Abstract
Aim. To study if gene alterations affecting renal sodium reabsorption associate with susceptibility to licorice-induced hypertension.
Methods. Finnish subjects (n = 30) with a previously documented incident of licorice-induced hypertension were recruited for the study using a newspaper announcement. Their previous clinical and family histories as well as serum electrolyte levels were examined. DNA samples from all individuals were screened for variants of the genes encoding 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) and α-, β-, and γ-subunits of the epithelial sodium channel (ENaC).
Results. Upon licorice predisposition, the patients had a mean blood pressure of 201/118 mmHg. Circulating potassium, renin, and aldosterone levels were low. No significant DNA variations were identified in the 11βHSD2 gene. Four subjects were heterozygous for β- and γENaC variants previously shown to be associated with hypertension. Furthermore, a novel G insertion (2004–2005insG) in the SCNN1A gene encoding the αENaC was identified in two subjects. The frequency of these ENaC variants was significantly higher in subjects with licorice-induced hypertension (6/30 i.e. 20%) than in blood donors (11/301 i.e. 3.7%, P = 0.002).
Conclusions. Defects of the 11βHSD2 gene do not constitute a likely cause for licorice-induced hypertension. Variants of the ENaC subunits may render some individuals sensitive to licorice-induced metabolic alterations and hypertension.
Key words::
| Abbreviations | ||
| AME | = | apparent mineralocorticoid excess |
| BMI | = | body mass index |
| BP | = | blood pressure |
| 11βHSD2 | = | 11β-hydroxysteroid dehydrogenase type 2 |
| ENaC | = | epithelial sodium channel |
Key messages
DNA variants of the 11βHSD2 gene do not constitute a common cause for licorice-induced hypertension.
Certain genetic defects of epithelial sodium channel subunits may increase the effects of licorice consumption and predispose some individuals to licorice-induced metabolic alterations and hypertension.
Introduction
Elevated blood pressure is a multifactorial trait that involves complex interactions between genetic and environmental factors. The exact molecular mechanisms underlying essential hypertension have remained obscure in spite of the large variety of attempts to identify DNA alterations associated with hypertension in genetic epidemiologic studies (reviewed in (Citation1)). A few monogenic disorders causing elevated blood pressure have been established, such as Liddle's syndrome (Citation2) and apparent mineralocorticoid excess (AME) (Citation3,Citation4), which both represent salt-sensitive forms of hypertension.
In the dominantly inherited Liddle's syndrome, gain-of-function mutations in the genes coding for beta- or gamma-subunits of the epithelial sodium channel (β- and γENaC) result in increased renal sodium reabsorption, with subsequent suppression of plasma renin and aldosterone levels, extracellular volume expansion, and hypertension (Citation5,Citation6). Several earlier findings support the assumption that ENaC variants may play a role in more common forms of hypertension (Citation7–10). Furthermore, our previous study showed that three common variants of the β- and γENaC genes (βENaC G589S, intron 12 -17CT, and γENaC V546I) occur approximately three times more frequently in Finnish patients with moderate-to-severe hypertension than in the general Finnish population (Citation11).
The recessively inherited AME is caused by a genetic deficiency of 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) (Citation12,Citation13). In normal kidney, 11βHSD2 converts cortisol to its inactive metabolite cortisone, thus protecting the mineralocorticoid receptor from binding of cortisol. In AME this protection is lost, and cortisol-mediated excessive mineralocorticoid action results in early-onset severe hypertension, failure to thrive, hypokalemia, and suppressed plasma renin and aldosterone levels. Genotype–phenotype analyses have revealed that, depending on the effect of the mutation on 11βHSD2 levels and/or activity, the phenotype of AME may vary widely from mild to severe forms (Citation14,Citation15). Although mutations causing classical AME are rare, subtle variations of the 11βHSD2 gene may play a role in the pathogenesis of salt sensitivity or essential hypertension (Citation16–18), possibly when interacting with some other genetic factors or living habits.
Excessive licorice ingestion is known to cause an acquired form of AME. Licorice contains glycyrrhizic acid which is hydrolyzed to glycyrrhetinic acid, an inhibitor of 11βHSD2 (Citation19–21), resulting in an AME-like phenotype, including hypokalemia, hypertension, and low plasma renin and aldosterone levels (Citation19,Citation22–24). Since licorice consumption is very common and not all of its consumers develop a hypertensive syndrome, we reasoned that genes influencing licorice action may at least partly determine susceptibility to its side-effects. Accordingly, we recruited subjects reporting licorice-induced hypertension to clinical and metabolic evaluation as well as molecular studies of their 11βHSD2 and ENaC subunit genes. Our study represents the first molecular genetic approach to licorice-induced hypertension.
Materials and methods
Patients and controls
We sought contact with people encountering significant blood pressure elevation in response to ingestion of licorice-containing confectionary using an advertisement published in the leading Finnish daily newspaper with a nation-wide market area. The invitation was directed to individuals who, subsequent to ingesting licorice-containing confectionary, had experienced one episode of an elevation of blood pressure to the extent necessitating treatment by a physician. A total of 52 subjects from different regions of Finland initially volunteered to participate. They were interviewed and given detailed information of the study. At this point, seven subjects declined to continue. After exclusion of subjects whose clinical data were equivocal, we ended up with a group comprising 30 subjects (23 females, 7 males) with a mean age of 44 ± 3 years (range 14–66 years). They had a medical documentation of an incident of substantially elevated blood pressure (≥150/100 mmHg), measured by a physician or a nurse, at the time of licorice ingestion. In addition, it was required that cessation of licorice consumption resulted in lowering of blood pressure levels and disappearance of any accompanying symptoms. Previous hospital and other medical records were collected, and all study subjects filled in a questionnaire regarding their health status, family history, and data on licorice consumption. All subjects were of Finnish origin and were apparently unrelated. Venous blood samples for DNA isolation were collected from all the study subjects. Some of the subjects (n = 17) also volunteered to give blood samples for measurement of serum sodium, potassium, creatinine, and cortisol levels, plasma renin activity and aldosterone levels, as well as urine samples for determination of the daily (24 h) excretion of potassium, sodium, and free cortisol. All the study subjects, except for one, were non-smokers. The study protocol was approved by the Ethics Review Committee of the Helsinki University Central Hospital and was in accordance with the Helsinki Declaration. All subjects gave their written informed consent.
For estimation of allele frequencies in the background population, we obtained blood samples from randomly selected healthy blood donors visiting the Finnish Red Cross Blood Transfusion Service. This group consisted of either 163 (76 women, 87 men, for the 11βHSD2 variations) or 312 (157 women, 155 men, for the αENaC insertion mutation) blood donors aged 40–50 years. The original study protocol did not permit disclosure of any health information of these blood donors. The occurrence of the αENaC insertion mutation was also examined in a group of treatment-resistant Finnish hypertensive patients (n = 346) characterized previously in detail (Citation11).
DNA analysis
DNA was isolated from venous blood samples using standard methods. The coding parts of all exons, exon/intron boundaries and introns 2 and 3 of the 11βHSD2 gene, and the exons 13 (in which all the currently identified mutations associating with Liddle's syndrome have been localized) of each of the α-, β-, and γENaC genes were amplified by polymerase chain reaction (PCR). In order to unequivocally assess the DNA sequence around the αENaC insertion allele, the mutant PCR product was subcloned into pDrive cloning vector using Qiagen PCR cloning kit (Qiagen Inc., Valencia, CA) according to the manufacturer's instructions.
To determine the frequency of the G insertion mutation of the αENaC gene among patients and controls, denaturing high-performance liquid chromatography (dHPLC) on the WAVE Nucleic Acid Fragment Analysis System HSM 3500A (Transgenomic, Omaha, NE) was used. First, a PCR product (198 bp) was amplified using primers 5′-GCC AGT TCC TCC ACC TGT C-3′ and 5′-AGC AAC TTC CTG AG CCT TAC-3′. The resulting amplicons were denatured and analyzed by dHPLC. All samples showing divergent chromatogram profiles were sequenced with ABI 3730 (Applied Biosystems, Foster City, CA).
Polymorphisms in the 11βHSD2 gene were detected using a PCR-based primer-induced restriction assay (PIRA).
In vitro mutagenesis and functional assays of the αENaC insertion mutation
A wild-type (wt) construct was first prepared from a construct containing human αENaC cDNA (NM_001038) in pBSK-SP6-globin vector. A PCR product of 554 bp in size was generated using a forward primer 5′-CTC CTC GGT GTT GTC TGT GGT-3′ and a reverse primer 5′-CAA GGG GTA CAG GGC TCG AG-3′ introducing an artificial XhoI site. This fragment was subcloned into the BlpI and XhoI sites of the original construct using standard methods. The resulting construct thus contained 200 nucleotides of ENaC sequence after the translation stop codon, and this construct was used in the in vitro experiments as the wt control. To obtain the mutant construct with the G insertion (αInsG), in vitro mutagenesis was carried out using QuickChange site-directed mutagenesis kit (Stratagene, LaJolla, CA) according to the manufacturer's instructions. The resulting construct (αInsG) was sequenced to verify the insertion mutation and to exclude any undesired sequence errors.
For functional studies, human ENaC wild-type and mutant α-subunits were co-expressed with β- and γ- human ENaC subunits in Xenopus laevis oocytes. cRNAs of the human ENaC αInsG mutant and wild-type α-, β-, and γ-subunits were synthesized in vitro and injected into stage V–VI Xenopus laevis oocytes as previously described (Citation11). After injection, whole oocyte currents were measured, using the two-electrode voltage clamp technique. The ENaC activity was assessed by measuring the amiloride-sensitive Na+ current, defined as the difference between the Na+ current recorded at a membrane potential of −100 mV in the absence and presence of 10 μM amiloride in the bath. For comparison, the amiloride-sensitive current (INa) for the αInsG mutants was expressed as INa relative to the mean ENaC wild-type current (INa InsG/INa wt).
In silico analysis of putative novel phosphorylation sites in the predicted novel 61 amino acid stretch in the αENaC was done using the programs ScanProsite (http://tw.expasy.org/tools/scanprosite) and NetPhos2.0 (http://www.cbs.dtu.dk/services/NetPhos). Search for cysteines possibly forming disulfide bridges was done using the program CYSPRED (http://cubic.bioc.columbia.edu/predictprotein/).
Statistical analysis
Data were analyzed using statistical SPSS program (version 11.0). Fisher's exact test was used for the frequency analysis of the variants.
Results
Patient characteristics
Thirty unrelated Finnish subjects, with a documented elevation of blood pressure levels at the time of licorice ingestion (1–25 years ago) volunteered for the present study (). The dosage and duration of their licorice consumption, as evaluated retrospectively from questionnaires, varied greatly, ranging from 25–50 g licorice candies/day for 1–3 days to 100–200 g/day for years. As the subjects had consumed variable amounts of mixed types of licorice products, it was impossible to assess the exact amounts of glycyrrhizic acid consumed.
Table I. Characteristics of the study group at the time of the initial episode of licorice-induced hypertension and during the present study. All laboratory values at the time of licorice-induced hypertension are based on the availability of previous medical records. Values are mean ± SEM.
Blood pressure (BP) levels at initial admission varied substantially, with a range of 150 to 240 mmHg (systolic BP) and 100 to 140 mmHg (diastolic BP), and an average value of 201 ± 4/118 ± 2 mmHg. Two-thirds (21/30) of the patients had sought medical help due to symptoms compatible with licorice side-effects, including headache, nausea, swelling of feet, anxiety or dizziness, while one-third (9/30) was found to have elevated blood pressure and/or hypokalemia at their routine doctor's check-up. One patient was admitted to the hospital due to pulmonary edema and weight gain of several kilograms. In all study subjects, as called for in the inclusion criteria, the blood pressure decreased (), and the accompanying symptoms disappeared after cessation of licorice consumption.
The average body mass index (BMI) of the study group was 25.3 ± 0.8 kg/m2; eight subjects presented with mild obesity (BMI 25–30 kg/m2), while five were significantly obese (BMI >30 kg/m2). Antihypertensive medication was in use in 5 subjects already at the time of initial admission, and 13 subjects used antihypertensive drugs at the time of the present study.
A total of 21 subjects (70%) reported a history of treatment-requiring hypertension in at least one first-degree relative. In addition, two subjects reported having close relatives with licorice-induced elevations of blood pressure.
Blood electrolyte, renin, and aldosterone levels
When studied, most of the subjects (80%) were hypokalemic, and in some cases hypokalemia was severe (lowest potassium level detected: 2.1 mmol/L) requiring potassium supplementation (). When measured (n = 4), plasma renin activity and aldosterone levels were low ().
We were able to carry out additional studies in 17 voluntary subjects 1–25 years after the initial evaluation (). Serum creatinine, sodium, chloride, and cortisol levels were normal in all but one subject who presented with increased serum cortisol level (data not shown). Urinary cortisol excretion was increased in two subjects. Serum potassium level was within the normal range in all but four subjects, who showed border-line low potassium levels (3.4–3.7 mmol/L), one of them also showing increased daily urinary potassium secretion. There was a wide variation in individual plasma renin activity and aldosterone concentrations, but altogether eight subjects had suppressed plasma renin activity (≤0.3 μg/L/h) and/or aldosterone (<183 pmol/L) levels. The average renin activity (1.2 ± 0.4 μg/L/h, n =14) was below the normal range. Four of the subjects had hypertensive medication, which may have affected the renin and aldosterone levels.
Variants of the 11βHSD2 gene
The ability of licorice to inhibit 11βHSD2 rendered this enzyme as the most attractive genetic candidate to contribute to licorice-induced hypertension. However, no nucleotide changes predicted to result in amino acid changes were found in this gene in any of the 30 individuals. Two previously identified nucleotide changes, i.e. a C to A nucleotide change in exon 2 (C468A) (NM_000196) and a G to A change in exon 3 (G534A) (Citation25), were detected in four of our study subjects who all were heterozygous for both variations, suggesting the variants were in linkage disequilibrium.
To examine further the possible significance of these two silent base changes, their occurrence was examined in a group of apparently healthy Finnish blood donors (n = 163). Altogether 14 carriers of each of these substitutions were identified, and again they were present simultaneously in the same individuals, thus confirming their complete linkage in the Finnish population. The frequency of the double-variant allele was not significantly different in subjects with licorice-induced hypertension (4/30 or 13.3%) and in blood donors (14/163 or 8.6%, P = 0.63).
Search for variations in the ENaC subunit genes
Sequencing of the exon 13 of each of the α-, β-, and γENaC genes revealed altogether six carriers of gene alterations. All subjects were women, and all were heterozygous for the gene variants identified ().
Table II. Characteristics of the subjects with α-, β-, or γENaC gene variants during the present study. Current licorice consumption could not be excluded for subjects 1 and 6.
An insertion of one extra G nucleotide into a stretch of seven consecutive Gs, located at the end of the αENaC gene (exon 13 encoding the carboxy-terminus of alpha ENaC) (αENaC 2004-2005insG, NM_001038; numbering from the translation start codon), was identified in two subjects. This novel G insertion is predicted to cause a frame shift at the terminal codon CCC, with resultant deletion of the last amino acid (proline) of the αENaC subunit protein. The DNA structure of the mutant allele predicts insertion of 61 novel amino acids to the carboxy-terminus of the αENaC protein ().
Figure 1. Partial amino acid sequence of the wild-type αENaC (αENaC wt) and predicted amino acid sequence corresponding to the αENaC G insertion mutation. The mutation affects the carboxy-terminal codon (corresponding to proline residue 669) predicted to result in insertion of 61 amino acids (underlined). Only sequences corresponding to the carboxy-terminal parts (starting from the PPPXY motif, indicated by a box) are shown. In silico prediction revealed a putative protein kinase C phosphorylation site (asterisk), a putative casein kinase II phosphorylation site (square), and a cysteine predicted to form a disulfide bond (triangle).

In addition, four subjects were carriers of some of the previously identified gene variants of the β- and γENaC genes: one subject had the βENaC G589S variant (Citation11,Citation26,Citation27), two subjects had the βENaC intron 12 -17C/T variant (Citation11), and one subject had the γENaC V546I variant (Citation11).
Clinical and family data of subjects with the ENaC variants
All six subjects with the ENaC variants had a family history of hypertension. Patients with the β- or γENaC variants presented with mild-to-moderate obesity, which may have contributed to their elevated blood pressure levels (Subjects 3–6, ). Their renin levels were below the normal range, and serum aldosterone levels varied greatly. Five had antihypertensive medication currently, which may affect the renin and aldosterone levels. Subjects 3 and 5 reported to be able to consume only small amounts of licorice without symptoms, and they reported to have family members showing similar sensitivity to licorice.
Of the two carriers of the αENaC G insertion mutation, one (Subject 1, ) was a 21-year-old woman, who at the age of 18 was found to be hypokalemic (2.9 mmol/L) and to have elevated blood pressure (200/130 mmHg). For years, she had been consuming licorice-containing candies in amounts of approximately 100 to 150 g twice a week. Treatment with amiloride/thiazide combination resulted in normalization of blood pressure levels. Her plasma renin and aldosterone levels were low but concurrent licorice use could not be excluded. The proband's identical twin sister (, Family I) also consumed licorice-containing candies, and her blood pressure levels were somewhat elevated (119–140/90–100 mmHg). The mother of the proband had had elevated blood pressure from the age of 30 years. The twin sister and the mother were found to be heterozygous for the G insertion mutation, and their renin and aldosterone levels were low. The other family members were non-carriers and normotensive, and their electrolytes as well as plasma renin and aldosterone levels were normal, with the exception of the father who had low plasma renin, aldosterone, and potassium levels (). Thus, in this family the αENaC mutation appeared to co-segregate with the hypertensive phenotype with low renin/aldosterone levels.
Figure 2. The pedigrees of the two families with the αENaC G insertion mutation. The probands are shown by arrow (the probands of Families I and II are the Subjects 1 and 2 of , respectively). The ages are given as recorded at the time of the present study (ht+ = treatment-requiring hypertension; ht− = no hypertension; ht(+) = elevated blood pressure without antihypertensive treatment; ? = blood pressure status unknown; ND = not determined; Ren = plasma renin, μg/L/h; Aldo = serum aldosterone, pmol/L; K = potassium, mmol/L).
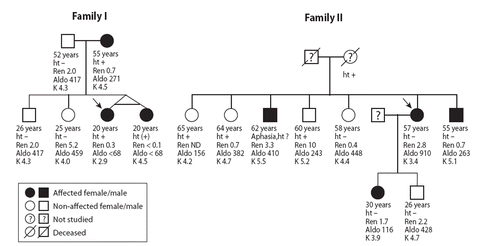
The other proband with the αENaC insertion mutation was a 57-year-old woman (Subject 2, ). She had consumed 50–75 g of licorice/day for a week; thereafter she experienced swelling of her feet and a weight gain of 5 kg. At that time, her blood pressure was 180/108 mmHg, but normalized after cessation of licorice consumption. She had mild hypokalemia, but her plasma renin and aldosterone levels were normal (). Family studies revealed three carriers of the αENaC mutation (, Family II), but none of them reported to have hypertension. The daughter of the proband had inherited the mutation and had relatively low plasma renin, aldosterone, and potassium levels. Two of the proband's brothers were likewise carriers of the mutation, and one of them was suffering from aphasia resulting from a stroke. Taken together, in this family no convincing co-segregation of the αENaC G insertion mutation with the low-renin hypertensive phenotype could be demonstrated.
Frequency of the αENaC insertion mutation in controls and hypertension patients
To further explore the pathophysiologic significance of the αENaC insertion mutation, we determined its frequency in a group of apparently healthy Finnish blood donors (n = 312) as well as in a selected group of Finnish hypertension patients (n = 346) (Citation11). Two of the blood donors were found to be heterozygous carriers of the G insertion, whereas no such carriers were found in the group of hypertensive patients. Thus, although the frequency of the αENaC mutation in our highly selected group of subjects with licorice-induced hypertension (2/30 or 6.7%) was higher than that observed in the blood donor group (2/312 or 0.6%, P = 0.04), this mutation does not appear to be a common cause of essential hypertension in Finland.
Functional properties of αENaC G insertion mutation
The functional consequences of the G insertion mutation of the αENaC gene were examined in vitro in Xenopus oocytes. The frame shift mutation αInsG, predicted to result in the insertion of 61 amino acid residues in the C-terminus of αENaC, did not increase ENaC-mediated Na current. The amiloride-sensitive current (INa) relative to control was 0.995 ± 0.186 (n = 33) versus 0.642 ± 0.097 (n = 41), wild-type versus αInsG mutant ENaC, respectively (mean ± SE, P = 0.07). Thus, when expressed in Xenopus oocytes, the αInsG mutant ENaC channel does not show detectable functional alterations.
Discussion
The present study is focusing on the hypothesis that a genetic susceptibility would underlie some cases with licorice-induced hypertension. We proposed that licorice ingestion, by inhibiting the 11βHSD2 enzyme, might potentiate the effect of subtle gene alterations which otherwise do not necessarily cause a clear-cut phenotype with elevated blood pressure. This type of mutation could be present, not only in the 11βHSD2 gene, but also in genes encoding subunits of ENaC, which would result in increased sodium reabsorption by the effect of altered cortisol metabolism induced by licorice. As licorice ingestion is fairly common in Finland, with the estimated consumption of licorice amounting to 1.3 kg/person/year (Citation28), we proposed that natural licorice ingestion could be used to identify subjects susceptible to blood pressure elevation. We were indeed able to recruit a group of 30 voluntary Finnish subjects with a previously documented incident of licorice-induced hypertension.
Licorice consumption, metabolic disturbances, and hypertension
Many of our study subjects presented with a typical licorice syndrome with hypokalemia, suppressed plasma renin activity, and serum aldosterone levels, accompanied with a variety of other symptoms including headache, edema, and nausea (Citation22,Citation29). In some cases the symptoms were severe, including pulmonary edema, but most of the clinical findings normalized after licorice cessation. Two subjects characterized themselves as ‘licorice-sensitive’. They had ingested only 25–50 g of licorice/day, an amount previously estimated to produce adverse effects for sensitive subjects (Citation30), when elevated blood pressure or other symptoms occurred. The levels of both systolic and diastolic blood pressures during licorice ingestion were greatly, and in some cases severely, elevated () and were substantially higher than usually described in the literature for licorice ingestion (Citation19,Citation22–24,Citation31). In patients with essential hypertension the blood pressure rise induced by licorice may be greater than in healthy volunteers (Citation32). After cessation of licorice consumption the reported blood pressure levels diminished in all study subjects, suggesting that licorice ingestion contributed to the observed high blood pressure levels ().
The low average plasma renin activity in our study subjects may, at least in part, be due to current surreptitious licorice consumption. It is of interest that altogether eight subjects had currently low plasma renin activity and/or aldosterone levels, which in some cases were accompanied by hypokalemia, and two subjects also presented with elevated daily urinary cortisol levels suggesting a potential subtle Liddle- or AME-like phenotype. Taken together, although variations in metabolism of glycyrrhizin/glycyrrhetic acid may account in part for the reported individual licorice sensitivity (Citation33), the observed metabolic changes and the positive family history of hypertension in 70% of our study subjects were suggestive of influence of additional genetic factors affecting sodium or cortisol metabolism.
ENaC gene variants and licorice-associated hypertension
In the present study we chose 11βHSD2 and α-, β-, and γENaC genes as the most attractive candidate genes in our search for genetic alterations predisposing to licorice-induced hypertension. Indeed, we were able to identify altogether six carriers of different ENaC gene variants. Two of the subjects were found to be carriers of a novel G insertion mutation in the αENaC gene, and four subjects were heterozygous for previously identified gene variations in the β- and γENaC genes () (Citation11,Citation26,Citation27). The prevalence of the gene variants, i.e. 20%, observed among subjects with licorice-induced hypertension is higher compared to the frequency among control subjects, i.e. 3.7% ((Citation11) and the present study), supporting the idea that gene alterations in ENaC genes may predispose to licorice-induced hypertension. Furthermore, five out of six variant carriers were currently requiring hypertensive treatment, and three of them responded favorably to a specific ENaC inhibitor, amiloride, suggesting that the underlying ENaC variations might play some role in their hypertension. This assumption is also supported by our previous study showing that the variant carriers presented with a tendency to increased urinary potassium loss in relation to plasma renin activity, suggesting that in the long run the variants may indeed affect the renin-aldosterone axis and contribute to hypertension (Citation11) .
Alpha ENaC insertion mutation and hypertension
No mutations associated with increased blood pressure have been identified in the coding region of the αENaC gene. Previously, a promoter polymorphism in the αENaC gene was suggested to associate with blood pressure in Japanese subjects (Citation34), and polymorphisms, such as αENaC A663T, have been shown to affect ENaC activity in vitro both in Xenopus oocytes and in mammalian cells (Citation35,Citation36). In the current study, we were able to identify a novel insertion of one G to a stretch of seven consecutive Gs located at the end of the αENaC. This insertion is predicted to result in a frame shift and incorporation of 61 novel amino acids to the carboxy-terminus of the αENaC protein.
With one exception (Citation37), all hitherto identified mutations shown to associate with Liddle's syndrome have been localized in exon 13 in the part coding for cytoplasmic C-terminus of either the β- or γ-subunit of ENaC. These alterations mutate or delete the proline-rich domain PPPXY (PY motif), a binding site for the ubiquitin ligase Nedd-4, which promotes endocytosis and lysosomal degradation of ENaC. The αInsG mutation leads to addition of 61 amino acid residues downstream of the PPPXY. At least in Xenopus oocytes, these additional residues do not seem to affect the integrity of the PY motif, as was shown by the absence of detectable functional alterations of the αInsG mutant in the Xenopus oocyte expression system. It remains possible that the αInsG mutation may lead to subtle changes in ENaC activity in aldosterone-sensitive epithelial cells that are undetectable in frog oocytes. Prediction in silico revealed a cysteine with a potential to form disulfide bonds in the C-terminus of the mutant αENaC protein (). In addition, the 61 amino acid region is predicted to harbor at least two novel phosphorylation sites, one for protein kinase C (PKC) and the other for casein kinase II (CKII) (). Phosphorylation may be an important mechanism for ENaC regulation, and PKC and CKII sites are also present in the C-terminus of β- and γENaC genes (Citation38,Citation39). Thus, it is possible that, although not shown in vitro in the oocyte system, subtle long-term effects in ENaC regulation may occur in the native kidney epithelium in the αInsG mutation carriers.
The phenotypes of the mutation carriers varied considerably. This may not be an unexpected finding, as even in cases with molecularly well defined Liddle's syndrome the penetrance of the disease phenotype has been shown to vary greatly (Citation2,Citation40), consistent with gene–environment interactions. It is tempting to speculate that other modifier genes or extrinsic factors, such as licorice consumption, salt intake, or obesity, might act in concert to modify the resulting phenotype of αENaC gene insertion mutation.
The finding that the αENaC gene mutation was present in higher frequency in our study subjects than in population controls (6.7% versus 0.6%, respectively, P < 0.05) suggested that it may play a role in licorice-induced hypertension. Furthermore, as hypertension does not prevent blood donation, some of the blood donors used as controls in our study may in fact have elevated blood pressure levels. Due to ethical limitations, however, the clinical data of the blood donors were not available.
11βHSD2 gene and licorice
Based on the observed increased urinary free cortisol and low plasma renin activity/aldosterone levels, two subjects were suspected to have disturbances in cortisol metabolism. However, no amino acid altering variations in the 11βHSD2 gene were found in any of the study subjects.
The relevance, if any, of the two base variations in exon 2 and 3 remains unclear. The G534A nucleotide variation has been previously shown to associate with end-stage renal disease but not with essential hypertension (Citation25). Studies regarding association of the marker with salt-sensitive blood pressure have also been controversial (Citation17,Citation41). The similar frequency of the 11βHSD2 variants observed in our study group and in the control group suggests that these alterations do not seem to play a significant role in licorice-induced hypertension. It remains yet possible that pathophysiologically significant alterations do exist in the 11βHSD2 gene in our study subjects, but they lie in the regions not sequenced in the present study, including the promoter region, as previously shown in one kindred with AME (Citation12).
Even other genes regulating renal sodium reabsorption may contribute to salt-sensitive forms of hypertension (Citation42) and thereby to licorice sensitivity. A specific form of salt-sensitive hypertension was recently described in circadian clock-defective Cry null mice (Citation43). The underlying defect was attributed to constitutively high activity of 3β-hydroxysteroid dehydrogenase type VI (Hsd3b6) in aldosterone-producing cells. The gene encoding the human counterpart of this enzyme, 3β-hydroxysteroid dehydrogenase type I (Hsd3b1), thus constitutes another interesting candidate influencing salt and licorice sensitivity.
Study limitations
The major limitation of the current study is the fairly small size (n = 30) of the study group. However, these subjects were very carefully selected fulfilling strict recruitment criteria, including not only medically documented licorice-induced hypertension, but also showing lowering of blood pressure after cessation of licorice intake. It seems that, at least in Finland, subjects fulfilling these criteria are fairly rare.
Conclusion
In conclusion, our study for the first time explores the possibility that the well documented licorice-related elevation of blood pressure could have a genetic background. Our results demonstrate that mutations of the 11βHSD2 gene do not appear to constitute a common cause for licorice-induced hypertension. Subtle variants of the α-, β-, and γ-subunits of the ENaC may contribute to licorice-induced side-effects in certain individuals, but further prospective studies are needed to fully clarify this issue.
Acknowledgements
We thank Dr Leena Mykkänen for help in collection of pedigree data and Mrs Susanna Saarinen for technical assistance. This study was supported by The Academy of Finland, Sigrid Juselius Foundation, and The Finnish Foundation for Cardiovascular Research. Authors HEM and KP contributed equally to this work.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56.
- Liddle GW, Bledsoe T, Coppage WSJ. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. 1963;76: 199–213.
- New MI, Levine LS, Biglieri EG, Pareira J, Ulick S. Evidence for an unidentified steroid in a child with apparent mineralocorticoid hypertension. J Clin Endocrinol Metab. 1977;44:924–33.
- Ulick S, Levine LS, Gunczler P, Zanconato G, Ramirez LC, Rauh W, . A syndrome of apparent mineralocorticoid excess associated with defects in the peripheral metabolism of cortisol. J Clin Endocrinol Metab. 1979;49:757–64.
- Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, . Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–14.
- Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, . Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82.
- Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, . Association of hypertension with T594M mutation in beta subunit of epithelial sodium channels in black people resident in London. Lancet. 1998;351:1388–92.
- Ambrosius WT, Bloem LJ, Zhou L, Rebhun JF, Snyder PM, Wagner MA, . Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertension. 1999;34:631–7.
- Rayner BL, Owen EP, King JA, Soule SG, Vreede H, Opie LH, . A new mutation, R563Q, of the beta subunit of the epithelial sodium channel associated with low-renin, low-aldosterone hypertension. J Hypertens. 2003;21:921–6.
- Wong ZY, Stebbing M, Ellis JA, Lamantia A, Harrap SB. Genetic linkage of beta and gamma subunits of epithelial sodium channel to systolic blood pressure. Lancet. 1999;353:1222–5.
- Hannila-Handelberg T, Kontula K, Tikkanen I, Tikkanen T, Fyhrquist F, Helin K, . Common variants of the beta and gamma subunits of the epithelial sodium channel and their relation to plasma renin and aldosterone levels in essential hypertension. BMC Med Genet. 2005;6:4.
- Mune T, Rogerson FM, Nikkila H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Nat Genet. 1995;10:394–9.
- van Uum SH, Hermus AR, Smits P, Thien T, Lenders JW. The role of 11 beta-hydroxysteroid dehydrogenase in the pathogenesis of hypertension. Cardiovasc Res. 1998;38:16–24.
- Dave-Sharma S, Wilson RC, Harbison MD, Newfield R, Azar MR, Krozowski ZS, . Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. J Clin Endocrinol Metab. 1998;83:2244–54.
- White PC, Agarwal AK, Nunez BS, Giacchetti G, Mantero F, Stewart PM. Genotype-phenotype correlations of mutations and polymorphisms in HSD11B2, the gene encoding the kidney isozyme of 11beta-hydroxysteroid dehydrogenase. Endocr Res. 2000;26:771–80.
- Watson B Jr, Bergman SM, Myracle A, Callen DF, Acton RT, Warnock DG. Genetic association of 11 beta-hydroxysteroid dehydrogenase type 2 (HSD11B2) flanking microsatellites with essential hypertension in blacks. Hypertension. 1996;28:478–82.
- Lovati E, Ferrari P, Dick B, Jostarndt K, Frey BM, Frey FJ, . Molecular basis of human salt sensitivity: the role of the 11beta-hydroxysteroid dehydrogenase type 2. J Clin Endocrinol Metab. 1999;84:3745–9.
- Agarwal AK, Giacchetti G, Lavery G, Nikkila H, Palermo M, Ricketts M, . CA-Repeat polymorphism in intron 1 of HSD11B2: effects on gene expression and salt sensitivity. Hypertension. 2000;36:187–94.
- Stewart PM, Wallace AM, Valentino R, Burt D, Shackleton CH, Edwards CR. Mineralocorticoid activity of liquorice: 11-beta-hydroxysteroid dehydrogenase deficiency comes of age. Lancet. 1987;2:821–4.
- Whorwood CB, Sheppard MC, Stewart PM. Licorice inhibits 11 beta-hydroxysteroid dehydrogenase messenger ribonucleic acid levels and potentiates glucocorticoid hormone action. Endocrinology. 1993;132:2287–92.
- Tanahashi T, Mune T, Morita H, Tanahashi H, Isomura Y, Suwa T, . Glycyrrhizic acid suppresses type 2 11 beta-hydroxysteroid dehydrogenase expression in vivo. J Steroid Biochem Mol Biol. 2002;80:441–7.
- Epstein MT, Espiner EA, Donald RA, Hughes H. Effect of eating liquorice on the renin-angiotensin aldosterone axis in normal subjects. BMJ. 1977;1:488–90.
- Forslund T, Fyhrquist F, Froseth B, Tikkanen I. Effects of licorice on plasma atrial natriuretic peptide in healthy volunteers. J Intern Med. 1989;225:95–9.
- Ferrari P, Sansonnens A, Dick B, Frey FJ. In vivo 11beta-HSD-2 activity: variability, salt-sensitivity, and effect of licorice. Hypertension. 2001;38:1330–6.
- Smolenicka Z, Bach E, Schaer A, Liechti-Gallati S, Frey BM, Frey FJ, . A new polymorphic restriction site in the human 11 beta-hydroxysteroid dehydrogenase type 2 gene. J Clin Endocrinol Metab. 1998;8383:1814–7.
- Melander O, Orho M, Fagerudd J, Bengtsson K, Groop PH, Mattiasson I, . Mutations and variants of the epithelial sodium channel gene in Liddle's syndrome and primary hypertension. Hypertension. 1998;31:1118–24.
- Persu A, Barbry P, Bassilana F, Houot AM, Mengual R, Lazdunski M, . Genetic analysis of the beta subunit of the epithelial Na+ channel in essential hypertension. Hypertension. 1998;32:129–37.
- Blomberg K, Hallikainen A. Suomessa kaupan olevien lakritsivalmisteiden glykyrritsiinipitoisuudet. Helsinki: Elintarvikeviraston julkaisuja; 1993.
- Conn J, Rovner JR, Cohen EL. Licorice-induced pseudoaldosteronism. Hypertension, hypokalemia, aldosteronopenia, and suppressed plasma renin activity. JAMA. 1968;205:80–4.
- Stormer FC, Reistad R, Alexander J. Glycyrrhizic acid in liquorice—evaluation of health hazard. Food Chem Toxicol. 1993;31:303–12.
- Sigurjonsdottir HA, Franzson L, Manhem K, Ragnarsson J, Sigurdsson G, Wallerstedt S. Liquorice-induced rise in blood pressure: a linear dose-response relationship. J Hum Hypertens. 2001;15:549–52.
- Sigurjonsdottir HA, Manhem K, Axelson M, Wallerstedt S. Subjects with essential hypertension are more sensitive to the inhibition of 11 beta-HSD by liquorice. J Hum Hypertens. 2003;17:125–31.
- Ploeger B, Mensinga T, Sips A, Seinen W, Meulenbelt J, DeJongh J. The pharmacokinetics of glycyrrhizic acid evaluated by physiologically based pharmacokinetic modeling. Drug Metab Rev. 2001;33:125–47.
- Iwai N, Baba S, Mannami T, Ogihara T, Ogata J. Association of a sodium channel alpha subunit promoter variant with blood pressure. J Am Soc Nephrol. 2002;13:80–5.
- Samaha FF, Rubenstein RC, Yan W, Ramkumar M, Levy DI, Ahn YJ, . Functional polymorphism in the carboxyl terminus of the alpha-subunit of the human epithelial sodium channel. J Biol Chem. 2004;279:23900–7.
- Tong Q, Menon AG, Stockand JD. Functional polymorphisms in the alpha-subunit of the human epithelial Na+ channel increase activity. Am J Physiol Renal Physiol. 2006;290:F821–7.
- Hiltunen TP, Hannila-Handelberg T, Petajaniemi N, Kantola I, Tikkanen I, Virtamo J, . Liddle's syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens. 2002;20:2383–90.
- Shi H, Asher C, Yung Y, Kligman L, Reuveny E, Seger R, . Casein kinase 2 specifically binds to and phosphorylates the carboxy termini of ENaC subunits. Eur J Biochem. 2002;269:4551–8.
- Shimkets RA, Lifton R, Canessa CM. In vivo phosphorylation of the epithelial sodium channel. Proc Natl Acad Sci U S A. 1998;95:3301–5.
- Findling JW, Raff H, Hansson JH, Lifton RP. Liddle's syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab. 1997;82:1071–4.
- Poch E, Gonzalez D, Giner V, Bragulat E, Coca A, de La Sierra A. Molecular basis of salt sensitivity in human hypertension. Evaluation of renin-angiotensin-aldosterone system gene polymorphisms. Hypertension. 2001;38:1204–9.
- Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, . Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–9.
- Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, . Salt-sensitive hypertension in circadian clock–deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16:67–75.