Abstract
Tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS) is a dominantly inherited autoinflammatory disease caused by heterozygous mutations in the TNFRSF1A gene encoding for the TNF receptor 1 (TNFR1). TRAPS is a multi-faceted and heterogeneous disease which commonly manifests as recurrent episodes of high fever accompanied by abdominal pain, pleurisy, migratory rash, and myalgia. Disease attacks occur spontaneously or may be elicited by minor triggers. Because of a vigorous and sustained acute-phase response it may be complicated by systemic AA amyloidosis. Therapeutically interleukin-1 blockade seems even more promising than TNF blockade. Studies on the pathogenesis of TRAPS have shown TNFα-dependent cellular signalling to be defective, an enigmatic finding considering the hyperinflammatory phenotype of the disease. Several studies indicate that most mutated receptors never reach the cell surface but are misfolded and trapped in the endoplasmic reticulum, where they may elicit an intracellular inflammatory response, and thus lead to constitutional expression of proinflammatory cytokines. The aim of this review is to describe the current understanding of the pathogenesis of TRAPS by integrating recent clinical and laboratory data.
| Abbreviations | ||
| BS | = | Blau syndrome |
| CAPS | = | cryopyrin-associated periodic syndromes |
| CIAS1 | = | cold-induced autoinflammatory syndrome 1 gene |
| CINCA | = | chronic infantile neurological, articular, and cutaneous syndrome |
| CRD | = | cysteine-rich domain |
| DD | = | death domain |
| DIRA | = | deficiency of interleukin-1 receptor antagonist |
| DISC | = | death-inducing signalling complex |
| ELISA | = | enzyme-linked immunosorbent assay |
| ER | = | endoplasmic reticulum |
| FADD | = | Fas-associated death domain |
| FCAS | = | familial cold autoinflammatory syndrome |
| FMF | = | familial Mediterranean fever |
| HIDS | = | hyperimmunoglobulinaemia D with periodic fever syndrome |
| IgD | = | immunoglobulin D |
| IKK | = | inhibitor of κB kinase |
| IL-1 | = | interleukin-1 |
| IL1RN | = | the gene encoding the interleukin-1 receptor antagonist |
| IL-6 | = | interleukin-6 |
| iPSCs | = | induced pluripotent stem cells |
| JNK | = | c-Jun N-terminal kinase |
| LPS | = | lipopolysaccharide |
| MAPK | = | mitogen-activated protein kinase |
| MEFV | = | Mediterranean fever |
| MIM | = | Mendelian inheritance in man |
| MMP | = | matrix metalloproteinase |
| MRI | = | magnetic resonance imaging |
| MVK | = | mevalonate kinase |
| MWS | = | Muckle–Wells syndrome |
| NADPH | = | nicotinamide adenine dinucleotide phosphate |
| NRLP3 | = | cryopyrin |
| NF-κB | = | nuclear factor kappa B |
| NOD | = | nucleotide-binding oligomerization domain |
| NOMID | = | neonatal-onset multisystem inflammatory disease |
| PAPA | = | pyogenic arthritis with pyoderma gangrenosum and acne |
| PLAD | = | pre-ligand assembly domain |
| PSTPIP | = | proline serine threonine phosphatase interacting protein |
| ROS | = | reactive oxygen species |
| SAA | = | serum amyloid A |
| SODD | = | silencer of death domain |
| TNF | = | tumour necrosis factor |
| TNFR | = | tumour necrosis factor receptor |
| TNFRSF1A | = | tumour necrosis factor receptor superfamily 1A gene |
| TRADD | = | TNFR1-associated death domain adaptor |
| TRAF | = | TNF-receptor associated factor |
| TRAPS | = | tumour necrosis factor receptor-associated periodic syndrome |
| UPR | = | unfolded protein response |
Key messages
In TRAPS, a dominantly inherited autoinflammatory disease caused by mutations in the TNFRSF1A gene encoding for the type 1 tumour necrosis factor receptor (TNFR1), there is paradoxically low TNF-α-induced cellular signalling observed in in-vitro studies.
Mutant TNFR1 fails to reach the cell surface and does not participate in TNF-α-induced signalling but is misfolded and trapped in the endoplasmic reticulum (ER), where it may induce ER stress and elicit an intracellular inflammatory response with constitutional expression of proinflammatory cytokines, amplified and prolonged by creation of a positive feedback loop.
Interleukin-1 signalling is implied to play a role in the pathogenesis of TRAPS by mounting evidence of therapeutic response to anakinra in patients, possibly induced by production of reactive oxygen species and inflammasome-activating mechanisms.
Introduction
Tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS; MIM 142680) is a rare dominantly inherited disease characterized by recurrent episodes of fever and generalized and localized inflammation (Citation1). It belongs to the group of hereditary autoinflammatory diseases previously named hereditary periodic fever syndromes (). The term autoinflammatory disease and the concept of autoinflammation were introduced in 1999 after the discovery of TRAPS (Citation1) and have subsequently been extended to cover not only rare monogenic disorders but also multifactorial diseases such as gout, systemic-onset juvenile idiopathic arthritis, adult-onset Still's disease, and Crohn's disease (Citation2–6). The new concept has been very useful for our understanding of the whole spectrum of immune-mediated diseases (Citation7). The autoinflammatory diseases differ from the classical autoimmune diseases such as systemic lupus erythematosus in that they lack high-titre autoantibodies and antigen-specific T cells (Citation1). In each of the hereditary autoinflammatory diseases a specific genetic defect which involves the regulation of innate immunity has been demonstrated () (Citation8–11). The discovery of the genetic basis of the hereditary autoinflammatory diseases has led to the identification of several novel inflammatory pathways, among them activation of interleukin-1β (IL-1β) through NRLP3 inflammasome formation and caspase-1 activation. This review represents an effort to integrate clinical and laboratory data to create a cohesive picture of the pathogenesis of TRAPS.
Table I. Hereditary autoinflammatory diseases. (Modified after (Citation6)).
Clinical features of TRAPS
TRAPS is caused by heterozygous mutations in the gene TNFRSF1A on chromosome 12p13, which encodes the type 1 TNF receptor (TNFR1, TNFRSF1A, p55, CD120A). The previous name of the disease was familial Hibernian fever (Citation12), but it was renamed in 1999 after its aetiology had been determined and because of its worldwide distribution (Citation1). In patients with TRAPS, recurrent inflammatory episodes occur either spontaneously or after minor triggers, such as local injury, minor infection, stress, exercise, and hormonal changes. The mean age of onset of TRAPS is 3 years, but many diagnoses have been made in adolescents or young adults. Clinically TRAPS presents one of the most variable and multi-faceted syndromes among the hereditary autoinflammatory diseases (Citation13). Symptoms include high fever (up to 40–41°C), intense abdominal pain, pleurisy, pericarditis, arthritis or arthralgia, migratory rashes and myalgia associated with underlying fasciitis, headaches, and conjunctivitis and periorbital oedema. Myocarditis, sacroiliitis, pharyngitis, stomatitis, testicular pain, and inguinal hernias have also been described. Attacks can last from a few days to months, with an average duration of 21 days. There is no fixed periodicity between attacks, and the severity and in some patients the frequency of attacks tend to decrease with increasing age. During attacks and sometimes even between them neutrophil leucocytosis, thrombocytosis, markedly elevated concentrations of the acute-phase reactants C-reactive protein and serum amyloid A (SAA), and an accelerated erythrocyte sedimentation rate can be recorded. The low-penetrance TNFR1 variants R92Q and P46L seem to contribute to atypical inflammatory responses in TRAPS, including myocarditis and pericarditis, and have recently been detected in patients with recurrent pericarditis as the only clinical manifestation (Citation14–16).
The diagnosis of TRAPS relies on mutational analysis of the TNFRSF1A gene combined with a compatible clinical picture. Recently, however, patients with a clinical picture of TRAPS in the absence of mutations in TNFRSF1A have been described (Citation17,Citation18). Like any disease accompanied by a sustained acute-phase reaction, including elevation of the amyloid precursor SAA, TRAPS can be complicated by systemic AA amyloidosis, usually manifesting as nephrotic syndrome and progressing to renal failure. Several different approaches to treatment of TRAPS have been tried (Citation13). Some patients show at least partial relief of their symptoms with non-steroidal anti-inflammatory drugs, but most need courses of oral corticosteroids (prednisone or equivalent starting from 20–30 mg/day) to control their symptoms. Some patients require prolonged therapy with corticosteroids because of a fluctuating or chronic disease course. Immunomodulators such as methotrexate, azathioprine, and cyclosporin have generally been ineffective, and colchicine, which benefits patients with familial Mediterranean fever, has no reported effect in TRAPS. TNF inhibition with etanercept (soluble TNFR2 fused to the Fc region of human IgG) has been reported to reduce the frequency and intensity of the inflammatory episodes (Citation13) but in the longer run has often been shown unable to totally eliminate inflammation (Citation19,Citation20). Infliximab (monoclonal anti-TNF-α antibody) has also been tried but usually without success, and in some patients it has even been associated with a flare of the disease (Citation21,Citation22). The IL-1 receptor antagonist anakinra has emerged as a promising therapeutic alternative even for those TRAPS patients who fail on etanercept (Citation23–25).
TNFR1-mediated signalling
TNFR1 mediates signalling of its ligand TNF-α, which is one of the major pleiotropic cytokines capable of activating proinflammatory and proapoptotic responses. TNFR1 signalling has been studied extensively and has shown marked complexity, thus full coverage of TNFR1 signalling is beyond the scope of this review, and we refer the reader to reviews on the subject for further information (Citation26–28). The best described signalling pathways activated by TNF-α binding to TNFR1 are the inflammatory and antiapoptotic nuclear factor kappa B (NF-κB) pathway and the apoptosis-inducing pathway, depicted in . The reactive oxygen species (ROS)-inducing pathway has been included to show variety of signalling and to highlight the emerging role of ROS in the inflammatory response (Citation32).
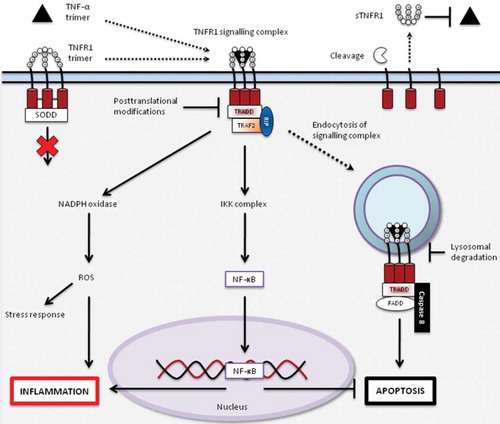
Figure 1. Signalling pathways activated by TNF-α binding to TNFR1. TNFR1 is expressed on the cell surface as a transmembrane receptor with four cysteine-rich domains (CRD) on the extracellular side and a death domain (DD) intracellularly. The first CRD is a pre-ligand assembly domain (PLAD) that interacts with PLADs from other TNFR1s to form receptor homotrimers, which without their ligand preferentially associate with inhibitory proteins in the cytoplasm, such as silencer of death domain (SODD) (Citation26,Citation28,Citation29). The binding of a TNF-α homotrimer leads to a change in relative positioning of the intracellular DDs, favouring assembly of a signalling complex by interaction of the TNFR1–TNF-α complex, TNFR1-associated death domain adaptor (TRADD) protein, receptor-interacting protein (RIP), and TNF-receptor-associated factor 2 (TRAF2) (Citation30). The interaction of this complex with other effector proteins leads to signalling through, among others, nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) and inhibitor of κB kinase complex (IKK complex), resulting in formation of reactive oxygen species (ROS) and liberation of nuclear factor kappa B (NF-κB) respectively, both of which contribute to the induction of the inflammatory response (Citation28,Citation31–34). The internalization of the signalling complex by endocytosis favours the assembly of a death-inducing signalling complex (DISC), consisting of the TNFR1–TNF-α complex, TRADD, Fas-associated death domain (FADD), and caspase-8, leading to the induction apoptosis, which in turn is inhibited by NF-κB activation (Citation30,Citation35). TNFR1 signalling is terminated by receptor endocytosis and degradation and post-translational modifications of the signal-transducing proteins (Citation36,Citation37). Cleavage of TNFR1 by matrix metalloproteinases (MMPs) on the cell surface results in the production of soluble TNFR1s (sTNFR1s) that are capable of binding free TNF-α trimers, possibly inhibiting TNF-mediated signalling (Citation37,Citation38).
This figure is partly based on the article by Kimberley et al. (Citation60).
TNFRSF1A mutations and TRAPS
More than 60 TNFRSF1A mutations have been reported in the INFEVERS database (http://fmf.igh.cnrs.fr/ISSAID/infevers/). All mutations hitherto described affect the extracellular part of the receptor, and most of them involve the first and second cysteine-rich domain (CRD). The first CRD is thought to be involved in the formation of the receptor trimer, whereas the second CRD contains the majority of contact residues between ligand and receptor. Many of the mutations involve cysteine residues and intermolecular disulfide bonds that are critical for the three-dimensional structure of the receptor and thus for the function of its extracellular portion. Mutations in the cysteine residues are generally associated with a more severe disease phenotype and carry the greatest risk of amyloidosis (Citation13).
Pathogenesis of TRAPS
Because the clinical picture and laboratory features of TRAPS are clearly those of a hyperinflammatory state, early investigators believed that the disease-related mutations in the TNFRSF1A gene would be gain-of-function mutations (Citation1). However, functional studies of mutated TNFR1 have presented researchers with an enigma. According to several studies there is in fact less binding of TNF-α to the mutated receptor (Citation2,Citation39), less surface expression of TNFR1 on cells from TRAPS patients compared with controls (Citation39–41), and less TNFR1-mediated activation of downstream mediators such as NF-κB (Citation41,Citation42). Many of the characteristics of the mutated TNFR1 would thus be expected to have an anti-inflammatory rather than a proinflammatory effect based on the current knowledge of TNFR1 signalling. What is the explanation for this apparent paradox?
The shedding hypothesis
Initial studies of a family with the C52F TNFR1 mutation demonstrated impaired activation-induced in-vitro shedding of TNFR1 (Citation1). Thus, TRAPS-associated mutations would lead to a defect in the ability of metalloproteases to cleave TNFR1 from the cell membrane with less potentially inhibitory soluble receptors available but still permitting stimulation of the cell through the normally functioning receptors (Citation1). The finding of low-soluble TNFR1 in the patients’ blood was consistent with this supposition. The hypothesis of impaired shedding indeed implicated the concept that cell membrane-bound mutant receptors would signal normally and that TNF signalling would be the central pathogenic inflammatory mechanism. Subsequently, shedding defects were demonstrated for a number of TNFR1 mutations but not for certain other disease-associated variants (Citation17,Citation43). In addition, the shedding defect seemed to be confined to certain cell types (Citation40). It gradually became evident that the hypothesis of defective shedding could not fully explain the inflammatory phenotype, and therefore alternative mechanisms have been sought by in-vitro analysis of mutant receptors. There are, however, rare patients with the TRAPS phenotype who have mutations near the cleavage site at the transmembrane region and whose leucocytes exhibit impaired receptor shedding (Citation44,Citation45). In these variants of TRAPS defective shedding may indeed be the central pathogenic mechanism.
The rationale behind the use of etanercept in TRAPS was to achieve compensation for the lack of soluble receptors capable of inhibiting circulating TNF-α. Although usually initially successful, the response to etanercept may wane with time, reinforcing the concept that other mechanisms than defective shedding of TNFR1 and TNF-α-induced inflammation would account for the inflammatory symptoms in TRAPS.
In-vitro stimulation tests with TNF-α
There have been several reports on decreased TNF-α-induced intracellular signalling in TRAPS patients. Siebert and co-workers (Citation41) used electrophoretic mobility shift assays to investigate the activation of NF-κB after TNF-α stimulation and demonstrated a decreased TNF-α-induced NF-κB activation in dermal fibroblasts with the C43S TNFR1 mutation. In a subsequent extended study, reduced NF-κB was a feature of all four TRAPS mutations investigated (C30R, C43S, T50M, and C52F). Reduced signalling correlated with reduced surface expression of receptors, as determined by flow cytometry and microscopy (Citation46).
In a study of ten Finnish patients with three different TNFR1 mutations (C73R, C88Y, F112I) and a fully developed TRAPS phenotype, the phosphorylation levels of NF-κB and p38 mitogen activated protein kinase (MAPK) were determined. Fresh peripheral blood leucocytes were stimulated with TNF-α in a whole-blood cytometry assay (Citation42). NF-κB and p38 phosphorylation levels of monocytes, lymphocytes, and neutrophils stimulated with TNF-α were significantly lower in the TRAPS patients than in healthy control subjects. That this defect was confined to TNF-α-mediated signalling was indicated by comparable levels of lipopolysaccharide (LPS)-and interferon-γ-induced phosphorylation levels in patient and control samples. In another study also involving patients with the C73R mutation, NF-κB activation in peripheral blood mononuclear cells was studied using electrophoretic mobility shift assay and ELISA of nuclear and cytoplasmic peripheral blood mononuclear cell extracts (Citation47). In this assay C73R cells showed a high concentration of functional TNFR1 on the plasma membrane, and both resting and TNF-α-stimulated C73R cells were more activated than cells without TNFR1 mutations and cells carrying the R92Q and P46L variants. Although the reasons for the discrepancy between the results concerning the C73R mutation in the two different studies are not known, they most probably involve differences between sample handling and methodology.
Defective apoptosis
An additional mechanism which could contribute to the hyperinflammatory phenotype is the decrease in TNF-α-induced apoptosis detected in neutrophils, monocytes, and fibroblasts from TRAPS patients (Citation46,Citation48) that could indicate overt NF-κB activation in these cells known to inhibit apoptosis, as suggested by our current knowledge of TNFR1 signalling (). This might result in prolonged survival of activated inflammatory cells, leading to an inappropriately prolonged inflammatory response.
Misfolding and trapping of mutant receptor
Normally TNFR1 after transportation from the endoplasmic reticulum is pooled in the Golgi apparatus before continuing to the cell surface. That TRAPS-related TNFR1 mutants behave abnormally was first demonstrated by Todd et al. (Citation39), who reported reduced translocation to the cell surface and retention within the cytoplasm of mutant receptors. Yousaf et al. (Citation49) found constitutive, TNF-α-independent, activation of NF-κB in mutant TNFR1 transfectants and proposed that such activation could derive from misfolding and retention of TNFR1 in the endoplasmic reticulum with subsequent activation of intracellular signalling. Indeed studies in different laboratories have shown that receptor misfolding and mislocalization is a universal feature of TNFR1 mutations in TRAPS (Citation39,Citation50,Citation51). Mutant TNFR1 cannot associate with the wild-type version but can form aggregates by self-interaction (Citation39,Citation50). Aggregation of mutant TNFR1 may cause ligand-independent TNFR1 signalling through formation of intracellular signalling complexes, the endoplasmic reticulum stress response, or an exaggerated unfolded protein response (UPR) (Citation52). This leads to induction of proinflammatory cytokines, generation of reactive oxygen species, and activation of other inflammatory pathways.
In a recent paper Simon et al. (Citation53) elaborates the findings of mutant receptor misfolding and intracellular trapping. They studied blood mononuclear cells from TRAPS patients and multiple cell types from two independent lines of knock-in mice harbouring TRAPS-associated TNFR1 mutations. Mutant TNFR1 did not function as a receptor for TNF-α, but it still induced inflammation by accumulating intracellularly and activating c-Jun N-terminal kinase (JNK) and p38 signalling, possibly by inducing reactive oxygen species. This activation sensitized cells to the effects of other innate stimuli such as LPS, resulting in enhanced production of inflammatory cytokines and chemokines at low concentrations of such stimuli. In cells that produce TNF-α, full-blown inflammation was seen only in heterozygous mutant cells that still expressed the normal TNFRSF1A allele. It thus seems that the inflammatory phenotype of TRAPS depends on the ability of intracellularly retained receptors to signal in a ligand-independent fashion and the functional co-operation between a disease-causing mutant receptor and its wild-type counterpart.
Interleukin-1 and TRAPS
Anakinra, a recombinant IL-1 receptor antagonist (IL-Ra), has been shown to be beneficial in TRAPS patients, including those resistant to treatment with anti-TNF agents (Citation22–24), which indicates that IL-1 signalling has a role in the pathogenesis of the disease. Indeed, in some patients the response to anakinra seems to be similar or only slightly inferior to that of patients with cryopyrin-associated periodic syndromes (CAPS), who generally respond swiftly and excellently to IL-1 blockade. The IL-1β secretion in CAPS and other diseases is dependent on the activation of inflammasomes (Citation32); however, there are no published studies linking inflammasome activation to TRAPS. Anakinra also blocks IL-1 receptor activation by IL-1α, which is secreted in an inflammasome-independent manner, representing another possible line for further studies. Reactive oxygen species (ROS) have been linked to TNFR1 signalling and inflammasome activation in recent studies (Citation31,Citation54), and endoplasmic reticulum (ER) stress has been linked to elevated ROS production (Citation55,Citation56), providing a possible mechanism for the induction of the inflammatory response by TNFRSF1A gene mutations. In contrast to the implications of clinical studies, a study of global gene expression in an endothelial cell line SK HEP-1 transfected with mutant TNFR1 (Citation57), genes encoding IL-1 family proteins or other members of the IL-1 pathway were not prominently featured in the measured expression profiles. The observed up-regulation of granulocyte macrophage colony-stimulating factor (GM-CSF) in this study, among other factors, could recruit monocytes to the site of inflammation that signal through the IL-1 pathway, explaining the efficacy of anakinra and raising the possibility of a multicellular inflammatory system involved in the pathogenesis of TRAPS. Indeed, monocytic fasciitis has been observed in a patient with TRAPS (Citation58). Though promising, the results obtained with IL-1 antagonists in TRAPS are, to date, limited to very few cases and should undergo further evaluation in larger cohorts of patients. On the whole, the role of IL-1α and IL-1β in the pathogenesis of TRAPS and, consequently, the role of IL-1 inhibition in the treatment of the disease clearly is worthy of more attention.
Interleukin-6 and TRAPS
IL-6 has been shown to be elevated in patients with TRAPS and implicated in the mouse model of TRAPS (Citation53,Citation59). IL-6 could represent the major pyrogen in TRAPS and readily explains many of the clinical and laboratory features of TRAPS. IL-6 production has been shown to be induced by, among others, IL-1α and IL-1β, which further underlines the role of IL-1 in TRAPS. Given the elevated IL-6 concentrations measured in TRAPS patients, an interesting alternative therapeutic approach could be IL-6 inhibition with tocilizumab (Citation53).
Concluding remarks
There is an emerging consensus among researchers that the hyperinflammation caused by mutant TNFR1 is mainly independent of its TNF-α-signalling function (Citation60,Citation61), as evidenced by the literature reviewed in this article. In summary, TRAPS can be seen as a disease clinically manifesting as a hyperinflammatory state, with a heterozygous loss-of-function mutation in TNFR1 resulting in reduced TNFR1 signalling due to paucity of functioning receptors on the cell surface, and an inflammatory response to internal cues, apparently exacerbated by the existence of a functioning TNFR1, depicted in . Whether the internal cues are related to death domain-dependent signalling of the mutant receptors, or relate to a death domain-independent mechanism by accumulation of mutant receptors in the ER leading to ER stress, or involve both, remains to be established. These intracellular events are likely to lead to a constitutive activation of proinflammatory pathways (Citation57), possibly resulting in production of ROS, secretion of IL-1α or IL-1β, and TNF-α itself. This could result in a positive feedback loop by the different cytokines and possibly between multiple cell types. The suggested mechanism would help to explain why subtle triggers may cause an exaggerated inflammatory reaction, and also represents a possible mechanism to account for the prolonged flares seen in TRAPS. A study using magnetic resonance imaging (MRI) suggests an acute-on-chronic nature of disease flares (Citation62), which could also point to a waxing and waning of local inflammation due to unknown stressors, capable of inducing occasional systemic inflammation during flares. Differences in signalling balance in cells from different TRAPS patients could also account for the differences in therapeutic response observed between patients (Citation24,Citation59).
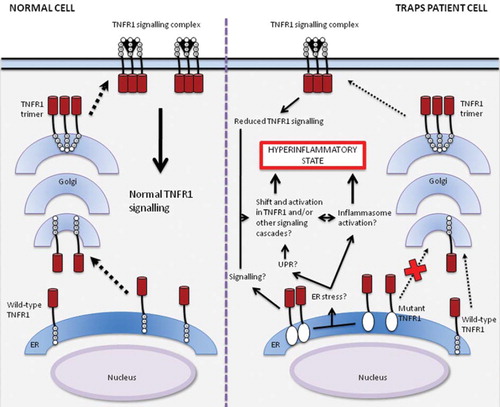
Figure 2. Model of signalling and trafficking pathways for wild-type and mutant TNFR1 in normal cells and cells from TRAPS patients. Wild-type receptors traffic from the endoplasmic reticulum (ER) through Golgi to the cell surface, allowing normal TNF-α-induced signalling. In cells from patients with TRAPS, mutant TNFR1s are retained in the ER (Citation39,Citation50,Citation51), where they may activate signalling pathways on their own or by inducing ER stress, leading to an unfolded protein response (UPR) (Citation52,Citation55,Citation56) or perhaps inflammasome activation (Citation23–25,Citation32), resulting in an inflammatory response. Trafficking and signalling of wild-type TNFR1s remains functional in cells of TRAPS patients, albeit reduced when compared to normal cells (Citation41,Citation42,Citation46). These mechanisms together could lead to a shift in balance of signalling and a self-perpetuating activation of TNFR1 or other signalling cascades by a positive feedback loop, producing the hyperinflammatory phenotype characteristic of TRAPS.
This figure is partly based on the article by Kimberley et al. (Citation60).
Drawing future directions of research, we acknowledge the fact that mutation-specific research material has been scarce, complicating comparison between inflammation induced by the different pathologic mutations. Also, the identity of the main cell type or types implicated in TRAPS pathogenesis remains elusive, although there is histopathological evidence of monocytic and lymphocytic inflammation in the skin and fascia (Citation58,Citation63). One answer to solve these issues could be the use of patient-specific induced pluripotent stem cells (iPSCs) for in-vitro studies of the different cell types expressing TNFR1, as demonstrated by the use of iPSCs in e.g. studying the LEOPARD syndrome (Citation64). Clinical studies with TRAPS patients, in the form of cytokine profiling during flares, MRI scanning, and possibly histological samples, could help to further delineate the main type and location of inflammation. The molecular cues needed to tip the balance into a hyperinflammatory state remain to be identified. Studies of therapeutic efficacy using anakinra, other IL-1 antagonists, or tocilizumab could also provide further insight into the pathogenesis of TRAPS.
It is certainly possible, as has been argued, that certain pathogenic mechanisms are more relevant to some mutations than to others, and that more than one mechanism may be operative for some, if not all mutations (Citation8). Further research is warranted, not only due to the devastating effects of inflammatory flares and amyloidosis on TRAPS patients, but also due to the implications of the proposed mechanisms on research of other diseases and the comprehension of signalling by TNFR1. If the mechanism by which mutant receptors induce inflammation can be proven to be caused by protein misfolding and endoplasmic reticulum stress due to protein aggregation, one can envisage a rather universal pathologic mechanism of inflammation that could explain a multitude of diseases of hitherto unclarified pathogenesis, including polygenic and multifactorial diseases. Indeed, endoplasmic reticulum stress has been linked to many diseases in the past years, including inflammatory diseases, cancer, and metabolic diseases such as obesity and type II diabetes (Citation65), making the research on TRAPS ever more compelling.
Declaration of interest: The scientific work partly underlying this review has been funded by the Finska Läkaresällskapet. The authors declare no conflicts of interest.
References
- McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, . Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44.
- Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–86.
- Hull KM, Shoham N, Chae JJ, Aksentijevich I, Kastner DL. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61–9.
- Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. 2005;17:586–99.
- Ryan JG, Goldbach-Mansky R. The spectrum of autoinflammatory diseases: recent bench to bedside observations. Curr Opin Rheumatol. 2008;20:66–75.
- Simon A, van der Meer JWM. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol. 2007; 292:R86–98.
- McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Medicine. 2006;8: 1242–8.
- Brydges S, Kastner DL. The systemic autoinflammatory diseases: Inborn errors of the innate immune system. Curr Top Microbiol Immunol. 2006;305:127–60.
- Glaser RI, Goldbach-Mansky R. The spectrum of monogenic autoinflammatory syndromes: understanding disease mechanisms and use of targeted therapies. Curr Allergy Asthma Rep. 2008;8:288–98.
- Bodar EJ, Drenth JPH, van der Meer JWM, Simon A. Dysregulation of innate immunity: hereditary periodic fever syndrome. Br J Haematol. 2009;144:279–302.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27: 621–68.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med. 1982;51:469–80.
- Hull KM, Drewe E, Aksentijevich I, Singh HK, Wong K, McDermott EM, . The TNF receptor-associated periodic syndrome (TRAPS). Emerging concepts of an autoinflammatory disorder. Medicine (Baltimore). 2002;81: 349–68.
- Dodé C, André M, Bienvenu T, Hausfater P, Pêcheux C, Bienvenu J, . The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2002;46:2181–8.
- Cantarini L, Lucherini OM, Cimaz R, Baldari CT, Bellisai F, Rossi Paccani S, . Idiopathic recurrent pericarditis refractory to colchicine treatment can reveal tumor necrosis factor receptor-associated periodic syndrome. Int J Immunopathol Pharmacol. 2009;22:1051–8.
- Cantarini L, Lucherini OM, Baldari CT, Laghi Pasini F, Galeazzi M. Familial clustering of recurrent pericarditis may disclose tumour necrosis factor receptor-associated periodic syndrome. Clin Exp Rheumatol. 2010;28:405–7.
- Aganna E, Hammond L, Hawkins PN, Aldea A, McKee SA, van Amstel HK, . Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003;48:2632–44.
- Cantarini L, Lucherini OM, Cimaz R, Rigante D, Baldari CT, Laghi Pasini F, . Typical and severe tumor necrosis factor receptor-associated periodic syndrome in the absence of mutations in the TNFRSF1A gene: a case series. Rheumatol Int. 2010 May 15 (Epub ahead of print).
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology. 2003;42:235–9.
- Cantarini L, Rigante D, Lucherini OM, Cimaz R, Laghi Pasini F, Baldari CT, . Role of etanercept in the treatment of tumor necrosis factor receptor-associated periodic syndrome: personal experience and review of the literature. Int J Immunopathol Pharmacol. 2010;23:701–7.
- Drewe E, Powell RJ. Novel treatments for tumor necrosis factor receptor-associated periodic syndrome (TRAPS): case history of experience with infliximab and sirolimus post etanercept. Clin Exp Rheumatol. 2002;4:S71.
- Jacobelli S, André M, Alexandra J-F, Dodé C, Papo T. Failure of anti-TNF therapy in TNF receptor 1-associated periodic syndrome (TRAPS). Rheumatology. 2007;46:1211–2.
- Simon A, Bodar EJ, van der Hilst JC, van der Meer JWM, Fiselier TJ, Cuppen MP, . Beneficial response to interleukin 1 receptor antagonist in TRAPS. Am J Med. 2004;117:208–10.
- Sacré K, Brihaye B, Lidove O, Papo T, Pocidalo M-A, Cuisset L, . Dramatic improvement following interleukin 1β blockade in tumour necrosis factor receptor-1-associated syndrome (TRAPS) resistant to anti-TNF-α therapy. J Rheumatol. 2008;35:357–8.
- Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, . Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–20.
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501.
- MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–92.
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–4.
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential complexes. Cell. 2003; 114:181–90.
- Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, . Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159-63.
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27: 229–65.
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
- Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–93.
- Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, . Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity. 2004; 21:415–28.
- Li L, Hailey DW, Soetandyo N, Li W, Lippincott-Schwartz J, Shu HB, . Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochim Biophys Acta. 2008;1783:1140–9.
- Müllberg J, Durie FH, Otten-Evans C, Alderson MR, Rose-John S, Cosman D, . A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J Immunol. 1995;155:5198–205.
- Engelmann H, Aderka D, Rubinstein M, Rotman D, Wallach D. A tumor necrosis factor-binding protein purified to homogeneity from human urine protects cells from tumor necrosis factor toxicity. J Biol Chem. 1989;264:11974–80.
- Todd I, Radford PM, Draper-Morgan K-A, McIntosh R, Bainbridge S, Dickinson P, . Mutant forms of tumour necrosis factor receptor I that occur in TNF-receptor-associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology. 2004;113:65–79.
- Huggins ML, Radford PM, McIntosh RS, Bainbridge SE, Dickinson P, Draper-Morgan K-A, . Shedding of mutant tumor necrosis factor receptor superfamily 1A associated with tumor necrosis factor-receptor associated periodic syndrome. Differences between cell types. Arthritis Rheum. 2004;50:2651–9.
- Siebert S, Amos N, Fielding CA, Wang EC, Aksentijevich I, Williams BD, . Reduced tumor necrosis factor signalling in primary human fibroblasts containing a tumor necrosis factor receptor superfamily 1A mutant. Arthritis Rheum. 2005;52:1287–92.
- Stjernberg-Salmela S, Ranki A, Karenko L, Siitonen S, Mustonen H, Puolakkainen P, . Low TNF-induced NF-κB and p38 phosphorylation levels in leucocytes in tumour necrosis factor receptor-associated periodic syndrome. Rheumatology. 2010;49:382–90.
- Aksentijevich I, Galon J, Soares N, Mansfield E, Hull K, Oh HH, . The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69:301–14.
- Kriegel MA, Hüffmeier U, Scherb E, Scheidig C, Geiler T, Kalden JR, . Tumor necrosis factor receptor-associated periodic syndrome characterized by a mutation affecting the cleavage site of the receptor implications for pathogenesis. Arthritis Rheum. 2003;48;2386–8.
- Stojanov S, Dejaco C, Lohse P, Huss K, Duftner C, Belohradsky BH, . Clinical and functional of a novel TNFRSF1A c.605T>A/V173D cleavage site mutation associated with tumor necrosis factor receptor/associated periodic fever syndrome (TRAPS), cardiovascular complications and excellent response to etanercept treatment. Ann Rheum Dis. 2008;67:1292–8.
- Siebert S, Fielding CA, Williams BD, Brennan P. Mutations of the extracellular domain of tumour necrosis factor receptor I causes reduced NF-κB activation due to decreased surface expression. FEBS Lett. 2005;579:5193–8.
- Nedjai B, Hitman GA, Yousaf N, Chernajovsky Y, Stjernberg-Salmela S, Pettersson T, . Abnormal tumor necrosis factor receptor I cell surface expression and NF-κB activation in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:273–83.
- D'Osualdo A, Ferlito F, Prigione I, Obici L, Meini A, Zulian F, . Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 2006;54:998–1008.
- Yousaf N, Gould DJ, Aganna E, Hammond L, Miriakian RM, Turner MD, . Tumor necrosis factor receptor I from patients with tumor necrosis factor receptor-associated periodic syndrome interacts with wild-type tumor necrosis factor receptor I and induces ligand-independent NF-κB activation. Arthritis Rheum. 2005;52:2906–16.
- Lobito AA, Kimberley FC, Muppidi JR, Komarow H, Jackson AJ, Hull KM, . Abnormal disulfide-linked oligomerization results in ER retention and altered signalling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood. 2006;108:1320–7.
- Rebelo SL, Bainbridge SE, Amel-Kashipaz MR, Radford PM, Powell RJ, Todd I, . Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–87.
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115: 2656–64.
- Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, . Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A. 2010;107:9801–6.
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40.
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–93.
- Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–27.
- Rebelo SL, Amel-Kashipaz MR, Radford PM, Bainbridge SE, Fiets R, Fang J, . Novel markers of inflammation identified in tumor necrosis factor receptor-associated periodic syndrome (TRAPS by transcriptomic analysis of effects of TRAPS-associated tumor necrosis factor receptor type I mutations in an endothelial cell line. Arthritis Rheum. 2009;60:8–11.
- Hull KM, Wong K, Wood GM, Chu WS, Kastner DL. Monocytic fasciitis: a newly recognized clinical feature of tumor necrosis factor receptor dysfunction. Arthritis Rheum. 2002;46:2189–94.
- Nowlan ML, Drewe E, Bulsara H, Esposito N, Robins RA, Tighe PJ, . Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology. 2006;45:31–7.
- Kimberley FC, Lobito AA, Siegel RM, Screaton GR. Falling into TRAPS—receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res Ther. 2007;9:217.
- Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: A clinical perspective. Cell. 2010;140:784–9.
- Quillinan N, Mohammad A, Mannion G, O'Keeffe D, Bergin D, Coughlan R, . Imaging evidence for persistent subclinical fasciitis and arthritis in tumour necrosis factor receptor-associated periodic syndrome (TRAPS) between febrile attacks. Ann Rheum Dis. 2010;69:1408–9.
- Toro JR, Aksentijevich I, Hull K, Dean J, Kastner DL. Tumor necrosis factor receptor-associated periodic syndrome: a novel syndrome with cutaneous manifestations. Arch Dermatol. 2000;136:1487–94.
- Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, . Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–12.
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17.