Abstract
The circadian clock generates oscillations in physiology and behavior, known as circadian rhythms. Links between the circadian clock genes Periods, Bmal1, and Cryptochromes and aging and cancer are emerging. Circadian clock gene expression is changed in human pathologies, and transgenic mice with mutations in clock genes develop cancer and premature aging. Control of genome integrity and cell proliferation play key roles in the development of age-associated pathologies and carcinogenesis. Here, we review recent data on the connection between the circadian clock and control of the cell cycle. The circadian clock regulates the activity and expression of several critical cell cycle and cell cycle check-point-related proteins, and in turn cell cycle-associated proteins regulate circadian clock proteins. DNA damage can reset the circadian clock, which provides a molecular mechanism for reciprocal regulation between the circadian clock and the cell cycle. This circadian clock-dependent control of cell proliferation, together with other known physiological functions of the circadian clock such as the control of metabolism, oxidative and genotoxic stress response, and DNA repair, opens new horizons for understanding the mechanisms behind aging and carcinogenesis.
Key messages
Recent progress on the role of circadian clock in cell cycle control connects the circadian clock, aging, and cancer.
Introduction
The circadian clock is an endogenous system that acts as an internal time-keeping device. The circadian clock generates approximately 24-hour oscillations in physiology and behavior known as circadian rhythms (Citation1). Circadian rhythms most likely developed as an adaptation to the 24-hour periodicity in the Earth's rotation. An important function of the circadian clock is to synchronize different metabolic processes in an organism, as well as to synchronize an organism to its environment in order to guarantee the optimal performance of different organ systems (Citation2). Circadian rhythms have been described in both prokaryotes and eukaryotes, and the many molecular pathways associated with the circadian clock are evolutionarily conserved (Citation3). In mammals, the central commanding center of circadian rhythms is located in the group of hypothalamic neurons termed the suprachiasmatic nucleus (SCN). The SCN synchronizes the activity of peripheral oscillators, which operate in practically every cell in every tissue of an organism. These peripheral oscillators generate rhythms in gene expression, metabolism, and hormone secretion which ultimately result in rhythmic changes in physiology and behavior (Citation4).
The importance of the circadian system has been demonstrated in model organisms such as fungi, flies, and mice, and epidemiological studies have highlighted the various disorders that are connected with defects in the circadian machinery. In humans, these disorders include depression and bipolar disorders (Citation5), sleep disorders such as familial advanced sleep phase syndrome (FASPS), as well as metabolic (Citation6) and cardiovascular diseases (Citation7). Evidence indicates that shift-workers have an increased risk of developing cardiovascular diseases, diabetes, and cancer (Citation8–10). Transgenic animals with mutations in circadian genes develop metabolic syndromes, cancer, and an accelerated aging phenotype (Citation11–13). The control of genome integrity and cell proliferation plays a critical role in the development of age-associated pathologies and carcinogenesis. Therefore, recently emerging links between the circadian clock, the cell cycle, and genotoxic stress response may shed new light onto the molecular connections between aging, cancer, and the circadian clock.
Molecular organization of circadian clock in mammals
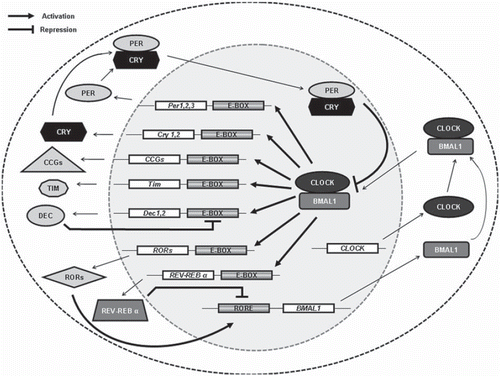
Circadian rhythms are genetically determined, and several genes important for circadian oscillation have been identified. The molecular circadian oscillator is organized into interconnected positive and negative transcription–translation feedback loops (Citation14). While many components of the molecular circadian oscillator are evolutionarily conserved, differences exist in the detailed organization between different organisms. In mammalian cells, the circadian oscillatory mechanism is achieved at the molecular level through interlocking autoregulatory feedback loops that control the daily rhythms of many proteins (). At the core of the circadian machinery lie two basic helix-loop-helix (bHLH) PAS (Period, ARNT, SIM) domain-containing proteins, brain and muscle ARNT-like 1 (BMAL1) (Citation15,Citation16) and circadian locomotor output cycles kaput (CLOCK) (Citation17,Citation18). Through the interaction of their PAS domains, BMAL1 and CLOCK heterodimerize in the cytoplasm and translocate to the nucleus where they bind to circadian E box elements located in the promoter region of various genes and activate the expression of circadian clock genes such as the three periods (PER1, PER2, and PER3) and two cryptochromes (CRY1 and CRY2) (Citation19). The BMAL1:CLOCK complex also binds to the circadian E box elements in the promoter of differentially expressed in chondrocytes (Dec1 and Dec2), the nuclear receptor REV-ERBα, and the retinoid-related orphan receptor (ROR). PERs and CRYs accumulate in the cytoplasm and bind each other, and upon their dimerization they translocate to the nucleus to complete the autoregulatory feedback loop by acting as negative regulators of the BMAL1:CLOCK complex (Citation20,Citation21). REV-ERBα and ROR compete for the binding to ROR elements present in the promoter of Bmal1, with ROR being a positive regulator of Bmal1 transcription (Citation22) and REV-ERBα being a negative regulator (Citation23). DECs recognize the same regulatory E box sites that the BMAL1:CLOCK complex recognizes but, in contrast to BMAL1:CLOCK, inhibit the target gene expression, thus creating an additional loop (Citation24). Therefore, several intertwined regulatory feedback loops control the circadian oscillator to produce rhythmic expression. An additional level of complexity supplements the multiple negative feedback loops because of the extensive regulation carried out through post-translational control of core circadian clock gene expression (Citation25). In addition, the BMAL1:CLOCK complex can bind to the E box regulatory elements present in many clock-controlled genes (CCGs). CCGs are genes that can exert control over a myriad of physiological processes, such as metabolism, the sleep–wake cycle, locomotor activity, and body temperature.
Figure 1. Overview of the positive and negative transcriptional–translational feedback loops controlling the circadian machinery. BMAL1 and CLOCK proteins heterodimerize and drive the expression of the target genes Pers, Crys, Decs, Rev-Erbα, and RORs through binding to the circadian E box element in their promoters. PERs and CRYs form a complex which inhibits the BMAL1:CLOCK complex transcriptional activity, and hence their own expression. The nuclear receptor REV-ERBα represses and the retinoid-related orphan receptor ROR promotes the transcription of BMAL1 though binding to ROR elements in the Bmal1 promoter (Citation2). DECs compete with the BMAL1:CLOCK complex at the E box located in the DEC promoter and so inhibit their own transcription. The BMAL1:CLOCK complex also regulates the expression of various clock-controlled genes (CCGs), which play important roles in many physiological functions.
Circadian clock and cell cycle
The cell cycle and circadian cycle are two periodic processes, and the connection between these two systems was observed decades ago in unicellular organisms (Citation26). It was proposed that circadian clock control of the cell cycle evolved as a protective mechanism against DNA-damaging UV light in order to restrict the phase of DNA replication to a time when UV inducing damage is minimal (during the night). It was also suggested that circadian gating helps to separate in time processes which are difficult to separate in space, as illustrated with the example of time separation of oxidative metabolism and DNA replication in yeast (Citation27). In metazoans, this connection is less obvious and harder to demonstrate. Indeed, in adult animals, cells in organs such as the brain, liver, kidneys, and heart do not proliferate and are probably maintained in a quiescent state. In other organs, such as the skin and intestinal epithelium, cells are constantly in a state of active proliferation, while connective tissue cells are mostly in a quiescent state. Thus, in practically every tissue, there are cells in both quiescent and proliferative states. However, for proliferative tissues, circadian and daily variations in the distribution of proliferating cells between different phases of the cell cycle have been demonstrated for lymphocytes, digestive tract epithelium, and others (Citation28,Citation29). Another line of evidence, which suggests a connection between the circadian clock and the cell cycle in mammalian cells, comes from analysis of primary cells isolated from circadian mutant mice and from down-regulation of circadian protein expression in immortalized cell lines. Circadian clock proteins are also involved in genotoxic stress response and cell cycle check-point control. Several recent reviews address the detailed role of the circadian clock in genotoxic stress response and carcinogenesis (Citation30–34); therefore, we will concentrate on the most recent data discussing the role of circadian transcription factors BMAL1 and CLOCK in cell cycle control. We propose that the BMAL1:CLOCK complex serves as a major linker between the circadian clock and the cell cycle.
Role of BMAL1:CLOCK complex in cell cycle and check-point control
Several recent studies, which address the effects of circadian clock gene mutations/deletions on cell proliferation both in vivo and in cell culture, connect the CLOCK and BMAL1 proteins with cell cycle control. Embryonic fibroblasts isolated from Clock mutant mice demonstrate a delayed cell cycle entrance after serum starvation, but there is no difference in the proliferation rate between wild-type and mutant cells under normal cell-culturing conditions (Citation35). Clock mutation results in a protein with an internal deletion, which strongly affects its ability to activate target gene expression; indeed, the expression of many genes, such as cell cycle progression inhibitors p21 and p27, was up-regulated, while Cdk2 and cyclins D3 and E1 were down-regulated in Clock mutant cells, which may be responsible for the observed effect on their proliferation. Expression of apoptosis-related genes such as Akt1, Bcl2, and Pbef was also changed (Citation35). Thus, the most likely scenario is that CLOCK transcriptional activity is important for cell proliferation.
Both CLOCK and BMAL1 are important for hair follicle cycling. Hair growth is a cyclic process with several well defined phases: anagen, telogen, and catagen. Anagen is a phase of active keratinocyte proliferation and differentiation, providing the basis for hair growth. Deficiency of CLOCK or BMAL1 activity in mice results in delayed anagen progression, specifically, delayed cell cycle progression in secondary hair germ of hair follicles. The anagen delay leads to an overall shift of the hair growth cycle; however, the entire hair growth cycle duration was not affected, and no morphological abnormalities in the hair follicles were detected (Citation36). In other studies, impaired proliferation of Bmal1-/- primary hepatocytes was detected by thymidine incorporation experiments (Citation37). Freshly isolated hepatocytes demonstrate spontaneous proliferation in culture, and this proliferation was significantly delayed in primary hepatocytes isolated from BMAL1-deficient mice compared with hepatocytes isolated from wild-type mice. It is important to note that no pathological changes in the liver of BMAL1-deficient mice have been observed. Thus, the observations made for Clock mutant embryonic fibroblasts (Citation35), hair follicles in CLOCK and BMAL1 mutant mice (Citation36), and for hepatocytes from BMAL1-deficient mice (Citation37) suggest that in normal cells the activity of the BMAL1:CLOCK complex is unnecessary for progression through the cell cycle but, rather, important for the transition from a quiescent state to a proliferative state. In both studies, significant up-regulation of p21 expression, described below, upon BMAL1 deficiency has been reported, and this up-regulation has been proposed as a potential mechanism for the delay in cell cycle progression.
p21 (also known as waf1/Cip1/CDKIN1) is a cyclin-dependent kinase inhibitor, which regulates G1 phase progression through its interaction with CDK2 complexes (Citation38). A BMAL1 deficiency results in an up-regulation of p21 expression in the liver (Citation37) and hair follicles of mice (Citation36). This induction of p21 is most likely responsible for the above-mentioned impairment of hepatocyte proliferation and hair follicle cycling. Indeed, down-regulation of Bmal1 expression in cultured hepatocytes with a Bmal1-specific siRNA eliminates the proliferation delay between wild-type and BMAL1-deficient cells (Citation37). There is no BMAL1:CLOCK-responsive element in p21 promoter, and most likely the BMAL1:CLOCK complex regulates p21 transcription indirectly, through transcriptional control of RORs and REV-ERBα nuclear receptors. RORs are positive and REV-ERBα negative regulators of p21 promoter, therefore, p21 expression is determined by mutual balance of ROR and REV-ERBα proteins (Citation37), which in turn are under BMAL1:CLOCK control.
In contrast to these observations, Bmal1 was isolated from several candidate genes in the screening of siRNA libraries for modulators of p53 function in human cells (Citation39). In experiments with human cancer cell lines, BMAL1 acts as a negative regulator of cell proliferation upon p53 induction or after exposure of cells to DNA-damaging agents. p53 activation leads to an increased level of p21, which is a major mediator of p53-dependent arrest. Down-regulation of Bmal1 expression with siRNA attenuates p53-dependent p21 induction and releases the cells from cell cycle arrest. Down-regulation of p21 with siRNA has the same effect, which suggests that arrest depends on p53-dependent induction of p21 (Citation39). Thus, these data suggest that BMAL1 is an important modulator of p53-dependent induction of p21 and check-point activation after DNA damage.
Similarly to the situation with wild-type cells, CLOCK activity is necessary for tumor cell proliferation, and inhibition of Clock expression with a Clock-specific siRNA significantly reduces the proliferation rate of Lewis lung carcinoma cells (LLC1) in culture, as well as tumor growth rates after inoculation in mice (Citation40). While CLOCK is important for the proliferation of the carcinoma cell line LLC1 (Citation40), BMAL1 acts as a tumor suppressor protein in some hematologic malignancies (Citation41). In several lymphomas and leukemias, Bmal1 gene expression is transcriptionally silenced by hypermethylation of promoter CpG islands. Re-introduction of BMAL1 into the BMAL1-deficient RAJI lymphoma cell line results in a significant inhibition of tumor cell growth both in cell culture and after injection into nude mice. In agreement with that, siRNA-mediated depletion of BMAL1 in the BMAL1-expressing leukemia cell line MOLT-4 stimulates cell proliferation (Citation41). It is known that in early tumors, probably as a consequence of oncogene activation, DNA damage check-point pathways are activated, but as a tumor progresses these normal check-point pathways are impaired through different mechanisms. Down-regulation of Bmal1 expression in hematologic tumors is a possible mechanism. These results are in good agreement with data on the role of BMAL1 in p53-dependent DNA damage-induced arrest.
Thus, CLOCK and BMAL1 are positive regulators of cell cycle progression in normal cells, while in tumor cells the same proteins act both as negative and positive regulators (Citation35–37,Citation40,Citation41). In order to explain the differences between the above-cited works, we propose that the BMAL1:CLOCK complex has different roles in cell cycle control depending on whether the cell is under normal or genotoxic stress conditions: the BMAL1:CLOCK complex stimulates the transition from a quiescent state to G1, and at the same time BMAL1 (and probably CLOCK) activity is necessary for check-point arrest/apoptosis after stress induced by DNA damage or oncogene expression.
Thus, the BMAL1:CLOCK complex demonstrates dualistic properties: acting as a positive regulator of cell cycle progression under non-stress condition and as a negative regulator after stress. Additionally, the effects exerted by CLOCK and BMAL1 can be tissue and cell type-specific, and the pattern of changes in target gene expression upon a deficiency of CLOCK or BMAL1 is different in different tissues; therefore, reported discrepancies in results may be due to difference in cell types used in different studies. Future studies will help elucidate the exact role that the BMAL1:CLOCK transcriptional complex has in the cell cycle, which, in turn, will contribute to our understanding of the reported role of circadian clock proteins in tumorigenesis and aging.
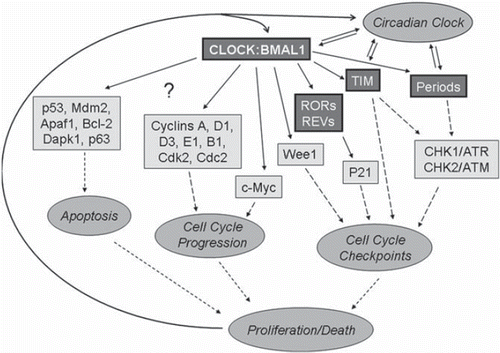
What is a possible molecular mechanism of BMAL1:CLOCK-dependent cell cycle control? summarizes the proteins and molecular pathways linking the circadian clock and the cell cycle. Microarray analysis and direct measurement of cell cycle-associated gene expression has revealed a group of important cell cycle regulators, which have been identified as potential targets of the circadian clock. Among them are cyclin-dependent kinase inhibitors p21, p27, Cdk2, cyclins A, B1, D1, D3, and E1, cyclin-dependent kinases Cdk2 and Cdc2, transcription factors c-myc, Wee1 kinase, as well as others. For Wee1 and c-myc, direct transcriptional control by the BMAL1:CLOCK complex has been demonstrated. Wee1 kinase expression is positively regulated by the BMAL1:CLOCK complex, and circadian variation in Wee1 expression is disrupted in circadian clock mutants (Citation42). Wee1 regulates the G2/M transition through phosphorylation of the CDC2/Cyclin B1 complex, and most likely is responsible for circadian variation in liver regeneration (Citation42). c-Myc is a transcription factor that is a critical regulator of cell cycle progression. c-Myc demonstrates a circadian oscillation in its expression, and this oscillation is impaired in circadian mutants. The BMAL1:NPAS2 complex suppresses the activity of the c-myc promoter in transient transfection experiments (Citation43). NPAS2 is the closest homolog of the CLOCK protein, which suggests that the BMAL1:CLOCK complex can regulate c-Myc expression too; however, this regulation has not been demonstrated. p21 expression is regulated by the BMAL1:CLOCK complex indirectly through control of the expression of ROR and REV-ERBα nuclear receptors, as well as by interference with p53-dependent activation. Mechanisms of circadian control for other cell cycle-related targets are currently unknown. The expression of several apoptosis-related genes, such as p53, mdm2, Apaf1, Dapk1, p63, and Bcl2, is changed in the circadian mutants, and while there are no data related to the mechanisms of these changes, it is possible that the BMAL1:CLOCK complex is involved in the control.
Figure 2. Control of cell death and proliferation by the circadian clock transcriptional complex. All circadian clock proteins are shown in bold, direct transcriptional control is shown by thick arrows, transcriptional control through unknown mechanisms is shown by thin arrows, other interaction and regulations are shown by dashed arrows. Transcriptional factor BMAL1:CLOCK regulates the expression of circadian clock proteins TIM, PERs RORs, REV-ERBα, and cell cycle-associated proteins WEE1 kinase and transcriptional factor c-Myc. TIM and PERs regulate cell cycle check-points through interaction with CHK1/ATR and CHK2/ATM complexes. RORs and REV-ERBα directly regulate the expression of cyclin-dependent kinase inhibitor p21. Several other cell cycle-associated proteins such as cyclins, cyclin-dependent kinases, and pro- and anti-apoptotic proteins are under circadian clock control, but the exact mechanism of regulation is unknown. The fate of the cell (proliferation or death) depends on the mutual balance of all these proteins. In turn cell proliferation and check-point proteins can regulate circadian clock function.
Cell cycle progression requires passing through the so-called cell cycle check-points. There are three major cell cycle check-points: the G1/S check-point, the S phase check-point, and the G2/M check-point. These check-points serve as a surveillance mechanism in order to maintain the integrity of the genome. DNA damage activates signaling pathways related to certain cell cycle check-points and delays cell cycle progression. The delay in cell cycle progression occurs in order to allow a cell to decide if it should repair the DNA damage, or eliminate the cell through apoptosis if the damage cannot be repaired. Dysfunctional cell cycle check-points can lead to premature aging and cancer (Citation44–46).
Recently, several groups demonstrated some interesting connections between DNA damage response pathways and circadian clock proteins. The circadian clock proteins PER1 (PERIOD1) and TIM (TIMELESS) have been linked with the cell cycle check-points through two important check-point associated protein kinases: ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) (Citation33).
ATM is critical for activating the cell cycle check-points in response to DNA double-stranded breaks. In response to DNA damage, ATM autophoshorylates, which results in further phosphorylation of downstream substrates involved in cell cycle arrest, DNA damage repair, and other cellular processes (Citation47). One of the most commonly studied and well known direct substrates of ATM is p53 (Citation48). p53 is a tumor suppressor gene that promotes cell cycle arrest or apoptosis (Citation49). Under normal, non-stress conditions, p53 exists in a complex with MDM2, a ubiquitin ligase which promotes p53 degradation, and helps maintain a low intracellular concentration of p53. In response to DNA damage, p53 is phosphorylated and dissociates from MDM2, resulting in the stabilization of p53 and activation of p53-controlled pathways. In the early response DNA damage, activated p53 induces the expression of the p21 cyclin-dependent kinase inhibitor, which mediates G1 cell cycle arrest by inhibiting the activity of CDK2 and CDK4.
p53 is also known to regulate many genes involved in G2/M cell cycle progression (Citation49). Depending upon the extent of DNA damage, p53 will promote cell survival, cell senescence, or cell death. The protein kinase CHK2 is another important substrate of ATM. CHK2 is activated by phosphorylation, and in turn phosphorylates and inhibits CDC25C phosphatase. CDC25C-dependent dephosphorylation and activation of CDC2 is required for cell cycle progression from the G2 to M phase (Citation48). Therefore, inhibition of CDC25C by CHK2 results in cell cycle arrest. It was observed that PER1 can interact with the ATM/CHK2 complex, and irradiation (IR) treatment enhances that interaction. This interaction is critical for CHK2 activation because the inhibition of Per1 expression by siRNA leads to impaired phosphorylation of CHK2. In agreement with these data, cellular response to DNA damage depends on the level of Per1 expression, and Per1 expression is down-regulated in some human cancers (Citation34).
A second important mediator of DNA damage stress response is ATR kinase, which acts in a heterodimeric complex with its partner ATRIP (ATR interacting protein). Unlike ATM, ATR mainly responds to replication stress and is activated by stalled replication forks (Citation45). ATR interacts with and activates CHK1 kinase through phophorylation. CHK1 activation results in phosphorylation and inhibition of CDC25C and CDC25A phosphatases. CDC25A dephosphorylates the CDK1 and CDK2 complexes, which mediates the G1 to S phase transition and S phase progression. Inhibition of CDC25A activity by CHK1 results in growth arrest at the G1/S and S phase check-points. Concurrently, similar to the ATM/CHK2 pathway, inhibition of CDC25C activity leads to G2/M arrest (Citation45).
The circadian clock protein TIM interacts with both the ATR/ATRIP complex and with CHK1, and DNA-damaging agents such as ultraviolet (UV) light or hydroxyurea stimulate these interactions (Citation50). Similar to PER1/ATM/CHK2 interaction, TIM/ATR/CHK1 interaction is critical for the activation of the ATR/CHK1 pathway (Citation50), and indeed, down-regulation of Tim expression using siRNA leads to a reduction in basal and damage-induced phosphorylation levels of CHK1. Down-regulation of Tim also results in defects in replication check-points, resulting in early entry into M phase before DNA replication (Citation51). TIM also regulates the cell cycle through direct interaction with components of the replication fork and through complex formation with TIPIN (Tim interacting protein) (Citation51). A detailed role of TIM in DNA replication has been recently reviewed (Citation33).
Post-translational modifications of the CLOCK/BMAL1 complex—possible feedback from cell cycle to the circadian clock
The circadian clock controls cell cycle progression, and likewise the phase of the cell cycle influences the progression through the circadian cycle. This creates a reciprocal feedback mechanism for regulation. Indeed, mitosis can affect circadian oscillation, causing positive and negative phase shifts (Citation2), and the treatment with cell cycle progression-interfering agents such as irradiation can reset the phase of the circadian clock both in cell culture and in vivo (Citation52,Citation53). A potential molecular mechanism for this feedback is post-translational regulation of circadian clock proteins. Post-translational modifications play an important role in circadian biology (Citation25). The molecular clock-work that directs an organism's daily rhythms are subject to various post-translational modifications that are important for robust circadian oscillatory activity (Citation19). The function of post-translational modifications differs based on the type of modification as well as the location of the modification. The effects include protein degradation and translocation, among others. In particular, BMAL1, CLOCK, PERs, and CRYs are able to be modified meticulously by various kinases, as well as being able to be altered by modifications such as acetylation, sumoylation, and ubiquitination (Citation25).
Post-translational modifications of PERs and CRYs have been recently reviewed (Citation19,Citation25,Citation54). Here, we discuss recent advances in circadian biology with regard to the post-translational regulation of BMAL1 and CLOCK. It is interesting to note that the formation of the BMAL1:CLOCK complex is necessary for phosphorylation (Citation55), acetylation (Citation56), sumoylation (Citation57), and ubiquitination (Citation58) to occur. It has been shown that different kinases, such as casein kinase I (CKI), mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), and glycogen synthase kinse (GSK)3β, are able to phosphorylate BMAL1 (Citation59–62). Each serves a different function. GSK3β phosphorylates BMAL1 specifically on Ser17 and Thr21 through direct interaction with BMAL1 (Citation60). The phosphorylation event results in ubiquitination of BMAL1 and leads to 26S proteasome-dependent degradation. MAPK interacts with BMAL1 and phosphorylates it at multiple sites. These sites include Ser527, Thr534, and Ser599. Phosphorylation by MAPK reduces transcriptional activity of BMAL1 (Citation61). In sharp contrast to MAPK, phosphorylation by casein kinase Iδ and casein kinase Iε positively regulates BMAL1 transcriptional activity (Citation59). Finally, it was demonstrated recently that PKCα controls the circadian clock through phosphorylation of BMAL1 (Citation63).
Acetylation also regulates circadian protein activity. CLOCK has an intrinsic histone acetyltransferase (HAT) activity and can acetylate histones and BMAL1 (Citation64). Histone acetylation by CLOCK stimulates transcription of CCGs, whereas CLOCK-mediated acetylation of BMAL1 acts on the negative limb of the circadian feedback loop (Citation56,Citation64). The acetylation of BMAL1 is controlled by the SIRT1 histone deacetylase (HDAC) (Citation65). SIRT1 belongs to the family of mammalian class III HDACs, and SIRT1 physically associates with the BMAL1:CLOCK heterodimer and deacetylates BMAL1 (Citation65). SIRT1 also interacts with PER2 and deacetylates this protein (Citation66). Post-translational regulation of BMAL1 proteins is summarized in .
Table I. Post-translational modifications of BMAL1.
Taken together, these data suggest that post-translational modifications fill an important role in controlling the BMAL1:CLOCK complex activity and regulation of circadian oscillation. It is possible that through these modifications the intracellular circadian clock may possibly sense the phase of cell cycle. Therefore, an intricate network between the circadian machinery and different kinases creates a basis for connection and reciprocal regulation between the circadian clock and the cell cycle.
Circadian clock and aging
Aging is a genetically and environmentally driven decay of organism function. Multiple systems are involved in the regulation of aging, and most systems are affected by aging. The fact that circadian clock function can be affected by aging has been known for decades. Furthermore, changes in the sleep–wake pattern and oscillations of physiological parameters in aging humans and other organisms have been reported (for recent review see (Citation67)). The hypothesis that the circadian clock can equally influence the aging process has been suggested only recently based on observations made in model organisms (Citation68). Mice deficient in BMAL1 represent the most striking example of the effect of circadian clock proteins on aging. These mice demonstrate an accelerated aging phenotype, displaying an average life-span of 8 months compared with 26 months for wild-type mice. Furthermore, BMAL1-deficient mice develop multiple age-associated diseases over time (Citation12). Clock mutant mice and Period2 mutant mice do not demonstrate accelerated aging under normal conditions, but some features of accelerated aging do develop in these mice after exposure to non-lethal doses of radiation (Citation11,Citation40). Accelerated aging has not been reported for Cry1,2 double-deficient mice (Citation69). This difference between different circadian mutants suggests a complex relationship between the circadian clock and aging, and that the effect of the circadian clock on the aging process depends on balance between different circadian proteins.
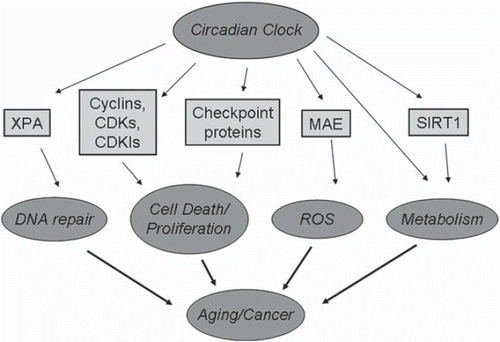
The above-discussed circadian control of the cell cycle and genotoxic stress response can contribute to the clock-dependent regulation of aging, but as illustrated in the circadian clock controls other systems which are known to be associated with aging, such as the control of metabolism (Citation70), oxidative stress response (Citation68,Citation71), and DNA repair (Citation72).
Figure 3. Cell cycle control links the circadian clock with aging and carcinogenesis. Circadian clock, through transcriptional control and protein–protein interaction, regulates cellular metabolism, proliferation, ROS homeostasis, and DNA repair. Disruption of circadian clock function or deficiency of circadian clock proteins will interfere with the activity of these systems, which are known as critical regulators of aging and carcinogenesis.
Increased levels of reactive oxygen species (ROS) in tissues are correlated with aging. High levels of ROS result in oxidative stress damage to DNA, lipids, and proteins, and progressive accumulation of this damage was proposed as a cause or major contributor to many age-related diseases (Citation73) and, probably, to aging itself (Citation74). BMAL1 controls ROS levels in cells, as a BMAL1 deficiency results in chronic oxidative stress; and although the molecular mechanism of regulation is unknown, several major antioxidant enzymes are potential transcriptional targets of the BMAL1:CLOCK complex (Citation75). Thus, through control of intracellular production and detoxification of ROS, BMAL1 and probably the BMAL1:CLOCK complex contribute to the circadian clock-dependent control of aging.
Genome integrity is critical in connection with aging and carcinogenesis. Therefore, damage to DNA is considered very hazardous (Citation46). DNA repair is a major mechanism which prevents the accumulation of DNA damage, and various systems for repairing different types of DNA damage exist. Defects in these systems are often associated with an increased incidence of tumor development and accelerated aging (Citation44), which is in agreement with the proposed role of these systems in the control of aging. Recently it was demonstrated that circadian clock control of nucleotide excision repair occurs through transcriptional control of the rate-limiting enzyme xeroderma pigmentosum A (XPA) protein expression (Citation72). The circadian oscillation of DNA excision repair is responsible for the oscillation in removal of cisplatin-produced adducts and can contribute to the circadian control of genome integrity.
Another emerging connection between the circadian clock, metabolism, and aging is the interaction and mutual regulation between the circadian clock proteins and SIRT1. As it was mentioned above, the BMAL1:CLOCK complex and BMAL1:CLOCK: PER2 complex physically interact with SIRT1 in vivo (Citation65,Citation66). SIRT1 belongs to the evolutionarily conserved family of protein deacetylases (known as sirtuins), which are critical regulators of metabolism and, at least in model organisms, are implicated in the control of aging and in the response to caloric restriction, which is the only known intervention that increases the life-span of different species, including primates. The exact mechanism of SIRT1 and other sirtuin family members’ function is a subject for intense study (Citation76). SIRT1 is in turn regulated by the circadian system. SIRT1 is an NAD+-dependent histone deacetylase, and its activity depends on the concentration of its natural co-factor NAD+. One of the systems that determine NAD+ levels in cells is the NAD+ salvage pathway. The enzyme nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in the salvage pathway, and nampt is under direct transcriptional control of the BMAL1: CLOCK complex. The expression of nampt and NAD+ levels demonstrate circadian oscillation, which suggests circadian control of SIRT1 activity (Citation65,Citation77).
Shift-work and risk of cancer
In humans, a disruption in circadian rhythms can result in changes in physiology and in the development of certain pathologies, including cardiovascular disease and cancer (Citation30). Shift-work has been associated with various types of cancers (). For example, studies by Schernhammer et al. and others have shown an increased risk of breast cancer in shift-workers (Citation78–80). In addition, other studies are proposing a modest increase in breast cancer prevalence in flight attendants, who are likely to incur jet lag as a result of their occupation (Citation81,Citation82). Moreover, there is evidence for an increase in the prevalence of prostate cancer in airline pilots (Citation83,Citation84), as well as other professionals who have a tendency to do shift-work, such as fire-fighters (Citation85) and police officers (Citation86). A recent study in Japan has shown correlation between prostate cancer and rotating shift-work (Citation87). However, there is an inconsistency on breast cancer formation as a result of shift-work, such as a recent study in Chinese women that failed to show an association between night shift-work and an increased incidence of breast cancer (Citation88). Therefore, the problem is complex, and many additional factors such as exposure to cosmic rays and gamma radiation need to be considered (Citation89).
Table II. Incidence of cancer risk as a result of shift-work.
Conclusions
The circadian regulation of aging and carcinogenesis is a fast-growing field of research, and there are several recent reviews on this subject (Citation27,Citation34). Cell cycle control and genotoxic stress response play critical roles in both processes. Recent research data suggest a mutual influence between the circadian clock and the cell cycle, and provides a functional link between the circadian clock, aging, and cancer. Details of the molecular organization involved in the control of cell cycle and the circadian clock are well studied, while intracellular pathways connecting these two processes are just emerging. Circadian transcription factors BMAL1 and CLOCK, either directly or through their transcriptional targets PERs, TIM, and REV-ERBα/ROR, control the expression and activity of critical regulators of cell metabolism, proliferation, and death, and, as such, represent a potential molecular link between these systems. Many questions still need to be addressed, such as the exact roles of individual components of the clock, the cause of and importance of tissue specificity, the different effects occurring in normal and stressed cells, and the function of circadian clock proteins in tumor cells. Further studies will resolve these problems and contribute to our understanding of the mechanisms controlling cancer and aging, and will help identify novel targets for therapy.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–41.
- Gachon F, Nagoshi E, Brown SA, Ripperger J, Schibler U. The mammalian circadian timing system: from gene expression to physiology. Chromosoma. 2004;113:103–12.
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, . Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–20.
- Dardente H, Cermakian N. Molecular circadian rhythms in central and peripheral clocks in mammals. Chronobiol Int. 2007;24:195–213.
- McClung CA. Circadian genes, rhythms and the biology of mood disorders. Pharmacol Ther. 2007;114:222–32.
- Laposky AD, Bass J, Kohsaka A, Turek FW. Sleep and circadian rhythms: key components in the regulation of energy metabolism. FEBS Lett. 2008;582:142–51.
- Reilly DF, Westgate EJ, FitzGerald GA. Peripheral circadian clocks in the vasculature. Arterioscler Thromb Vasc Biol. 2007;27:1694–705.
- Filipski E, Li XM, Levi F. Disruption of circadian coordination and malignant growth. Cancer Causes Control. 2006; 17:509–14.
- Curtis AM, Fitzgerald GA. Central and peripheral clocks in cardiovascular and metabolic function. Ann Med. 2006; 38:552–9.
- Lowden A, Moreno C, Holmback U, Lennernas M, Tucker P. Eating and shift work—effects on habits, metabolism and performance. Scand J Work Environ Health. 2010;36: 150–62.
- Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50.
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–73.
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, . Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–5.
- Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111:919–22.
- Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, . Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280: 1564–9.
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, . Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–17.
- Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, . Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89:655–67.
- King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, . Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–53.
- Hirayama J, Sassone-Corsi P. Structural and functional features of transcription factors controlling the circadian clock. Curr Opin Genet Dev. 2005;15:548–56.
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, . mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205.
- Griffin EA Jr, Staknis D, Weitz CJ. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;286:768–71.
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, . A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–37.
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, . The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002; 110:251–60.
- Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, . Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–4.
- Harms E, Kivimae S, Young MW, Saez L. Posttranscriptional and posttranslational regulation of clock genes. J Biol Rhythms. 2004;19:361–73.
- Edmunds LN Jr, Funch RR. Circadian rhythm of cell division in Euglena: effects of random illumination regimen. Science. 1969;165:500–3.
- Chen Z, McKnight SL. A conserved DNA damage response pathway responsible for coupling the cell division cycle to the circadian and metabolic cycles. Cell Cycle. 2007;6: 2906–12.
- Gomes JR, Pereira AA, Barth L, Silva JS, Leite ML, Wille AC, . Circadian variation of the cell proliferation in the jejunal epithelium of rats at weaning phase. Cell Prolif. 2005;38:147–52.
- Potten CS, Owen G, Roberts SA. The temporal and spatial changes in cell proliferation within the irradiated crypts of the murine small intestine. Int J Radiat Biol. 1990;57:185–99.
- Levi F, Filipski E, Iurisci I, Li XM, Innominato P. Cross-talks between circadian timing system and cell division cycle determine cancer biology and therapeutics. Cold Spring Harb Symp Quant Biol. 2007;72:465–75.
- Hunt T, Sassone-Corsi P. Riding tandem: circadian clocks and the cell cycle. Cell. 2007;129:461–4.
- Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010;106:68–78.
- Kondratov RV, Antoch MP. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007;17:311–7.
- Gery S, Koeffler HP. Circadian rhythms and cancer. Cell Cycle. 2010;9(6):1097–103.
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, . Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104:3342–7.
- Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, Smyth P, . Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009;5:e1000573.
- Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. 2008;283:4535–42.
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82: 675–84.
- Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One. 2009;4:e4798.
- Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, . Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008;7:1197–204.
- Taniguchi H, Fernandez AF, Setien F, Ropero S, Ballestar E, Villanueva A, . Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009;69:8447–54.
- Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–9.
- Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, . Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol. 2002;12:540–50.
- Laiho M, Latonen L. Cell cycle control, DNA damage checkpoints and cancer. Ann Med. 2003;35:391–7.
- Li L, Zou L. Sensing, signaling, and responding to DNA damage: organization of the checkpoint pathways in mammalian cells. J Cell Biochem. 2005;94:298–306.
- Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9.
- Khanna KK, Lavin MF, Jackson SP, Mulhern TD. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001;8:1052–65.
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst). 2004;3:889–900.
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31.
- Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–16.
- Gotter AL, Suppa C, Emanuel BS. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol. 2007;366:36–52.
- Oklejewicz M, Destici E, Tamanini F, Hut RA, Janssens R, van der Horst GT. Phase resetting of the mammalian circadian clock by DNA damage. Curr Biol. 2008;18:286–91.
- Gamsby JJ, Loros JJ, Dunlap JC. A phylogenetically conserved DNA damage response resets the circadian clock. J Biol Rhythms. 2009;24:193–202.
- Mehra A, Baker CL, Loros JJ, Dunlap JC. Post-translational modifications in circadian rhythms. Trends Biochem Sci. 2009;34:483–90.
- Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17:1921–32.
- Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, . CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. 2007;450:1086–90.
- Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309:1390–4.
- Lee J, Lee Y, Lee MJ, Park E, Kang SH, Chung CH, . Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol Cell Biol. 2008;28:6056–65.
- Eide EJ, Vielhaber EL, Hinz WA, Virshup DM. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase Iepsilon. J Biol Chem. 2002;277: 17248–54.
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS One. 2010;5:e8561.
- Sanada K, Okano T, Fukada Y. Mitogen-activated protein kinase phosphorylates and negatively regulates basic helix-loop-helix-PAS transcription factor BMAL1. J Biol Chem. 2002;277:267–71.
- Spengler ML, Kuropatwinski KK, Schumer M, Antoch MP. A serine cluster mediates BMAL1-dependent CLOCK phosphorylation and degradation. Cell Cycle. 2009;8: 4138–46.
- Robles MS, Boyault C, Knutti D, Padmanabhan K, Weitz CJ. Identification of RACK1 and protein kinase Calpha as integral components of the mammalian circadian clock. Science. 2010;327:463–6.
- Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125: 497–508.
- Grimaldi B, Nakahata Y, Kaluzova M, Masubuchi S, Sassone-Corsi P. Chromatin remodeling, metabolism and circadian clocks: the interplay of CLOCK and SIRT1. Int J Biochem Cell Biol. 2009;41:81–6.
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, . SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–28.
- Hofman MA, Swaab DF. Living by the clock: the circadian pacemaker in older people. Ageing Res Rev. 2006;5:33–51.
- Kondratov RV. A role of the circadian system and circadian proteins in aging. Ageing Res Rev. 2007;6:12–27.
- Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2841–6.
- Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134:728–42.
- Hardeland R, Coto-Montes A, Poeggeler B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol Int. 2003;20:921–62.
- Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci U S A. 2009;106:2864–7.
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300.
- Kondratov RV, Vykhovanets O, Kondratova AA, Antoch MP. Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging (Albany NY). 2009;1:979–87.
- Guarente L, Picard F. Calorie restriction—the SIR2 connection. Cell. 2005;120:473–82.
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, . Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324: 651–4.
- Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. Night work and risk of breast cancer. Epidemiology. 2006;17:108–11.
- Davis S, Mirick DK, Stevens RG. Night shift work, light at night, and risk of breast cancer. J Natl Cancer Inst. 2001;93: 1557–62.
- Hansen J. Increased breast cancer risk among women who work predominantly at night. Epidemiology. 2001;12:74–7.
- Rafnsson V, Hrafnkelsson J, Tulinius H. Incidence of cancer among commercial airline pilots. Occup Environ Med. 2000;57:175–9.
- Rafnsson V, Tulinius H, Jonasson JG, Hrafnkelsson J. Risk of breast cancer in female flight attendants: a population-based study (Iceland). Cancer Causes Control. 2001;12:95–101.
- Pukkala E, Aspholm R, Auvinen A, Eliasch H, Gundestrup M, Haldorsen T, . Incidence of cancer among Nordic airline pilots over five decades: occupational cohort study. BMJ. 2002;325:567.
- Pukkala E, Aspholm R, Auvinen A, Eliasch H, Gundestrup M, Haldorsen T, . Cancer incidence among 10,211 airline pilots: a Nordic study. Aviat Space Environ Med. 2003; 74:699–706.
- Krstev S, Baris D, Stewart P, Dosemeci M, Swanson GM, Greenberg RS, . Occupational risk factors and prostate cancer in U.S. blacks and whites. Am J Ind Med. 1998;34: 421–30.
- Zeegers MP, Friesema IH, Goldbohm RA, van den Brandt PA. A prospective study of occupation and prostate cancer risk. J Occup Environ Med. 2004;46:271–9.
- Kubo T, Ozasa K, Mikami K, Wakai K, Fujino Y, Watanabe Y, . Prospective cohort study of the risk of prostate cancer among rotating-shift workers: findings from the Japan collaborative cohort study. Am J Epidemiol. 2006;164: 549–55.
- Pronk A, Ji BT, Shu XO, Xue S, Yang G, Li HL, . Night-shift work and breast cancer risk in a cohort of Chinese women. Am J Epidemiol. 2010;171:953–9.
- Pukkala E, Auvinen A, Wahlberg G. Incidence of cancer among Finnish airline cabin attendants, 1967–92. BMJ. 1995;311:649–52.
- Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, . The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–40.
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–7.
- Viswanathan AN, Hankinson SE, Schernhammer ES. Night shift work and the risk of endometrial cancer. Cancer Res. 2007;67:10618–22.
- Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, . Night-shift work and risk of colorectal cancer in the Nurses’ Health Study. J Natl Cancer Inst. 2003;95:825–8.