Abstract
Melperone is an atypical antipsychotic agent that has shown a wide spectrum of neuroleptic properties, particularly effective in the treatment of senile dementia and Parkinson’s-associated psychosis, and is marketed in Europe as an immediate-release (IR) tablet and syrup. An orally disintegrating tablet (ODT) dosage form would be advantageous for patients who experience difficulty in swallowing large tablets or capsules or those who experience dysphagia. Controlled-release (CR) capsule and ODT formulations containing melperone HCl were developed with target in vitro release profiles suitable for a once-daily dosing regimen. Both dosage forms allow for the convenient production of dose-proportional multiple strengths. Two ODT formulations exhibiting fast and medium release profiles and one medium release profile capsule formulation (each 50 mg) were tested in vivo using IR syrup as the reference. The two medium release formulations were shown to be bioequivalent to each other and are suitable for once-daily dosing. Based on the analytical and organoleptic test results, as well as the blend uniformity and in-process compression data at various compression forces using coated beads produced at one-tenth (1/10) commercial scale, both formulations in the form of CR capsules and CR ODTs have shown suitability for progression into further clinical development.
Introduction
Melperone is an atypical antipsychotic with neuroleptic properties and is structurally related to haloperidol. It is used to treat schizophrenia, senile dementia, and the psychosis that typically develops with long-term use of anti-Parkinsonian agents. Oral doses are rapidly absorbed (Tmax 1.5–3.0 hours upon oral tablet administration) with a bioavailability of 50–70%. Following oral administration of a 100 mg melperone tablet, the pharmacokinetics, expressed as AUC or Cmax, showed non-linearity, possibly due to saturation of the hepatic elimination system. The elimination half-life is approximately 3 hours after a single dose oral administration and approximately 8 hours at steady stateCitation1–5.
Formulations of melperone permitting a once- or twice-daily dosage regimen are desirable in order to improve compliance in patients with schizophrenia or in the treatment of psychosis associated with Parkinson’s Disease (PD). An orally disintegrating tablet (ODT) formulation is most desirable in view of the swallowing dysfunction seen in early stages of PD. However, developing ODT dosage forms comprising sustained-release (SR) melperone beads having a sufficiently small mean particle size not to feel gritty in the mouth (≤300 µm), while exhibiting a sufficiently extended-release profile to be suitable for a once-daily dosing regimen, is very challenging due to melperone’s pKa and extreme solubility in the physiologically relevant pH range (>800 mg/mL). The purpose of this paper is to describe the development of both once-daily controlled-release (CR) dosage forms – capsule and ODT formulations of melperone – to provide convenience and/or improve patient compliance with dosing regimen.
Materials and methods
Materials
The following materials were procured: Melperone hydrochloride from Lundbeck (Padova, Italy); Sugar Spheres, NF (25–30 mesh, 45–60 mesh, or 60–80 mesh) from Paulaur Coporation (Cranbury, NJ); hydroxypropylcellulose NF (Klucel® LF) from Ashland Aqualon (Hopewell, VA); ethylcellulose, NF (Ethocel Standard 10 Premium) from Colorcon (West Point, PA); Hypromellose phthalate (HP-55) from Shin Etsu Chemical Company (Tokyo, Japan); triethyl citrate and dibutyl sebacate from Vertellus Performance Chemicals (Greensborro, NC); polyethylene glycol (PEG 400) from Dow Chemicals (Manro, LA), a plasticizer; Dibasic Sodium Phosphate Anhydrous from Jost Chemicals (St. Louis, MO); D-mannitol (Pearlitol 25) from Roquette (France); Crosspovidone, NF (Polyplasdone XL-10) from GAF (Wayne, NJ); Sucralose, NF from Tate & Lyle (Singapore, China); magnesium stearate from Tai Hei Chemical (Tokyo, Japan); Wintergreen and peppermint from Synergy (Wauconda, IL); Gelatin Capsules, NF from Capsugel® (Greenwood, SC).
Pharmacokinetic modeling
Melperone exhibits non-linear pharmacokinetics particularly above 50 mg. Since the target dose strength of melperone for the treatment of psychosis in patients associated with PD is 50 mg, the PK data available for 25 and 50 mg dosesCitation6,Citation7 (unpublished data, H. Larsson, Department of Biochemistry, AB Ferrosan, Malmo, Sweden 1976) were used to derive target release profiles for a sustained release formulation suitable for a once-daily dosing regimen (). These pharmacokinetic profiles were utilized to estimate the pharmacokinetic parameters using WinNonlin version 4.1 (Pharsight Corporation, Mountain View, CA). Despite the absence of a well defined distribution phase in the case of melperone as reportedCitation6, the intravenous (IV) and oral data could be fitted well to a two compartment model described by the following equation:
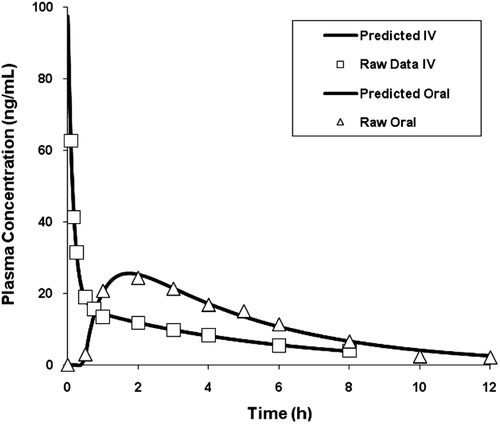
Figure 1. Actual and predicted plasma concentration – time profiles for 20 mg intravenous (IV) and 50 mg oral administrations of melperone.
Where C (ng/mL) is the concentration in the central compartment, Ka (hr−1) is the first order absorption constant, F is the bioavailability, D (ng) is the dose, V (mL) is the volume of distribution, K12 (hr−1) is the rate constant into the peripheral compartment, K21 (hr−1) is the rate constant from the peripheral compartment, α (hr−1) is the distribution rate constant, β (hr−1) is the elimination rate constant, T (hr) is time, and Tlag (hr) is the lag time.
The fitting and parameter estimates are shown in and . The predicted plasma profiles are very similar to the actual plasma concentration-time profiles following oral and IV administrations of melperone.
Table 1. Paramater estimates of 20 mg I.V. and 50 mg P.O. melperone.
Determination of Cav,ss for melperone
Using the estimates of the plasma concentration, profiles for melperone were generated. AUC0-t,ss was obtained from simulated data at steady state were τ = dosing interval = 12 hours. The Cav,ss was calculated using the following relationship:
Simulation of melperone levels
Based on the above initial estimates, drug levels were simulated to evaluate the feasibility of a sustained release dosage form, assuming a two compartment open model with:
| 1. | First order dosage form release | ||||
| 2. | First order absorption | ||||
| 3. | First order elimination | ||||
| 4. | Linear pharmacokinetics (through 50 mg) | ||||
| 5. | Unit dose (50 mg) every 24 hours | ||||
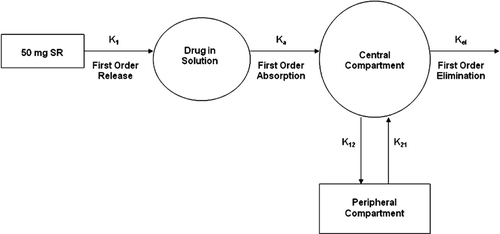
A schematic representing this model is shown in . The model can be defined by the following set of equations:
Figure 2. Schematic of the 2-compartment PK modeling for melperone based on first order kinetics of drug release from SR beads into the environment of use, systemic absorption of the released drug, and distribution and elimination of the absorbed drug.
Where:
A = Dose
B = Drug concentration in the GI tract
C = Concentration in central compartment
D = Concentration in peripheral compartment
K1 = Drug release rate constant
Ka = Absorption rate constant
Kel = Elimination rate constant
V = Volume of distribution
K12 = Rate constant into peripheral compartment
K21 = Rate constant from the peripheral compartment
A set of theoretical release rates (K1 values) was then used in the simulations to determine theoretical potential in vitro drug release profiles. The anticipated drug release profiles are shown in while the corresponding values of T95 (time to reach 95% of drug release) are shown in . These profiles were to represent the desired release profile such that a variety of formulations could be manufactured and assessed in a clinical study. lists the simulated AUC0–24 hr values for 25- and 50-mg doses at the above release rate constants and as an immediate release (IR). The plasma concentration-time profiles at 25- and 50-mg doses were calculated by simulation using the above model and are shown in –.
Figure 3. Predicted in vitro melperone release profiles of prototype once-daily dosage forms: (A) fast, medium, or slow release dosage forms, 25 mg and (B) fast, (C) medium, and (D) slow release dosage forms, 25 and 50 mg.
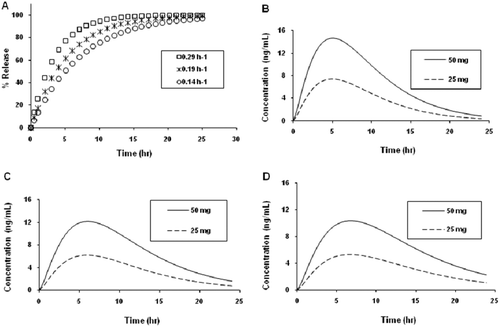
Table 2. Time for release of 95% of the drug load (T95).
Table 3. AUC of melperone at different release rates.
Solubility of melperone as a function of pH
An excess of melperone HCl was suspended in 0.1N HCl, USP Water, or in a phosphate buffer at a pH of 4.5, 6.8, 7.0, 7.1, or 7.8 for about 30 min while stirring, and the filtered aliquot was diluted with HPLC mobile phase and the solubility was determined by HPLC (see for solubility values).
Table 4. Solubility (mg/mL) of melperone HCl.
Melperone SR beads, CR capsules, ODT-CR
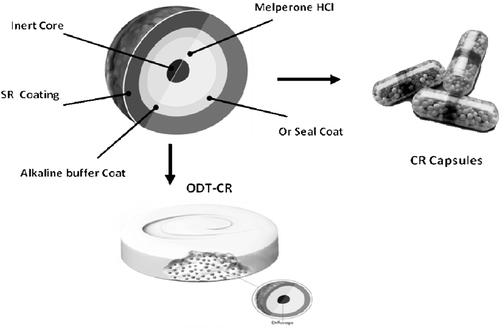
A schematic for the SR bead and corresponding CR capsules or ODT-CR tablet comprising such SR beads is shown in . For the preparation of CR capsules, melperone hydrochloride is layered onto 20–25 mesh sugar spheres producing IR beads at a drug load of 25% w/w and 2% w/w seal coat of Klucel® LF (hydroxypropylcellulose). These IR beads are coated with water-insoluble ethylcellulose (Ethocel Standard 10 Premium) with polyethylene glycol (PEG 400) as the plasticizer, and filled into hard gelatin capsules (50 mg dose) on a capsule filler, MG-2 Futura as discussed in detail elsewhereCitation8. For preparing ODT-CR tablets, melperone hydrochloride is layered onto 60–80 mesh or 45–60 mesh sugar spheres producing IR beads at a drug load of 25% w/w and 2% w/w seal coat of Klucel LF (hydroxypropylcellulose). These IR beads are coated with an alkaline buffer coat comprising anhydrous dibasic sodium phosphate and further coated with water-insoluble ethylcellulose (Ethocel Standard 10 Premium) with dibutyl sebacate or triethyl citrate as the plasticizer to produce SR beads. The SR beads prepared by either of the processes described above are referred to as Diffucaps® beads. These SR beads blended with rapidly dispersing microgranules (AdvaTab® base granules), prepared as described in detail elsewhereCitation9,Citation10, microcrystalline cellulose, sucralose, a flavor (peppermint or wintergreen) and compressed into ODT-CR tablets (50 mg dose weighing 1000 mg) on a rotary tablet press equipped with an external lubrication system (e.g., Hata Tablet Press- Matsui ExLub System) and 15 mm round, flat faced, radius edge toolingCitation11.
Figure 4. Schematic of SR bead (Diffucaps® bead comprising an inert core sequentially coated with a drug layer, a protective seal coat, an alkaline buffer layer, and an outer SR polymer coat).and CR Capsules and ODT tablet comprising SR beads.
Preparation of melperone IR beads
Melperone hydrochloride was slowly added to 50/50 acetone/water while stirring until dissolved. A Glatt GPCG 5 equipped with a 9” bottom spray Wurster 13” column, 25 mm partition gap, 14 mm tubing, bottom air distribution ‘D’ plate with 200 mesh screen, atomization air pressure of 1.30 bar and 1.0 mm nozzle diameter was charged with 3000 g of sugar spheres (e.g. 20–25 mesh, 25–30 mesh, 45–60 mesh, or 60–80 mesh; for incorporation into ODT formulations, the starting sugar spheres need to be as small as possible to avoid grittiness, while for filling into capsules the starting sugar sphere size is not critical, and hence larger sugar spheres can be used in order to better control the dissolution rate.) that were sprayed with the melperone coating solution (15% solids) at 30–36 mL/min while maintaining the product temperature at 34 ± 2°C and fluidization air volume at 100 CFM for a drug load of 20% or 25% by weight. A seal coat of Klucel® LF dissolved in acetone/water was applied for a weight gain of 2%, and the resulting IR beads were dried to remove residual moisture and acetone.
Preparation of alkaline buffer coated melperone IR beads
Dibasic sodium phosphate anhydrous was dissolved in purified water and spray coated onto IR beads at a spray rate of 2 mL/min with a stepwise increase to 20 mL/min for a weight gain of 3.0% in a Glatt GPCG 3 equipped with a 6” bottom spray Wurster column, ‘C’ air distribution plate covered with a 200 mesh product retention screen, and 1.0 mm port size nozzle. Following completion of spraying and rinsing the spray system with acetone, a protective seal coat solution (Klucel LF dissolved in 85/15 acetone/water, 7.0% solids) was coated onto the disodium phosphate coated beads for a 2% coating weight gain at a product temperature of 33 ± 2°C and a flow rate of 10 mL/min. The dried disodium phosphate coated IR beads were sieved to discard oversized beads and fines, if any.
Preparation of melperone SR beads
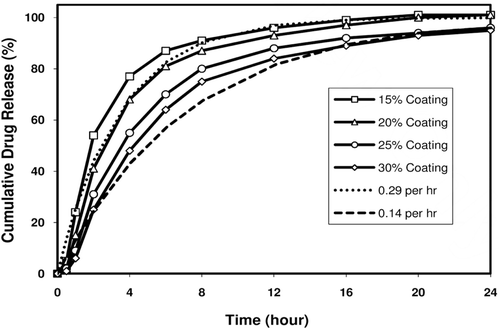
The alkaline buffer-coated IR beads were fluid-bed coated with an SR functional polymer coating formulation of ethylcellulose plasticized with dibutyl sebacate (DBS) at a ratio of 92.5/7.5 ethylcellulose/DBS dissolved in 85/15 acetone/water mixture (10% solids) at a product temperature of 33 ± 1°C, atomization air pressure of 1.25 bar, inlet air volume of 15 CFM, and an initial flow rate of 8 mL/min with a stepwise increase to 20 mL/min for a SR coating level of 20%, 25%, or 30% by weight. Hydroxypropylcellulose (Klucel® LF) dissolved in acetone/water was also coated onto the SR-coated beads to function as a compressible coating thereby minimizing the potential for fracture during compression of SR beads. The resulting SR beads were dried in the Glatt unit for five minutes to drive off residual solvents and sieved using 30 and 80 mesh sieves to discard oversized beads and fines (see for drug release profiles). Oversized beads are typically doubles, if at all formed, and may behave differently from normal coated beads in terms of lease profiles, especially upon compression, while fines are usually coating polymer fragments, if formed due to spray drying.
Figure 5. Melperone HCl release profiles from prototype SR bead formulations. Figure shows theoretical fast and slow release profiles and melperone release profiles from prototype SR beads coated at 15, 20, 25, and 30% by weight of the SR beads.
Preparation of melperone TPR beads
Melperone hydrochloride IR beads were coated with a TPR (timed, pulsatile release) coating composition comprising EC-10/HP-55/TEC (ethylcellulose 10 cps/hypromellose phthalate/triethyl citrate at a ratio of 70/20/10) dissolved in 90/10 acetone/water (10% solids) for a weight gain of 35% (relative to the weight of the coated bead) in a Glatt GPCG 3 equipped with a 7” bottom spray Wurster insert at an inlet temperature of 40–44°C and inlet air volume at 35 CFM. The TPR beads were further coated with the compressible coating of Klucel® LF at 2% by weight.
Preparation of rapidly dispersing microgranules
Rapidly dispersing microgranules were prepared following the procedure disclosed elsewhereCitation13,Citation14. D-mannitol (152 kg) with an average particle size of approximately 20 µm or less (Pearlitol 25 from Roquette, France) was blended with cross-linked povidone (Crospovidone XL-10 from ISP) in a high shear granulator (GMX 600 from Vector), granulated with purified water (approximately 32 kg), wet-milled using a Comil from Quadro, and finally tray-dried to provide microgranules having an LOD (loss on drying) of about 0.8%. The dried granules were sieved, and oversize material was again milled to produce rapidly dispersing microgranules with an average particle size in the range of approximately 175–300 µm.
Preparation of melperone ODT-CR
Melperone hydrochloride SR beads (see for compositions of ODT-CR1 and ODT-CR2) were blended with rapidly dispersing microgranules, and other pre-blended pharmaceutically acceptable ingredients (i.e., peppermint or wintergreen flavor, sucralose, crospovidone, and microcrystalline cellulose) at a ratio of rapidly dispersing microgranules to melperone HCl SR beads of about 3:2 in a twin shell V-blender for a sufficient time to provide a homogeneously distributed blend for compression. The Hata tablet press was set up with appropriate punches and dies (15 mm flat face radius edge with no embossing) and the Matsui Ex-Lube system was set up to uniformly spray magnesium stearate onto the material contacting surfaces of the punches and dies at a desired spray rate. The hopper was filled with the compression mix. The compression parameters were adjusted to fill the dies with correct weights of the compression mix and compressed into tablets having a desired friability. After the main compression force equilibrated, 10 tablets were collected and tested for individual weights, thickness, hardness and friability. Once meeting the target weight (1 g), hardness (>30 N), and friability (≤0.6%), the press was operated in the ‘Auto’ mode to begin tableting. Tablets were periodically collected as a ‘composite’ and all compression attributes were recorded in the in-process compression data sheet of the batch record. Melperone HCl ODT-CR (50 mg) thus produced, rapidly disintegrated in the oral cavity creating a smooth, easy-to-swallow suspension comprising coated melperone HCl beads, and met the disintegration time specification of not more than 30 sec when tested by USP <701> method as well as provided a target in vitro release profile suitable for a once-daily dosing regimen ().
Figure 6. Melperone HCl release profiles from prototype ODT-CR formulations. Figure shows similar melperone release profiles from prototype ODT CR 161 and ODT CR 55 together with the corresponding SR beads.
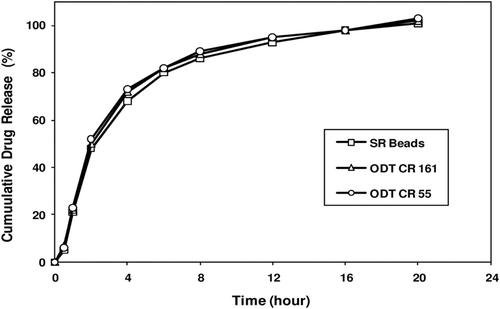
Table 5. Compositions and tableting properties of ODTs.
Preparation of melperone CR capsules
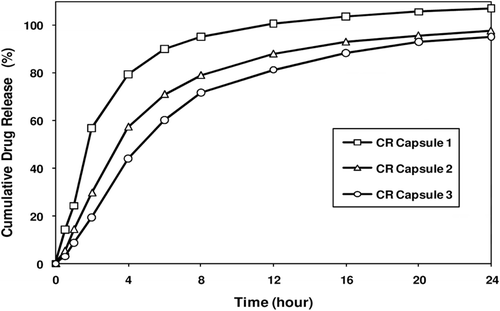
Melperone hydrochloride IR beads were coated with a solution of EC-10 and PEG 400 at a ratio of 90/10 dissolved in 85/15 acetone/water (10% solids) for a weight gain of 20%, 25%, and 30% following the procedures as disclosed above, and dried in the Glatt GPCG 5 at approximately 33°C for 5 minutes to drive off residual moisture and acetone. Dried beads were sieved to discard oversize beads, if formed. An appropriate amount of the SR beads at a coating weight of 20% (247.5 mg), 25% (267.4 mg) and 30% (287.4 mg) were filled into hard gelatin capsules (approximately 6000 capsules of each formulation) using an MG-2 Futura 8400 equipped with a 4.8 mm dosing ring to produce 50 mg melperone hydrochloride CR capsules. demonstrates the in vitro drug release profiles from these capsules, matching the release profiles calculated from the two-compartment model as shown in .
Figure 7. Melperone HCl release profiles from prototype CR Capsule formulations. Figure shows varying release profiles from fast, medium, and slow release capsule formulations containing SR beads coated at different levels.
Analytical test methods
The analytical methods developed to test intermediate and finished products include the tests for identification, dissolution, assay/impurities, uniformity of dosage forms, moisture content, and residual acetone content. The SR beads, TPR beads, CR capsules and ODT-CR tablets were dissolution tested using USP Apparatus 2 (paddles@ 75 RPM) and a 2-stage dissolution media (testing at 37.0 ± 0.5°C in 700 mL of 0.1N HCl for the first 2 hrs followed by testing for additional up to 22 hrs in 900 mL of pH 6.8 obtained by adding 200 mL of a pH modifier). Samples (1.5 mL) pulled at predetermined time points were tested for melperone by HPLC methodology (detector set at 248 nm) using Varian Pursuit XRs C18, 5 µm, 150 × 4.6 mm with guard column, 70:30 KH2PO4: acetonitrile (pH = 3.0 ± 0.05) mobile phase, 1.5 mL/min flowrate, and 10 µL injection volume. Beads, capsule contents, or ODT tablets were dissolved in acetonitrile, diluted with USP water, centrifuged and tested for melperone by HPLC. Individual unknown impurities were quantified by dividing the integrated areas of the individual unknown impurity peaks by the total area (i.e., melperone + sum of the individual impurities) while the individual specified identified impurities were quantified by dividing the individual area by the relative response factor of the corresponding impurity standard. The moisture content was determined by Karl Fischer titration method. Residual acetone contents were estimated by gas chromatograph equipped with a helium purged packed column and flame ionization detector. Disintegration times of ODT tablets were determined by current USP <701> without the use of disks and with USP water as the media.
Preparation of pilot CTM supplies
Melperone HCl ODT-CR tablets Test 1 (drug load: 20% w/w and SR coating: 30% w/w, see for composition) and Test 2 (drug load: 25% w/w and SR coating: 30% w/w, see for composition) comprising 50-mg melperone HCl as SR beads were manufactured under cGMP conditions by applying a drug layering, onto 45-60 sugar spheres, a protective seal coating, an alkaline buffer coating, and a protective seal coating (if present), and further applying an SR coating of ethylcellulose and dibutyl sebacate (92.5/7.5 Ethocel 10/DBS) for a weight gain of 25% w/w followed by a compressible coating of Klucel® LF at 2% by weight, blending with appropriate amounts of rapidly dispersing microgranules and other excipients (see for compositions), and compressing the blend as described previously.
Melperone HCl CR Capsules (Test 3) containing 50-mg melperone HCl as SR beads were manufactured under cGMP conditions by coating IR beads at a drug load of 25% by weight using 25-30 mesh sugar spheres for drug layering/protective seal coat of Klucel LF at 2% by weight, with ethylcellulose and polyethylene glycol 400 (92.5/7.5 Ethocel 10/PEG 400) for a weight gain of 25% w/w and filling appropriate amount of SR beads into size # 1 hard gelatin capsules, as described previously (see for the release profiles).
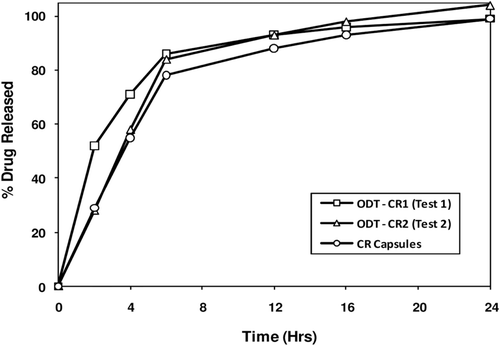
Figure 8. In vitro drug release profiles of pilot CTM supplies – ODT-CR (Test 1 (fast release) & Test 2 (medium release) contain an alkaline buffer layer and smaller inert cores) and CR Capsule (Test 3 contain larger inert cores without an alkaline buffer layer).
Pilot PK studies
A single-dose, randomized, crossover study was conducted using healthy adult subjects under fasting conditions to assess the pharmacokinetics of four treatments: 25 and 50-mg syrup, 50-mg Test 1 (ODT-CR 1 [fast]), 50 mg Test 2 (ODT-CR 2 [medium]), and 50-mg Test 3 (CR capsule). The melperone plasma concentration vs. time profiles are illustrated in . Dose normalized least square mean PK parameters and 90% confidence intervals for reference (melperone syrup), and Test 1, test 2, and Test 3 are presented in and .
Figure 9. Plasma concentration-time profiles of pilot CTM supplies – Syrup 25-mg, ODT-CR (Test 1 (fast release) & Test 2 (medium release)), and CR Capsule (Test 3).
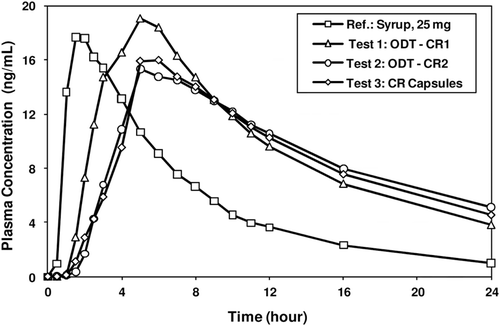
Table 6. PK parameters for melperone treatments A (25-mg Syrup; 50-mg Syrup), B (50-mg ODT-CR 1), C (50-mg ODT-CR 2), and D (50-mg CR Capsule).
Table 7. Melperone plasma PK data - least-squares means and 90% CIs.
Product Stability
Pilot CTM supplies including melperone HCl CR capsules, ODT-CR 1 (fast) and ODT-CR 2 (medium) packaged in 60 cc (capsules) or 75 cc (ODTs) induction-sealed, HDPE bottles (30 counts) were subjected to stability evaluation per International Committee on Harmonization guidance for 6 months. The stability data are presented in .
Table 8. Stability data for Melperone CR Capsules and ODT-CR 2.
Manufacture of melperone SR beads at one-tenth commercial scale
Melperone hydrochloride (15.0 kg) was dissolved in 50/50 mixture of acetone and water (50 kg each), with constant stirring. A Glatt GPCG 120 equipped with an 18” bottom spray Wurster 23.75” column; outer “G” and inner “C” bottom air distribution plate covered with a 100 mesh product retention screen, partition height: 50 mm; nozzle port size: 3.0 mm with HS Collar; was charged with 43.8 kg of 45–60 mesh sugar spheres. The above melperone solution was sprayed onto the sugar spheres at an initial rate of 100 g/min with ramp up (range: 75–700 g/min); inlet air volume: 450 CFM (range: 500–900 CFM); set process air temperature: 45°C (range: 39–65°C); atomization air pressure: 2 bar; target product temperature: 32°C (range: 29–36°C). Following rinsing of the spray system with 50/50 acetone/water, a sealant solution (Klucel LF dissolved in 85/15 acetone/water, 7.5% solids) was sprayed onto the melperone-coated beads at an initial rate of 200 g/min and a stepwise increase to 300 g/min, and process air temperature of 45°C (range: 35–50°C). The resulting sealant-coated beads (Klucel® LF coating at 2% w/w) were then dried in the Glatt unit for 5 min. to drive off residual solvents. The resulting IR beads were sieved to discard oversized (>500 µm or 35 mesh) beads and fines (<80 mesh).
Dibasic sodium phosphate anhydrous (DPA; 6.2 kg) was dissolved in 124 kg of purified water while stirring. Melperone hydrochloride IR beads (52.6 kg) were coated with the aqueous alkaline buffer solution at a fluidization air volume of 650 CFM (range: 400–800 CFM); atomization air pressure: 2.5 bar, target product temperature: 53°C (range: 49–60°C); spray rate: 100 g/min with ramp up to 400 g/min (range: 75–700 g/min). Following the buffer layering, a protective seal coat with an Opadry Clear aqueous solution (6% solids) was applied to the DPA coated beads for a weight gain of about 2% (relative to the weight of the seal-coated beads) at a product temperature of 50°C and a flow rate of 200 g/min (range: 100–350 g/min). The seal-coated beads were then dried in the Glatt unit for not more than 5 min. to drive off residual solvents.
Ethylcellulose, EC-10 (13.9 kg) was slowly added to 85/15 acetone/water mixture (214.2 kg), with stirring, until dissolved. Dibutyl sebacate (1.1 kg) was then slowly added to the polymer solution and stirred for 30 min. The buffer-coated melperone IR beads (34.0 kg) were charged into a Glatt GPCG 120 equipped with an 18” bottom spray Wurster 18” column, 3.0 mm nozzle port with HS Collar and bottom inner “G” and outer “C” distribution plate, and coated with the above SR coating solution (7% solids) at an initial rate of 100 g/min with ramp up to 500 g/min (range: 75–700 g/min); inlet air volume: 500 CFM (range: 400–900 CFM); process air temperature: 64°C (range: 40–70°C); atomization air pressure: 2.5 bar; target product temperature: 33°C (range: 29–40°C). Following rinsing with acetone, the SR-coated beads were sprayed with a compressible coating solution (Klucel® LF dissolved in 85/15 acetone/water, 7.5% solids) at an initial rate of 200 g/min (range: 100–350 g/min); process air temperature: 45°C (range: 35–50°C); product temperature: 32°C (range: 28–40°C), and then dried in the Glatt unit for 5 min. to drive off residual solvents (including moisture).
Pharmaceutically acceptable ingredients (i.e., peppermint flavor, sucralose, crospovidone, and microcrystalline cellulose; see ) were first blended in a V blender to achieve a homogeneously blended pre-mix. Rapidly dispersing microgranules were blended with the melperone HCl SR beads and the pre-mix in a twin shell V-blender for sufficient time to obtain a homogeneously blended compression mix, which was compressed into ODT tablets using the production scale Hata tablet press equipped with an external lubrication system (Matsui ExLub System) and round, flat faced, radius edge tooling, under tableting conditions fine-tuned for optimal performance. Two ODT batches were produced as described above, having different tablet weights and relative amounts of melperone SR beads and rapidly dispersing granules, and these tablets exhibited the tableting properties shown in and .
Figure 10. Drug release profiles of ODT formulations and corresponding SR beads manufactured at one-tenth commercial scale. Figure shows minimum membrane fracture when compressible coated SR beads tableted with rapidly dispersing microgranules at different ratios of SR coated beads to RD microgranules, ODT tablets with a higher fraction of RD microgranules providing slightly faster release.
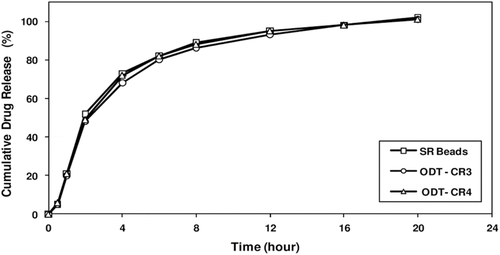
Table 9. Compositions and tableting properties of ODTs.
Results and discussion
Drug release profiles from SR beads and ODT-CR
demonstrates the extreme solubility of melperone HCl in the physiologically relevant pH range of 1 to 7. Due to this extreme solubility, dose dumping is very likely to occur from SR-coated melperone layered multiparticulates with a mean particle size of ≤400 µm required for incorporating in ODTs. However, melperone is poorly soluble above pH = 7.5, especially above pH = 8. This poor solubility was used to create once-daily ODT formulations comprising non-gritty multiparticulates by creating an alkaline pH microenvironment within the coated bead. In the dissolution media or upon oral administration, water imbibed into SR bead creates an alkaline pH of 8 or higher wherein melperone is poorly soluble, thereby prolonging drug release. This is evident from showing varying drug release profiles with increasing coating levels. The drug release profiles from SR beads at a coating of 20% by weight and the ODT tablets containing the SR beads are similar, suggesting the compressible coating with Klucel® LF applied over the rigid SR coating was largely effective in minimizing, if not eliminating membrane fracture during compression of the SR beads. These release profiles are similar to the fast release dosage form created at K1 = 0.29/hr. also demonstrates that the formulation is robust and reproducible. The development/use of a TPR bead population in combination with an immediate-release bead population enables it easier to achieve the target physicokinetic profile than possible with the use of an SR bead population alone. However, in spite of having an alkaline buffer coating which controlled pH of the microenvironment inside the coated SR or TPR beads developed starting from 60–80 mesh sugar spheres (i.e., smallest bead size) to be incorporated into ODT tablets, the drug release from both SR and TPR bead populations was too rapid due to extremely high aqueous solubility of the alkaline buffer itself. Hence, further development was focused on 45–60 mesh sugar spheres and SR coating.
Drug release profiles from CR capsules
shows that the CR capsules containing SR beads (i.e., drug layered on 20–25 mesh sugar spheres and coated with ethylcellulose without the intermediate alkaline buffer coating) at 20, 25, and 30% by weight exhibit release profiles which mimic fast, medium, and slow release profiles predicted from the PK modeling.
Process optimization at pilot scale
During the initial feasibility development, composition of the formulations for drug layering, protective seal-coat, alkaline buffer coating, SR or TPR coating, capsule filling, and ODT-CR, including the use of appropriate size sugar spheres for drug layering or alternate excipients for tableting, type of binder and its concentration, spray rate, and process parameters were varied and optimized to achieve highest possible useable yield of melperone beads with a desired particle size distribution. Dissolution was performed on the varied products to determine a range of ideal profiles that would be chosen for use in the pilot PK study. During the preparation of compression mixes of melperone beads, which were judged to have acceptable organoleptic properties, type and concentration of flavor, concentration of sucralose, composition of the compression mix, initial blending of the flavor, crospovidone, and sucralose were varied and optimized based on the organoleptic properties, dissolution data, disintegration time and acceptable tableting properties. Tablet size was chosen to achieve adequate dose while also having suitable organoleptic attributes. Tablets of 25, 50, and 100 mg strengths were compressed at 3-5 compression forces (e.g., 50 mg ODT weighing 1000 mg at a compression force of 12–16 kN). The physical attributes of the tablet were tested and compression parameters were optimized to achieve trouble-free tableting. Tablets were created on the commercial equipment to assess the processing parameters that would be used during the GMP manufacturing of pilot PK study material.
Pilot PK study results
shows the in vitro melperone release profiles from ODT-CR 1 (fast), ODT-CR 2 (medium), and CR capsules (medium). The SR beads used in these formulations have the same SR coating level, i.e., 30% by weight. The starting inert cores are 45–60 mesh and 25–30 mesh, respectively for the SR beads used in the ODT and capsule formulations, respectively. While the ODT-CR 1 (fast) has no alkaline buffer layer to retard drug dissolution/release into the surrounding environment, the alkaline buffer layer in the SR beads incorporated into the medium release ODT formulations significantly retarded drug dissolution so that both the medium release ODT and capsule formulations exhibit similar in vitro release profiles. illustrates the melperone plasma concentration vs. time profiles for the 25-mg syrup and three CR dosage forms. The PK results of the 25- and 50-mg studies are summarized in . Comparing Treatments A (25 mg syrup) to B–D, it is clear that Cmax values for Treatments B to D are about half of that for the 50-mg syrup indicating treatments B to D exhibit bioexposures equivalent to 25-mg syrup formulation A at normalized doses. Furthermore, treatments C (ODT-CR 2) and D (CR capsule) are bioequivalent and PK parameters for the syrup formulations, 25-mg and 50-mg are not dose proportional.
Stability study results
The stability data for the pilot CTMs, CR capsule, ODT-CR 2 are presented in . Both once-daily capsule and ODT formulations have demonstrated acceptable stability when stored in induction-sealed HDPE bottles. For example, the growth of specified and unspecified (until then unknown) impurities was limited, and dissolution profiles also remained largely unchanged during storage under ICH conditions.
Process scale-up and optimization
The manufacturing processes in the Glatt GPCG 120 equipped with the bottom spray 18” Wurster column including the drug-layering, alkaline buffer coating, seal or compressible coating were evaluated extensively to demonstrate robustness of the SR bead manufacturing process. The RD microgranules are manufactured using the validated process at a batch size of 160 kgs. The tablet manufacturing process as well as the tablet composition were evaluated using SR beads manufactured at one-tenth commercial scale. Pre-blending of the pharmaceutically acceptable ingredients as well as final compression blending was first optimized. ODTs tableting parameters including the magnesium stearate spray rate were optimized. Two ODT batches were produced as described above, having different tablet weights and relative amounts of melperone SR beads and rapidly dispersing granules (compare ODT-CR 3 to ODT-CR 4, ). shows the drug-release profiles of Melperone HCl SR beads and Melperone HCl ODT-CR 3 and ODT-CR 4. The release profiles of the SR beads and the corresponding ODT-CR tablet batches containing the compressible coated SR beads are comparable, suggesting minimal membrane fracture when compressible coated SR beads are tableted with rapidly dispersing microgranules at different ratios of SR coated beads to RD microgranules. Tablets containing a higher amount of rapidly dispersing granules had a marginally better mouthfeel and shorter disintegration time.
A comparison of the initial portions of the drug release profiles of the ODT-CR (comprising SR beads) with the corresponding SR beads in , it is apparent that membrane integrity is largely maintained. Furthermore, the melperone remains in a stable unaltered form in the solid dosage form.
These experiments demonstrate that the microparticles comprising melperone HCl, incorporating an alkaline buffer layer, optionally separated from the melperone by a sealant layer, and further coated with one or more CR layer(s) significantly sustains and/or delays the release of melperone.
Most desired tablet properties - conventional tablets versus ODTs
It is estimated that 50% of the population have problems swallowing conventional capsules/tablets due to dysphagia or other reasonsCitation15,Citation16. The higher the dose, the larger the tablet, the greater the difficulty in swallowing. Conventional tablets are typically compressed at high forces to achieve hardness and low friability. Furthermore, these tablets are normally film-coated so are not intended to disintegrate in the oral cavity (these tablets take several minutes to disintegrate when tested according to USP disintegration test method <701>) and are swallowed with water. In contrast, ODT formulations have an acceptable in vitro disintegration time of not more than 30 sec when tested using USP method <701> and rapidly disintegrate in the oral cavity. However, rapid disintegration alone does not ensure compliance and/or commercial success. The tablet must also have acceptable taste and mouthfeel characteristics. However, the taste-masking membrane applied onto bitter drug particles should not adversely affect their pharmacokinetic properties, resulting in non-bioequivalent product and requiring an expensive efficacy study for regulatory approval. Taste-masking approaches incorporating gastrosoluble pore formers are known which provide adequate taste-masking in the oral cavity while promoting faster drug release upon ingestion, thereby ensuring bioequivalence to reference listed drug. Such an ODT formulation would be ideal not only to improve patient acceptability/compliance and to prevent the medication from being discarded after “cheeking” (particularly in children, the elderly, and institutionalized patients with bipolar or psychiatric disorder), but also to ensure commercial success.
Furthermore, the tablets must have acceptable tableting properties (e.g., sufficiently high hardness and low friability) to maintain structural integrity during packaging in blisters and/or bottles, bulk storage, transportation, commercial distribution, and end-use. The conventional tablets are typically formed at high compression forces while ODTs are typically made at lower compression forces, but each has to maintain sufficiently high hardness and low friability. While traditional tablets, including ODTs based on other technology platforms (excepting based on lyophilization technology), are produced using an internal lubrication system in which lubricant is incorporated throughout the tablet blend to provide lubrication between the tablet surface and the metal surfaces of the tablet press, Melperone ODT-CR and others using AdvaTab® technology are manufactured with an external lubrication system in which the lubricant is only applied to the material contacting surfaces of punches and diesCitation10,Citation11. The presence of an internal lubricant in traditional tablets makes the tablet hydrophobic and necessitates the use of higher tablet compression forces for a given tablet hardness; with the external lubrication system the tablets do not require high tablet compression forces during manufacturing, and hence, the lower compression forces together with the presence of minimal lubricant on tablet surface insures rapid disintegration on contact with dissolution media or saliva in the oral cavity or when tested by USP <701> DT methodCitation11,Citation12. Irrespective of the lubrication method employed, it is most desirable to establish the robustness of the ODTs by in-process testing of tablets compressed under different tableting conditions as well as by conducting a shipping study.
Conclusion
CR capsule and ODT formulations containing melperone HCl were developed with in vitro release profiles suitable for once-daily dosing. Both dosage forms are dose-proportional allowing for the convenient production of multiple strengths. The ODT-CR and capsule medium release formulations are bioequivalent to each other and are suitable for once-daily dosing. Based on the analytical and organoleptic test results, as well as the blend uniformity and tableting in-process data at various compression forces using coated beads produced at one-tenth commercial scale, both formulations in the form of CR capsules and ODT-CR have shown suitability for progression into further clinical development.
The authors would like express their heartfelt thanks to Craig Kramer, Julius King, Oscar Batista, Bennie Young, Jeffrey Overfield, Ken Easter, Jennifer Fielder, Mark Dahlin, Dan Hensley, Jennifer Neuforth, for their technical and analytical support.
Declaration of interest
The product was co-developed in partnership with a pharmaceutical company which was responsible for designing and performing multiple clinical studies using the clinical trial materials (CTMs) manufactured by Aptalis Pharma. However, the partner does not want to be publicly acknowledged.
References
- Christensen I, Geismar L, Kirkegaard A, Kirkegaard G. (1986). Additional studies on side effects of melperone in long-term therapy for 1-20 years in psychiatric patients. Arzneimittelforschung, 36:855–860.
- Barbato L, Monge A, Stocchi F, Nordera G. (1996). Melperone in the treatment of iatrogenic psychosis in Parkinson’s disease. Funct Neurol, 11:201–207.
- Meltzer HY, Sumiyoshi T, Jayathilake K. (2001). Melperone in the treatment of neuroleptic-resistant schizophrenia. Psychiatry Res, 105:201–209.
- Fernandez HH, Trieschmann ME, Friedman JH. (2003). Treatment of psychosis in Parkinson’s disease: safety considerations. Drug Saf, 26:643–659.
- Sumiyoshi T, Jayathilake K, Meltzer HY. (2003). A comparison of two doses of melperone, an atypical antipsychotic drug, in the treatment of schizophrenia. Schizophr Res, 62:65–72.
- Borgström L, Larsson H, Molander L. (1982). Pharmacokinetics of parenteral and oral melperone in man. Eur J Clin Pharmacol, 23:173–176.
- Molander L, Borgström L. (1983). Sedative effects and prolactin response to single oral doses of melperone. Psychopharmacology (Berl), 79:142–147.
- Venkatesh, G., 2009, Diffucaps® technology for controlled release drug delivery, in: Youan, Bi-Botti C. (ed), Chronotherapeutics: Science and Technology for biological rhythm-guided therapy and prevention of Diseases. John Wiley & Sons, Inc., New York, pp. 121–144.
- Venkatesh, G.M., Qian, K.K., Vangala, S., Clevenger, J.M., Guenther, D., 2005. Orally disintegrating tablets and methods of manufacture. US patent # “20050232988 A1”.
- Venkatesh, G., 2010, Granulation approaches for orally disintegrating formulations, in: Parikh, D.M. (ed), Handbook of Pharmaceutical Granulation Technology, Third Edition. Informa Healthcare, Inc., New York, pp. 401–434.
- Morimoto K., Watanabe Y., Sanada Y., Miwa T., Masada T., 1997. Rotary type tableting machine with lubricant spraying means, assigned to Kyowa Hakko Kogyo Co. US patent “5,700,492”.
- Papp, M.K., Venkatesh, G.M., Harmon, T. 2010. Specialized techniques for developing ODT dosage forms. On Drug Delivery. April, 8-10, (http://www.ondrugdelivery.com/products/current_issues.htm).
- Pfister, W.R., Ghosh, T.K., 2005. Orally disintegrating tablets: products, technologies, and development issues Pharm Technol 29, 136–150.
- SQC Online. ANSI/ASQ Z1.4 Sampling plan for attribute data. (http://www.sqconline.com/mil-std-105.html). Accessed November 2010.
- Yarwood, R., 1990, Zydis – a novel fast dissolving dosage form. Manuf Chem. 61, 36–37.
- Lindgren, S., Janzon, L., 1993. Dysphagia: prevalence of swallowing complaints and clinical findings. Med Clin North Am. 77, 3–5.