Abstract
Background and aims. Carbonic anhydrases (CA) play a central role in osteoclast function and bone remodeling by catalyzing the formation of bicarbonate and proton from carbon dioxide. According to previous histochemical studies, advanced atherosclerotic plaques share similarities with bone. However, whether CAs are expressed in plaques is not known.
Methods and results. Whole genome expression array of arterial samples (n = 24) confirmed that several genes indicating osteoblastogenesis and osteoclastogenesis were up-regulated in plaques when compared to control vessel samples from internal thoracic arteries (n = 6), including CA2 and CA12, expression of which was also verified with quantitative reverse transcription polymerase chain reaction (RT-PCR). In atherosclerotic plaques there was 11.6-fold (P < 0.0001) and 11.4-fold (P < 0.0001) up-regulation of CA2 and CA12, compared to controls, respectively. According to quantitative PCR, CA2 expression was elevated in carotid (12.3-fold, P < 0.0001), femoral (13.2-fold, P < 0.01), and aortic plaques (7.5-fold, P < 0.0001). CA12 expression was elevated in carotid (11.6-fold, P < 0.0001), femoral (11.5-fold, P < 0.01), and aortic plaques (9.7-fold, P < 0.0001). CAII, CAXII, and CD68 and tartrate-resistant acid phosphatase (TRAP), a marker of osteoclast-like cells, were found to be co-localized in multinucleated giant cells in the atherosclerotic plaques using immunohistochemistry and double-staining immunofluorescence analysis.
Conclusions. The present findings provide evidence for the involvement of CAs in advanced atherosclerosis in osteoclast-like cells of monocyte-macrophage lineage.
Key messages
This study reports a comprehensive characterization of the expression of genes involved in osteoblastogenesis and osteoclastogenesis in human advanced atherosclerotic plaques in all major arterial beds.
For the first time, up-regulated expression of carbonic anhydrases 2 and 12 mRNA and protein in osteoclast-like cells of monocytic origin are implicated in advanced human atherosclerosis, and this suggests their involvement in the plaque remodeling.
A precise picture of carbonic anhydrase expression is a prerequisite for future mechanistic studies elucidating the role of carbonic anhydrases in plaque development and stability and potential development of novel therapeutic strategies.
Introduction
Atherosclerotic plaques contain collagen, cholesterol and its esters, hematopoietic tissue, calcium hydroxyapatite, and carbonate apatite (Citation1–3). The calcified atherosclerotic arterial plaque is histomorphologically indistinguishable from bone tissue (Citation2,Citation4,Citation5). At present, this process is considered an active and organized process with striking similarities to osteogenesis in bone with added chronic plaque inflammation. A passive model has also been suggested with mineral deposition in the plaque due to lack of inhibitory processes (Citation6).
In bone, the balance between bone formation and resorption is defined by the balance between osteoblasts and osteoclasts (Citation7,Citation8). Osteoclasts resorb bone and are capable of creating an acidic microenvironment during resorption (Citation9). Whether similar mechanisms are operative in arteries is not known, although arteries contain cells that exhibit similar phenotype as the major cell types involved in endochondral bone formation, i.e. chondrocytes, osteoblasts, and osteoclasts (Citation2,Citation10–13).
The resorptive cells in the atherosclerotic plaques are known as osteoclast-like cells (OCL) derived from hematopoietic mononuclear phagocyte progenitor cells. These cells are hypothesized to determine whether calcium is deposited in the plaque or not (Citation1), thus opposing the action of arterial osteoblast-like cells. The central stimulus for OCL formation is macrophage colony-stimulating factor-1 (M-CSF-1) and receptor activator of nuclear factor kappa beta (NF-κB) ligand (RANKL) which are sufficient for osteoclastogenesis from plaque mononuclear phagocytes and mineral resorption in bone (Citation1). RANKL is a cell surface receptor for receptor activator of nuclear factor kappa beta (NF-κB) (RANK) and its decoy receptor osteoprotegerin (OPG) (Citation14). Interestingly, OPG, RANK, and RANKL are found to be expressed in calcified arterial lesions of OPG-deficient mice (Citation15). In addition to these, tumor necrosis factor alpha (TNF-α) activates osteoclasts directly and potentiates the RANKL signaling (Citation16). Whether OCLs function in normal mineral homeostasis or are involved in specific sites in the plaque is not known.
Carbonic anhydrases (CAs) are expressed by osteoclasts (Citation9) and are a family of enzymes catalyzing formation of bicarbonate and protons from water and carbon dioxide (Citation17). Carbonic anhydrases are present in 16 different isoforms (Citation17) of which 13 are enzymatically active. The active isozymes are classified as cytosolic (CAI, CAII, CAIII, CAVII, and CAXIII), membrane-bound (CAIV, CAIX, CAXII, CAXIV, and CAXV) (Citation17,Citation18), mitochondrial (CA VA and CA VB), and secretory forms (CAVI). CAIX and CAXII are hypoxia-inducible forms, regulated by hypoxia-inducible factor 1α (HIF-1α) (Citation19). Both CAII and CAXII are expressed in osteoclasts (Citation9), and CAII is the primary cytosolic enzyme responsible for hydration of carbon dioxide. Subsequent dissociation of carbonic acid to bicarbonate and hydrogen ion is a prerequisite for osteoclast function and bone resorption, and, on the other hand, it provides bicarbonate for the formation of calcium carbonate. CAXII is another enzyme which probably acts to regulate pH homeostasis in the osteoclast microenvironment.
Rationale of the study
At present, no direct data on involvement of CAs or their expression in OCL cells in atherosclerosis exist, although a single study hypothesized that CA- mediated regulation of calcification might explain the different susceptibility of certain species to atherosclerosis (Citation20). For the discovery of novel strategies against development of atherosclerosis, we previously utilized genome-wide expression array (GWEA), encompassing all the known 23,000 genes to study a unique sample material of advanced plaques in the major vascular territories and control vessels (Citation21) classified according to the American Heart Association classification (Citation22). Over 200 genes were found to be up-regulated, including the CA gene family and other genes involved in bone remodeling. Of these, CA2 and CA12 were selected for more detailed analysis because the corresponding enzymes have been associated with bone formation with distinct features similar to that of atherosclerosis. In addition, since development of CA inhibitors is actively pursued, we considered these potential targets for therapeutic interventions.
Methods
Vascular samples
Vascular plaque sample series from femoral arteries, carotid arteries, and abdominal aortas were obtained during open vascular procedures from 30 patients (Division of Vascular Surgery) as a part of the on-going Tampere Vascular Study (Citation21). Six control samples were taken from internal mammary arteries obtained during coronary artery by-pass grafting (Heart Centre, Tampere University Hospital). The study has been approved by the Ethics Committee of Tampere University Hospital. All clinical investigation was conducted according to declaration of Helsinki principles. The vascular samples were classified according to recommendation of American Heart Association (AHA) (Citation22).
RNA isolation and genome-wide expression analysis (GWEA)
The fresh tissue samples were soaked in RNALater solution (Ambion Inc., Austin, TX, USA) and RNA isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA) and the RNAEasy Kit (Qiagen, Valencia, CA, USA). The concentration and quality of the RNA was evaluated spectrophotometrically (BioPhotometer, Eppendorf, Wesseling-Berzdorf, Germany). More than 23,000 known and candidate genes were analyzed using Sentrix Human-8 Expression BeadChips, according to the manufacturer's instructions (Illumina, San Diego, CA, USA). In brief, a 200 ng aliquot of total RNA from each sample was amplified to cDNA using the Ambion's Illumina RNA Amplification kit according to the instructions (Ambion Inc., Austin, TX, USA). Each sample cRNA (1500 ng) was hybridized to Illumina's Sentrix Human-8 Expression BeadChip arrays (Illumina). Hybridized biotinylated cRNA was detected with 1 μg/mL Cyanine3-streptavidine (Amersham Biosciences, Pistacataway, NJ, USA). BeadChips were scanned with the Illumina BeadArray Reader. The method has been described in more detail in our previous work (Citation21). The accuracy of Illumina Sentrix® Human-8 Expression BeadChips microarray methodology to measure the gene expression was earlier verified by real-time quantitative TaqMan polymerase chain reaction (PCR) by quantitating the expression of 20 genes with both methods (Citation23).
Quantitative real-time PCR (qPCR)
From the 30 tissue samples, gene expression analyses were done with TaqMan low-density arrays (LDAs) (Applied Biosystems, Foster City, CA, USA). Total-RNA (500 ng) was reverse-transcribed to cDNA using High Capacity cDNA Kit according to the manufacturer's instructions (Applied Biosystems). For the PCR, LDAs were loaded with 4 μL undiluted cDNA, 46 μL H2O, and 50 μL PCR Universal Master Mix and run according to the manufacturer's instructions (Applied Biosystems). Samples were analyzed as duplicates, and both cDNA synthesis and PCR reactions were validated for inhibition. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene control, and the qPCR results were analyzed using SDS 2.2 Software (Applied Biosystems) and ΔΔCt method (Citation24).
Immunohistochemistry (IH) and immunofluorescence (IF)
Immunohistochemistry was performed using the N-Histofine® Simple Stain MAX PO staining method (Nichirei Biosciences Inc., Tokyo, Japan) and paraffin-embedded vascular samples without any counterstain. CAII and XII were detected with polyclonal rabbit antibodies characterized previously (diluted 1:1000 and 1:600, respectively) (Citation25,Citation26). The following primary antibodies were used to detect vascular cell markers in adjacent sections: CD68 (mouse anti-human CD68, clone PG-M1 (DakoCytomation, diluted 1:70)) was used as marker of monocytes and macrophages. For the detection of OCLs (Citation27–29) mouse anti-tartrate-resistant acid phosphatase (TRAP) (TrapC, clone-ZY-9C5, Invitrogen) antibody was used. The sections were subjected to microwave antigen retrieval treatment as described earlier (Citation30). Endogenous peroxidase activity was extinguished by treating the section with 0.3% H2O2 for 30 min. Subsequently the sections were incubated overnight at +4°C with primary antibodies followed with appropriate N-Histofine staining reagent for 30 min. ImmPACT™ (Vector Laboratories, Burlingame, CA, USA) diaminobenzidine solution was used as the chromogen. All antibodies were diluted in phosphate buffered saline (PBS) containing 1% bovine serum albumin (BSA) and 0.3% of Triton X-100. Controls included omitting the primary antibody or replacing it with non-immune sera. No staining was seen in the controls.
The co-localization of CD68 and CAII and CAXII in carotid arteries was studied with double-staining immunofluorescence (IF). The samples were fixed with 4% paraformaldehyde (in 0.1 M PBS, pH 7.3) for 6 h at +4°C and cryoprotected with 20% sucrose in PBS. Frozen sections (6 μm) were cut with Micron HM560 cryostat and thaw-mounted onto Polysine glass slides (Menzel, Braunschweig, Germany). The sections were incubated overnight with the mixture of CD68 (diluted 1:10) antibody and CAII (diluted 1:40) or CAXII antibodies (diluted 1:20) followed by a mixture of biotinylated sheep anti-mouse antibody (1:200, Amersham) and fluorescein-conjugated goat anti-rabbit antibody (1:100, Boehringer Mannheim, Mannheim) for 30 min at 37°C. Subsequently, the sections were incubated with rhodamine-conjucated avidin (diluted 1:100, Vector Laboratories) for 30 min. Sections were mounted in a mixture of glycerol and PBS (3:1) containing 0.1% paraphenylenediamine and examined in Nikon Microphot FXA microscope equipped with proper fluorescence filters. Photographs were obtained with PCO Sensicam digital camera (PCO, Kelheim, Germany). Alternatively the co-localization of CD68, TRAP, CAII, and CAXII was studied in 5 μm paraffin sections (mirror image sections). Sections were stained with N-Histofine method as described above.
Statistical analyses
Statistical analyses were performed using SPSS version 14.0 (SPSS Inc., Chicago, IL, USA). The non-parametric Kruskall-Wallis test was used to detect differences in gene expression between different vascular bed regions. The Mann-Whitney U-test was used for comparison of mRNA expression between atherosclerotic and control tissues and between different arterial regions and control samples. Data are presented as median and range.
Results
Characteristics of the subjects and studied vascular samples
The median age was 70.0 years (45–93 years), and 75.9% of the subjects were men. The prevalence of risk factors was as follows: dyslipidemia 40.7%, hypertension 77.8%, diabetes 18.5%, history of smoking 86.2%, and alcohol usage more than once a week 42.8%. Of the atherosclerotic arteries, 73.3% were classified as type V–VI advanced plaques. All the internal mammary arteries that were used as controls were microscopically healthy.
Characterization of the expression of osteoclast markers and osteogenesis associated genes in the atherosclerotic tissue
The expressions of various genes involved in osteogenesis (osteoblastogenesis and osteoclastogenesis) are listed in . Supporting the hypothesis of on-going osteoclastogenesis, the expression of colony-stimulating factor 1 receptor (c-fms) mediating the effect of M-CSF on osteoclastogenesis (Citation1,Citation15,Citation31–34) was up-regulated in plaques in all the vascular beds (P < 0.0001 for all) (). Receptor activator of NF-κB variant 2 (RANKL2), the crucial factor for osteoclastogenesis (Citation1,Citation14), was up-regulated only in the carotid arteries (P < 0.05) (). In addition, osteoprotegerin (OPG), the receptor for RANKL and regulator for osteoclast activity (Citation1,Citation35), demonstrated significant up-regulation in carotid (P < 0.0001) and aortic plaques (P < 0.0001) (). Indicative of osteoclast function (Citation15,Citation34), osteonectin (P < 0.05 for femoral and aortic plaques) and osteopontin (P < 0.0001 for all), matrix metalloproteinase 9 (MMP9) (P < 0.0001 for all), cathepsin K, the protease necessary for osteoclasts (P < 0.01 for carotid and femoral plaques), and acid phosphatase 5 tartrate-resistant (ACP5) were up-regulated in all the regions (P < 0.0001 for carotid and femoral plaques, P < 0.01 for aorta). Chloride channel 7 (CLCN7), the ion exchanger necessary for proton and chloride transport (P < 0.0001 for all), and T-cell immunoregulator 1 (TCIRG1) (P < 0.0001 for carotid, P < 0.05 for femoral and aortic plaques), as well as osteopetrosis-associated transmembrane protein 1 (OSTM1) (P < 0.0001 for carotid plaques) were significantly up-regulated in atherosclerotic plaques. All of these genes are considered as markers of osteoclast function (Citation15,Citation34). The genes indicating differentiation of osteoblasts were not found to be altered in plaques versus healthy arteries, except for transcription factor Dlx5 (distal-less homeobox 5) an important regulator of bone turnover (Citation36) (P < 0.05 for carotid and femoral plaques) ().
Table I. Genome-wide gene expression analysis of genes related to bone formation in arterial plaques relative to non-atherosclerotic internal thoracic artery control samples in Tampere Vascular Study.
Carbonic anhydrase gene expression in the atherosclerotic tissue
The expression of CA1–14 was analyzed using GWEA (). Of these, CA2 and CA12 were consistently and significantly up-regulated in all the vascular beds. Therefore, these genes were selected for a more detailed analysis. According to the Kruskall-Wallis test, the expression of both CA2 (P < 0.01) and CA12 (P < 0.01) differed with respect to vascular bed region (). The median CA2 expression was elevated relative to controls in the carotid (2.30-fold, P < 0.0001), femoral (2.24-fold, P < 0.01), and aortic plaques (1.87-fold, P < 0.01), respectively (). The median CA12 expression was elevated relative to controls in the carotid (3.38-fold, P < 0.0001), femoral (3.72-fold, P < 0.0001), and aortic plaques (2.68-fold, P < 0.0001) ().
Figure 1. CA2 and CA12 expression in control internal thoracic artery (ITA), carotid, femoral, and aortic plaque samples. Gene expression value is the normalized average gene intensity for each group measured in the Illumina Expression BeadChips. *P < 0.05, **P < 0.01, ***P < 0.001 relative to control (ITA), Mann-Whitney U-test.
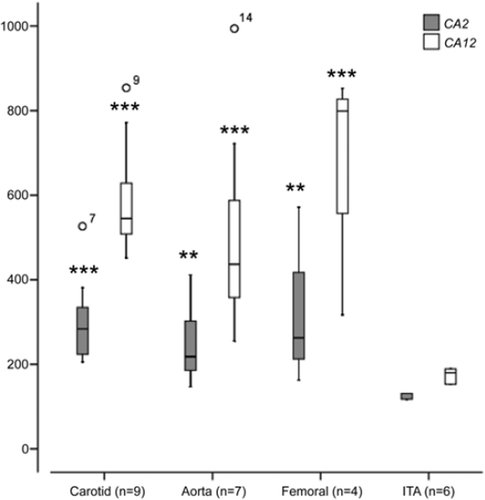
Table II. Genome-wide gene expression analysis of carbonic anhydrases in arterial plaques relative to non-atherosclerotic internal thoracic artery control samples in Tampere Vascular Study.
According to GWEA, the median CA2 expression level in atherosclerotic plaques (types V–VI) (n = 20) was 263 (146–571) and 131 (117–131) in control tissues (type 0, n = 6) (2.0-fold relative to control P < 0.0001). The median CA12 expression level in atherosclerotic plaques (types V–VI) (n = 20) was 535 (255–994) and 179 (152–189) in control tissues (type 0, n = 6) (3.0-fold relative to control P < 0.0001).
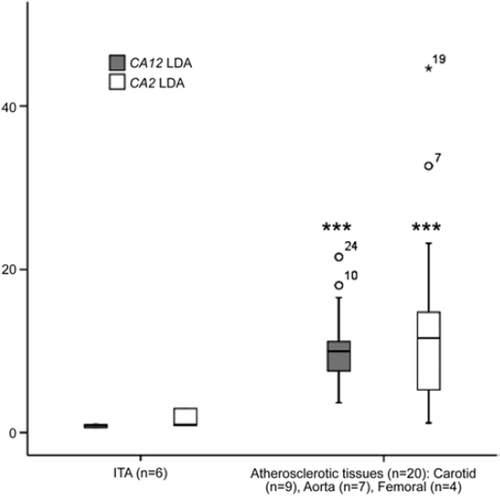
The Illumina GWEA array results were verified with quantitative PCR. The median CA2 expression level in atherosclerotic plaques (types V–VI) (n = 20) was elevated 11.6-fold compared with control tissues (type 0, n = 6) (P < 0.0001) (). The median CA12 expression level in atherosclerotic plaques (types V–VI) (n = 20) was elevated 11.4-fold compared with control tissues (type 0, n = 6) (P < 0.0001) ().
Figure 2. CA2 and CA12 expression in atherosclerotic plaque and control internal thoracic artery (ITA) samples measured with TaqMan Low-Density Array (LDA). *P < 0.05, **P < 0.01, ***P < 0.001 relative to control (ITA), Mann-Whitney U-test.
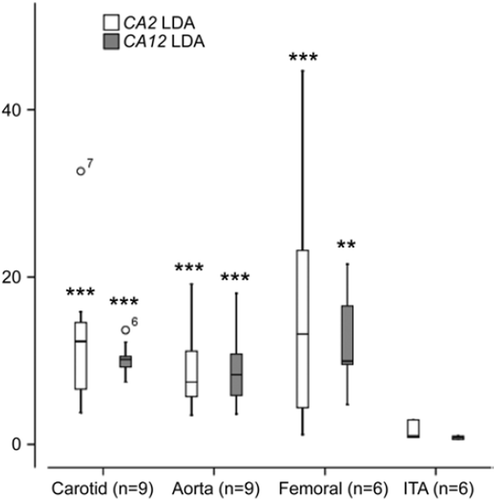
CA2 expression was elevated in carotid (12.3-fold, P < 0.0001), femoral (13.2-fold, P < 0.01), and aortic plaques (7.5-fold, P < 0.0001) compared with control tissues (). CA12 expression was elevated in carotid (11.6-fold, P < 0.0001), femoral (11.5-fold, P < 0.01), and aortic plaques (9.7-fold, P < 0.0001) compared with control tissues (). No apparent difference in the gene expression profiles was found between early (type V) and advanced plaques (type VI).
Figure 3. Expression of CA2 and CA12 in control internal thoracic artery (ITA), carotid, aortic, and femoral plaque samples measured with TaqMan Low-Density Array (LDA). *P < 0.05, **P < 0.01, ***P < 0.001 relative to control (ITA), Mann-Whitney U-test.
Carbonic anhydrase II and XII protein expression in the atherosclerotic tissue
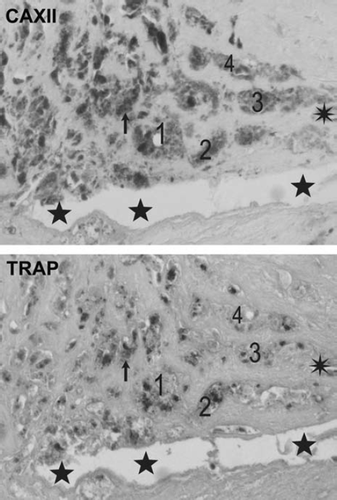
Immunohistochemistry of atherosclerotic lesions revealed that CAII and CAXII proteins were expressed in the atheromatous core and to some extent in all the vessel layers in early and advanced plaques of all the vessel beds, while only sparse cells were positive in control vessels (). Interestingly, expression of both CAII and CAXII concentrated on the same multinucleated giant cell clusters. Additional analyses of these cell clusters with double-staining immunofluorescence microscopy and mirror image immunohistochemistry confirmed co-localization of CAII and CAXII with CD68-positive cells indicating they are of monocytic origin (). CAII (data not shown) and CAXII co-localized with TRAP (tartrate-resistant acid phosphatase)-positive cells indicating the presence of osteoclast-like cells ().
Figure 4. Expression of CAII and CAXII proteins in human atherosclerotic plaques in human aortic (A), femoral (B), and carotid (C) plaques. Samples from internal thoracic artery served as controls (D). Arrows indicate typical positively stained cells. The stage of atherosclerosis was classified according to American Heart Association (AHA) classification (type I–VI). 100× magnification.
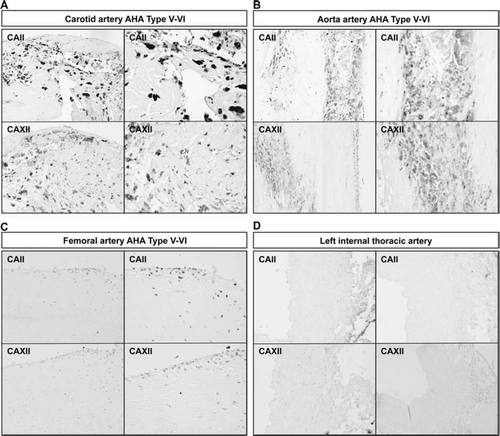
Figure 5. Double immunofluorescence and mirror image section images demonstrating co-localization of CAII and CAXII proteins with CD68 in human atherosclerotic plaques in representative carotid plaques. CAII labeling is cytosolic while CAXII labeling is strong in the cell membrane.
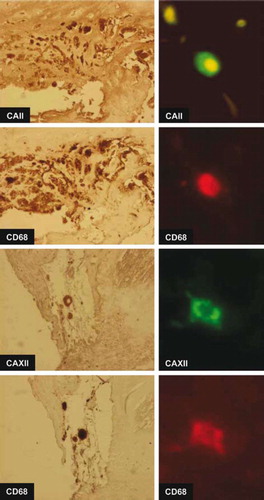
Figure 6. Mirror image section images demonstrating co-localization of CAXII protein with TRAP in human atherosclerotic plaques in representative carotid plaques. Corresponding cells, cell clusters, and microanatomic structures are indicated by an arrow-head, numbers 1–4, stars, and an asterisk.
Discussion
The present study is the first demonstration of up-regulation of CA2 and CA12 (Citation9) in human atherosclerotic plaques in the major vascular beds at both the transcriptional and protein levels. The results are novel as four different vascular territories were examined, and they suggest that the up-regulated expression of these CAs is likely to be associated with monocytic, osteoclast-like cells (OCLs) in the atherosclerotic plaques.
Our present demonstration of multinuclear CA-CD68-TRAP-positive giant cells in atherosclerotic lesions notably of OCL phenotype is supported by previous findings. The synergistic up-regulation of CA2 and CA12 in arterial plaques supports the hypothesis of presence of OCLs in atherosclerosis, since expression of both of them is needed in osteoclasts during bone resorption and bone remodeling (Citation9). Furthermore, up-regulation of carbonic anhydrase II expression in multinucleated giant cells and macrophage-like cells has been demonstrated (Citation37). Macrophages have been demonstrated to undergo osteoclastic transformation (Citation38). In addition, osteoclasts have been demonstrated to express CD68, a marker for monocytes (Citation39,Citation40). Similar CD68- positive OCLs induced by RANKL and M-CSF have been detected in bone in Langerhans histiocytosis (Citation41). Our present demonstration of CAII-, CAXII-, and CD68-positive cells co-expressing TRAP (Citation27–29) supports the notion that these cells have indeed adopted an OCL phenotype.
In the present study, osteopontin was expressed in the atherosclerotic plaques in all the vascular regions (42- to 65-fold), which is in line with the previous studies showing that it inhibits mineral formation in vitro and is not present in normal arteries (Citation42,Citation43). Up-regulation of osteopontin may be considered a defensive mechanism since osteopontin inhibits ectopic calcification and promotes mineral resorption by up-regulating expression of CAII by mononuclear phagocytic cells and it also induced multinucleated giant cells and macrophage-like cells (Citation37).
No previous data exist about the expression of CAs in atherosclerotic plaques. CAII has been demonstrated to be expressed in the endothelium of meningiomas (Citation44) and astrocytomas (Citation45), as well as in hematological malignancies (Citation46). CAXII may be involved in acidification of the extracellular milieu of tumor cells, and it has been demonstrated to be expressed in various tumors such as astrocytomas (Citation47) and ovarian (Citation48) and renal cancers (Citation49). In support for the critical role of CAII in osteoclast function, it has been demonstrated that osteoclasts lacking CAII are incapable of bone resorption (Citation50–52). In concert with this, mice deficient in CAII develop arterial calcification (Citation53), which suggests that the presence of CAII in OCLs in the arterial wall is required to oppose arterial mineral deposition promoted by arterial osteoblast-like cells.
Our findings of on-going osteoclastogenesis are supported by several observations. It has previously been shown that bone morphogenetic protein 2 (BMP-2) expression is increased in calcified arterial lesions (Citation12). In the present study, possible osteoclastogenesis and function are indicated by increased expression of cathepsin K which has been demonstrated to be expressed in osteoclasts and atherosclerotic plaques (Citation54). Increased expression of osteoprotegerin relative to RANKL isoforms and RANK potentially indicates that mechanisms inhibiting vascular calcification are activated in advanced atherosclerotic plaques. Substantial evidence supports the hypothesis that OPG protects against vascular calcification while RANKL promotes it (Citation55). Increased expression of c-fms, cathepsin K, osteonectin, ACP5, MMP9, OSTM1, and CLCN7 all together indicates that OCLs typically expressing these genes (Citation15,Citation34) might be active in the atherosclerotic plaque. Circulating osteoclast precursors, mononuclear phagocytic cells, and B-cells express c-fms which is the cell surface receptor for M-CSF (Citation31–33) in addition to the receptor of RANKL, i.e. RANK (Citation35).
The present findings might be of value in the prevention of atherosclerosis. Firstly, several modulators of carbonic anhydrase activity have been developed. Of these, sulfonamides are most widely used clinically (Citation17,Citation56,Citation57). Interestingly, celecoxib and valdecoxib are sulfonamide non-steroidal anti-inflammatory drugs and also potent inhibitors of carbonic anhydrases, especially CAII, IX, and XII (Citation58–61). Sulfonamide-type coxibs have been shown to inhibit CAII, differentiation and activity of osteoclasts in vitro, and bone destruction in vivo in an animal experiment (Citation62). Clinically, the problem with these sulfonamides is increased risk of cardiovascular events (Citation63). Whether their effect on carbonic anhydrases lies behind cardiovascular events is not currently known. To our knowledge there is no direct evidence of their effect on atherosclerotic plaque formation or their effect on arterial stenoses. In addition to inhibitors of CA activity, activators are also being developed (Citation17). At present it is not known whether inhibition of CAII or XII activity would result in increased arterial calcification, and augmentation of CAII or XII activity would result in decreased arterial calcification. In addition, an important clinical issue is whether inhibition of CAII or XII activity in unstable plaques would promote stabilization of the plaque.
As a potential limitation, it must be emphasized that due to ethical limitations we were not able to obtain any corresponding healthy arteries from carotid, aortic, and femoral regions as controls. Obviously, this would allow more accurate analysis. Due to the systemic nature of atherosclerosis, internal thoracic artery samples from patients subjected to coronary artery by-pass grafting may be affected by atherosclerosis and several humoral mediators although no microscopic evidence of plaques could be demonstrated. On the basis of the present findings, future mechanistic studies are urgently needed to establish the role of CAs in plaque pathophysiology.
In conclusion, our results imply that CA2 and CA12 are highly over-expressed in advanced atherosclerosis by OCLs of monocytic origin. As the therapeutic intervention of CAs in human diseases is actively pursued, our findings underline the potential of targeting the development of atherosclerosis through modulation of CA function.
Acknowledgements
The authors wish to thank Mrs Ulla Jukarainen, Nina Peltonen, Ms Elli Oksala, and Ms Rosanna Oksala for their skillful technical assistance.
Declaration of interest: This research is funded by European Union 7th Framework Program grant number 201668, Atheroremo. This study was supported with grants from the Medical Research Fund of Tampere University Hospital, the Emil Aaltonen Foundation (T.L.), the Pirkanmaa Regional Fund of the Finnish Cultural Foundation, the Research Foundation of Orion Corporation, the Jenny and Antti Wihuri Foundation, and the Academy of Finland (Grant no. 104821). Niku Oksala was supported by grants from the Finnish Angiology Association, Maire Taponen Foundation, and Paavo Nurmi Foundation. Niku Oksala and Mari Levula contributed equally to the work.
References
- Doherty TM, Uzui H, Fitzpatrick LA, Tripathi PV, Dunstan CR, Asotra K, . Rationale for the role of osteoclast-like cells in arterial calcification. FASEB J. 2002;16:577–82.
- Jeziorska M, McCollum C, Wooley DE. Observations on bone formation and remodelling in advanced atherosclerotic lesions of human carotid arteries. Virchows Arch. 1998;433:559–65.
- Schmid K, McSharry WO, Pameijer CH, Binette JP. Chemical and physicochemical studies on the mineral deposits of the human atherosclerotic aorta. Atherosclerosis. 1980;37: 199–210.
- Detrano RC, Doherty TM, Davies MJ, Stary HC. Predicting coronary events with coronary calcium: pathophysiologic and clinical problems. Curr Probl Cardiol. 2000;25: 374–402.
- Stary HC. Natural history of calcium deposits in atherosclerosis progression and regression. Z Kardiol. 2000;89 Suppl 2:28–35.
- Schinke T, McKee MD, Karsenty G. Extracellular matrix calcification: where is the action? Nat Genet. 1999;21:150–1.
- Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55.
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42.
- Riihonen R, Supuran CT, Parkkila S, Pastorekova S, Vaananen HK, Laitala-Leinonen T. Membrane-bound carbonic anhydrases in osteoclasts. Bone. 2007;40:1021–31.
- Dhore CR, Cleutjens JP, Lutgens E, Cleutjens KB, Geusens PP, Kitslaar PJ, . Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2001;21:1998–2003.
- Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, . Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386: 78–81.
- Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91: 1800–9.
- Hunt JL, Fairman R, Mitchell ME, Carpenter JP, Golden M, Khalapyan T, . Bone formation in carotid plaques: a clinicopathological study. Stroke. 2002;33:1214–9.
- Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5.
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, . osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–8.
- Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143:1108–18.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem. 2004; 19:199–229.
- Hilvo M, Tolvanen M, Clark A, Shen B, Shah GN, Waheed A, . Characterization of CA XV, a new GPI-anchored form of carbonic anhydrase. Biochem J. 2005;392(Pt 1):83–92.
- Halmi P, Lehtonen J, Waheed A, Sly WS, Parkkila S. Expression of hypoxia-inducible, membrane-bound carbonic anhydrase isozyme XII in mouse tissues. Anat Rec A Discov Mol Cell Evol Biol. 2004;277:171–7.
- Gamble W. Atherosclerosis: the carbonic anhydrase, carbon dioxide, calcium concerted theory. J Theor Biol. 2006;239:16–21.
- Oksala N, Levula M, Airla N, Pelto-Huikko M, Ortiz RM, Jarvinen O, . ADAM-9, ADAM-15, and ADAM-17 are upregulated in macrophages in advanced human atherosclerotic plaques in aorta and carotid and femoral arteries—Tampere vascular study. Ann Med. 2009;41:279–90.
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, . A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–74.
- Laaksonen R, Katajamaa M, Paiva H, Sysi-Aho M, Saarinen L, Junni P, . A systems biology strategy reveals biological pathways and plasma biomarker candidates for potentially toxic statin-induced changes in muscle. PLoS ONE. 2006;1:e97.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8.
- Parkkila AK, Parkkila S, Juvonen T, Rajaniemi H. Carbonic anhydrase isoenzymes II and I are present in the zona glomerulosa cells of the human adrenal gland. Histochemistry. 1993;99:37–41.
- Karhumaa P, Parkkila S, Tureci O, Waheed A, Grubb JH, Shah G, . Identification of carbonic anhydrase XII as the membrane isozyme expressed in the normal human endometrial epithelium. Mol Hum Reprod. 2000;6:68–74.
- Toyosawa S, Ogawa Y, Chang CK, Hong SS, Yagi T, Kuwahara H, . Histochemistry of tartrate-resistant acid phosphatase and carbonic anhydrase isoenzyme II in osteoclast-like giant cells in bone tumours. Virchows Arch A Pathol Anat Histopathol. 1991;418:255–61.
- Hayman AR. Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy. Autoimmunity. 2008;41:218–23.
- Halleen JM, Tiitinen SL, Ylipahkala H, Fagerlund KM, Vaananen HK. Tartrate-resistant acid phosphatase 5b (TRACP 5b) as a marker of bone resorption. Clin Lab. 2006;52:499–509.
- Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991;39:741–8.
- Rettenmier CW, Sacca R, Furman WL, Roussel MF, Holt JT, Nienhuis AW, . Expression of the human c-fms proto-oncogene product (colony-stimulating factor-1 receptor) on peripheral blood mononuclear cells and choriocarcinoma cell lines. J Clin Invest. 1986;77:1740–6.
- Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell. 1985;41:665–76.
- Sherr CJ, Rettenmier CW, Roussel MF. Macrophage colony-stimulating factor, CSF-1, and its proto-oncogene-encoded receptor. Cold Spring Harb Symp Quant Biol. 1988;53 Pt 1:521–30.
- Segovia-Silvestre T, Neutzsky-Wulff AV, Sorensen MG, Christiansen C, Bollerslev J, Karsdal MA, . Advances in osteoclast biology resulting from the study of osteopetrotic mutations. Hum Genet. 2009;124:561–77.
- Shalhoub V, Elliott G, Chiu L, Manoukian R, Kelley M, Hawkins N, . Characterization of osteoclast precursors in human blood. Br J Haematol. 2000;111:501–12.
- Samee N, Geoffroy V, Marty C, Schiltz C, Vieux-Rochas M, Levi G, . Dlx5, a positive regulator of osteoblastogenesis, is essential for osteoblast-osteoclast coupling. Am J Pathol. 2008;173:773–80.
- Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, . Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. 2002;161:2035–46.
- Athanasou NA, Quinn JM. Human tumour-associated macrophages are capable of bone resorption. Br J Cancer. 1992;65:523–6.
- Athanasou NA, Quinn J. Immunophenotypic differences between osteoclasts and macrophage polykaryons: immunohistological distinction and implications for osteoclast ontogeny and function. J Clin Pathol. 1990;43:997–1003.
- Athanasou NA, Alvarez JI, Ross FP, Quinn JM, Teitelbaum SL. Species differences in the immunophenotype of osteoclasts and mononuclear phagocytes. Calcif Tissue Int. 1992;50:427–32.
- da Costa CE, Annels NE, Faaij CM, Forsyth RG, Hogendoorn PC, Egeler RM. Presence of osteoclast-like multinucleated giant cells in the bone and nonostotic lesions of Langerhans cell histiocytosis. J Exp Med. 2005;201:687–93.
- Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan BL. Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest. 1998;101:1468–78.
- Fitzpatrick LA, Severson A, Edwards WD, Ingram RT. Diffuse calcification in human coronary arteries. Association of osteopontin with atherosclerosis. J Clin Invest. 1994;94: 1597–604.
- Korhonen K, Parkkila AK, Helen P, Valimaki R, Pastorekova S, Pastorek J, . Carbonic anhydrases in meningiomas: association of endothelial carbonic anhydrase II with aggressive tumor features. J Neurosurg. 2009;111: 472–7.
- Haapasalo J, Nordfors K, Jarvela S, Bragge H, Rantala I, Parkkila AK, . Carbonic anhydrase II in the endothelium of glial tumors: a potential target for therapy. Neuro Oncol. 2007;9:308–13.
- Leppilampi M, Koistinen P, Savolainen ER, Hannuksela J, Parkkila AK, Niemela O, . The expression of carbonic anhydrase II in hematological malignancies. Clin Cancer Res. 2002;8:2240–5.
- Haapasalo J, Hilvo M, Nordfors K, Haapasalo H, Parkkila S, Hyrskyluoto A, . Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro Oncol. 2008;10:131–8.
- Hynninen P, Vaskivuo L, Saarnio J, Haapasalo H, Kivela J, Pastorekova S, . Expression of transmembrane carbonic anhydrases IX and XII in ovarian tumours. Histopathology. 2006;49:594–602.
- Tureci O, Sahin U, Vollmar E, Siemer S, Gottert E, Seitz G, . Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci U S A. 1998;95:7608–13.
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8.
- Lehenkari P, Hentunen TA, Laitala-Leinonen T, Tuukkanen J, Vaananen HK. Carbonic anhydrase II plays a major role in osteoclast differentiation and bone resorption by effecting the steady state intracellular pH and Ca2+. Exp Cell Res. 1998;242:128–37.
- Laitala T, Vaananen HK. Inhibition of bone resorption in vitro by antisense RNA and DNA molecules targeted against carbonic anhydrase II or two subunits of vacuolar H(+)-ATPase. J Clin Invest. 1994;93:2311–8.
- Sundquist KT, Vaananen HK, Marks SC Jr. Carbonic anhydrase II and H+-ATPase in osteoclasts of four osteopetrotic mutations in the rat. Histochem Cell Biol. 1999;111:55–60.
- Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83.
- Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res. 2004;95:1046–57.
- Guzel O, Innocenti A, Scozzafava A, Salman A, Parkkila S, Hilvo M, . Carbonic anhydrase inhibitors: synthesis and inhibition studies against mammalian isoforms I-XV with a series of 2-(hydrazinocarbonyl)-3-substituted-phenyl-1H- indole-5-sulfonamides. Bioorg Med Chem. 2008;16: 9113–20.
- Innocenti A, Scozzafava A, Parkkila S, Puccetti L, De Simone G, Supuran CT. Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorg Med Chem Lett. 2008;18:2267–71.
- Dogne JM, Thiry A, Supuran CT. Carbonic anhydrase inhibition: insight into non-COX-2 pharmacological effect of some coxibs. Curr Pharm Des. 2008;14:679–84.
- Knudsen JF, Carlsson U, Hammarstrom P, Sokol GH, Cantilena LR. The cyclooxygenase-2 inhibitor celecoxib is a potent inhibitor of human carbonic anhydrase II. Inflammation. 2004;28:285–90.
- Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, . Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J Med Chem. 2004;47:550–7.
- Di Fiore A, Pedone C, D'Ambrosio K, Scozzafava A, De Simone G, Supuran CT. Carbonic anhydrase inhibitors: Valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II ‘selective’ inhibitor celecoxib. Bioorg Med Chem Lett. 2006;16:437–42.
- Katagiri M, Ogasawara T, Hoshi K, Chikazu D, Kimoto A, Noguchi M, . Suppression of adjuvant-induced arthritic bone destruction by cyclooxygenase-2 selective agents with and without inhibitory potency against carbonic anhydrase II. J Bone Miner Res. 2006;21:219–27.
- Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–9.