 8–10 mice in each group.
Discussion
In NOD mice first macrophages and later other immune cells including T-cells infiltrate the pancreatic islets leading to insulitis. As this process progresses with time, beta-cells are destroyed via direct cell-to-cell contacts between the beta-cells and T-killer cells (Citation25). This autoimmune-mediated destruction of beta-cells is a complex process, which at least in part seems to involve the activity of pro-inflammatory cytokines (Citation26). Because histamine, via the H1-receptor, in some cases promotes T-cell Th1 polarization, increases dendritic cell-mediated activation of cytotoxic CD8 T-cells, and augments inflammatory events in general (Citation1), we investigated its putative role in diabetes of NOD mice using the H1-receptor antagonist cetirizine. In our experimental set-up, in which the mice were given cetirizine in the drinking water from 9 to 30 weeks of age, cetirizine affected neither the overall diabetes incidence nor time of diabetes precipitation. This argues against a major role of immune- and islet cell H1-receptor activity in the late-stage autoimmune destruction of beta-cells that occurs after 9 weeks of age. However, we cannot exclude that H1-receptor activation affects earlier events in the NOD mouse, such as antigen priming and the initial expansion of beta-cell reactive T-cell clones occurring early in regional lymph nodes (Citation27), and that cetirizine supplementation to the drinking water from for example the fourth week might have given a different result. Indeed, allergic symptoms in infants have been reported to be associated with the development of type 1 diabetes-related auto-antibodies during the first years of life (Citation28). Nor can we exclude that doses of cetirizine other than the one presently used might have affected diabetes in NOD mice. The transferral of the H1-receptor knockout to a NOD background might bring more clarity to the putative role of histamine and the H1-receptor in type 1 diabetes.
We here report that the non-fasting blood glucose and the glucose tolerance of high-fat diet C57BL/6 mice were improved by a 2-week cetirizine treatment. At first glance, this appears to be at variance with previous studies using H1-receptor knockout mice. A genetic loss of H1-receptor activity promotes hyperleptinemia, visceral adiposity, and moderate insulin resistance in mice given a high-fat diet (Citation5,29). In addition, decreased weight and increased energy expenditure has been observed in mice infused with histamine to the central nervous system (Citation30). This is commonly explained by the leptin–histamine loop, a system in which the leptin-sensing neurons localized in hypothalamic nuclei require histamine-induced signaling for leptin to exert its effects. Histamine-facilitated leptin signaling in the central nervous system is known to decrease food intake and increase uncoupling in adipose tissue (Citation5,29,30). Accordingly, lack of H1-receptor signaling will result in increased leptin resistance, increased food intake, and decreased energy expenditure. The discrepancy between the present results using the H1-receptor antagonist cetirizine and the H1-receptor knockout mouse may be explained by the low permeability of cetirizine, a second-generation antihistamine, across the blood–brain barrier. Thus, it is reasonable to assume that cetirizine affects mainly histamine signaling in the periphery and to a lesser extent the central leptin–histamine loop. Nevertheless, we cannot completely rule out a modest effect of cetirizine on the central leptin–histamine loop. For example, our results might hint at a minor trend to increased body weight in response to cetirizine, and the high dose of cetirizine here used (25 mg/kg) could have facilitated some leakage across the blood–brain barrier.
Inhibition of the H1-receptor outside the central nervous system improved non-fasting blood glucose and the glucose tolerance of high-fat diet mice. As we did not measure insulin during the glucose tolerance test, it is not clear whether beta-cell function was actually improved. However, our results may suggest that peripheral inhibition of H1-receptor signaling improves beta-cell function, and that this occurs without affecting peripheral insulin resistance. In type 2 diabetes there is a shift in the profile of immune cells, both in islets and in adipose tissue, from an anti-inflammatory to a pro-inflammatory state (Citation31). Furthermore, islet macrophages, in response to hyperlipidemia, have been proposed to activate the NLRP3 inflammasome leading to IL-1β release, amyloid formation, and beta-cell dysfunction (Citation32). As we have recently observed that cetirizine partially counteracted the effects of the pro-inflammatory cytokines IL-1β and IFN-γ on beta-TC6 cell death (Citation14), possibly via modulating c-Jun N-terminal Kinase (JNK) activity and release of the chemokine macrophage migration inhibitory factor (MIF), it may be that H1-receptor signaling exacerbates islet inflammation and therefore also beta-cell dysfunction. Interestingly, other strategies to reduce systemic inflammation besides using cetirizine, for example by treatment with omega 3-fatty acids or an IL-1β neutralizing monoclonal antibody, have in some cases also failed to reduce peripheral insulin resistance (Citation33,34). Thus, anti-inflammatory treatments might have less effect on peripheral insulin resistance than on islet dysfunction, a notion that fits well with the results of the present investigation.
It is not yet known whether oral antihistamines affect glucose intolerance or type 2 diabetes in human subjects. The present findings open the possibility that targeted inhibition of the H1-receptor expressed on cells in pancreatic islets, and not targeted to adipose/neuronal tissues, can result in improved metabolic control.
Acknowledgements
This work was supported in part by the Swedish Diabetes Association, the Family Ernfors Fund, Barndiabetesfonden, and the Novo-Nordisk Foundation.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Neumann D, Schneider EH, Seifert R. Analysis of histamine receptor knockout mice in models of inflammation. J Pharmacol Exp Ther. 2014;348:2–11.
- Rajasekaran N, Solomon S, Watanabe T, Ohtsu H, Gajda M, Bräuer R, et al. Histidine decarboxylase but not histamine receptor 1 or 2 deficiency protects from K/BxN serum-induced arthritis. Int Immunol. 2009;21:1263–8.
- Bene L, Sápi Z, Bajtai A, Buzás E, Szentmihályi A, Arató A, et al. Partial protection against dextran sodium sulphate induced colitis in histamine-deficient, histidine decarboxylase knockout mice. J Pediatr Gastroenterol Nutr. 2004;39:171–6.
- Rozenberg I, Sluka SH, Rohrer L, Hofmann J, Becher B, Akhmedov A, et al. Histamine H1 receptor promotes atherosclerotic lesion formation by increasing vascular permeability for low-density lipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:923–30.
- Wang KY, Tanimoto A, Yamada S, Guo X, Ding Y, Watanabe T, et al. Histamine regulation in glucose and lipid metabolism via histamine receptors: model for nonalcoholic steatohepatitis in mice. Am J Pathol. 2010;177:713–23.
- Noubade R, Milligan G, Zachary JF, Blankenhorn EP, del Rio R, Rincon M, et al. Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J Clin Invest. 2007;117:3507–18.
- Jutel M, Watanabe T, Klunker S, Akdis M, Thomet OA, Malolepszy J, et al. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. 2001;413:420–5.
- Vanbervliet B, Akdis M, Vocanson M, Rozières A, Benetière J, Rouzaire P, et al. Histamine receptor H1 signaling on dendritic cells plays a key role in the IFN-γ/IL-17 balance in T cell-mediated skin inflammation. J Allergy Clin Immunol. 2011;127:943–53.
- Ma RZ, Gao J, Meeker ND, Fillmore PD, Tung KS, Watanabe T, et al. Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science. 2002;297:620–3.
- Saligrama N, Noubade R, Case LK, Poynter ME, Teuscher C. H1R expression by CD11B+ cells is not required for susceptibility to experimental allergic encephalomyelitis. Cell Immunol. 2012;278:27–34.
- Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;8:291–7.
- Cnop M, Welsh N, Jonas J, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54S2:S97–107.
- Campoli-Richards DM, Buckley MM, Fitton A. Cetirizine. A review of its pharmacological properties and clinical potential in allergic rhinitis, pollen-induced asthma, and chronic urticaria. Drugs. 1990;40:762–81.
- Anvari E, Fred RG, Welsh N. The H1-receptor antagonist cetirizine protects partially against cytokine- and hydrogen peroxide-induced β-TC6 cell death in vitro. Pancreas. 2014;43:624–9.
- Yoneda K, Yamamoto T, Ueta E, Osaki T. Suppression by azelastine hydrochloride of NF-kappa B activation involved in generation of cytokines and nitric oxide. Jpn J Pharmacol. 1997;73:145–53.
- Roumestan C, Henriquet C, Gougat C, Michel A, Bichon F, Portet K, et al. Histamine H1-receptor antagonists inhibit nuclear factor-kappaB and activator protein-1 activities via H1-receptor-dependent and –independent mechanisms. Clin Exp Allergy. 2008;38:947–56.
- Arnold R, Rihoux J, König W. Cetirizine counter-regulates interleukin-8 release from human epithelial cells (A549). Clin Exp Allergy. 1999;29:1681–91.
- Jinquan T, Reimert CM, Deleuran B, Zachariae C, Simonsen C, Thestrup-Pedersen K. Cetirizine inhibits the in vitro and ex vivo chemotactic response of T lymphocytes and monocytes. J Allergy Clin Immunol. 1995;95:979–86.
- Ashenager MS, Grgela T, Aragane Y, Kawada A. Inhibition of cytokine-induced expression of T-cell cytokines by antihistamines. J Investig Allergol Clin Immunol. 2007;17:20–6.
- Roch-Arveiller M, Tissot M, Idohou N, Sarfati G, Giroud JP, Raichvarg D. In vitro effect of cetirizine on PGE2 release by rat peritoneal macrophages and human monocytes. Agents Actions. 1994;43:13–16.
- Cheria-Sammari S, Aloui R, Gormand F, Chabannes B, Gallet H, Grosclaude M, et al. Leukotriene B4 production by blood neutrophils in allergic rhinitis--effects of cetirizine. Clin Exp Allergy. 1995;25:729–36.
- Charlesworth EN, Kagey-Sobotka A, Norman PS, Lichtenstein LM. Effect of cetirizine on mast cell-mediator release and cellular traffic during the cutaneous late-phase reaction. J Allergy Clin Immunol. 1989;83:905–12.
- Dridi D, Ben Attia M, Reinberg A, Boughattas NA. [Circadian rhythms in the toxic effects of the histamine antagonist cetirizine in mice]. Pathol Biol (Paris). 2005;53:193–8; French.
- Patel AC, Nunez NP, Perkins SN, Barrett JC, Hursting SD. Effects of energy balance on cancer in genetically altered mice. J Nutr. 2004;134:3394S–8S.
- Bach JF, Mathis D. The NOD mouse. Res Immunol. 1997;148:285–6.
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26.
- La Torre D, Lernmark A. Immunology of beta-cell destruction. Adv Exp Med Biol. 2010;654:537–83.
- Wahlberg J, Vaarala O, Ludvigsson J; ABIS Study Group. Asthma and allergic symptoms and type 1 diabetes-related autoantibodies in 2.5-yr-old children. Pediatr Diabetes. 2011;12:604–10.
- Masaki T, Yoshimatsu H, Chiba S, Watanabe T, Sakata T. Targeted disruption of histamine H1-receptor attenuates regulatory effects of leptin on feeding, adiposity, and UCP family in mice. Diabetes. 2001;50:385–91.
- Masaki T, Yoshimatsu H, Chiba S, Watanabe T, Sakata T. Central infusion of histamine reduces fat accumulation and upregulates UCP family in leptin-resistant obese mice. Diabetes. 2001;50:376–84.
- McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48.
- Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105:141–50.
- Spencer M, Finlin BS, Unal R, Zhu B, Morris AJ, Shipp LR, et al. Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes. 2013;62:1709–17.
- Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–48.