Abstract
In the post-reperfusion era, molecular and genetic mechanisms of cardioprotection and regeneration represent new therapeutic challenges to limit infarct size and minimize post-ischemic remodeling after acute myocardial infarction (AMI). Activation of cell survival mechanisms can be promoted by the administration of external drugs, stimulation of internal mechanisms, and genetic manipulation to delete or replace pathological genes or enhance gene expression. Among internal cardiovascular regulatory mechanisms, thyroid hormones (THs) may play a fundamental role. TH has a critical role in cardiovascular development and homeostasis in both physiological and pathological conditions. In experimental AMI, TH has been shown to affect cardiac contractility, left ventricular (LV) function, and remodeling. Several experimental studies have clearly shown that THs participate in the regulation of molecular mechanisms of angiogenesis, cardioprotection, cardiac metabolism, and ultimately myocyte regeneration, changes that can reverse left ventricular remodeling by favorably improving myocyte shape and geometry of LV cavity, thus improving systolic and diastolic performance. This review is focused on the role of thyroid on AMI evolution and on the potential novel option of thyroid-related treatment of AMI.
| Abbreviations | ||
| AKT | = | protein kinase B |
| AMI | = | acute myocardial infarction |
| CDP | = | Coronary Drug Project |
| D (1, 2, 3) | = | iodothyronine deiodinase |
| DITPA | = | 3,5-diiodothyropropionic acid |
| HSP | = | heat shock protein |
| LV | = | left ventricle |
| MAPK | = | mitogen-activated protein kinases |
| MHC | = | myosin heavy chain |
| mTOR | = | mammalian target of rapamycin |
| mtTFA | = | mitochondrial transcription factor |
| PGC | = | PPAR-gamma co-activator |
| PKC | = | protein kinase C |
| PLB | = | phospholamban |
| SERCA | = | sarcoplasmic reticulum calcium-activated ATPase |
| (r)T3 | = | triiodothyronine (r = reverse) |
| T4 | = | thyroxine (D- dextro; L- levo) |
| TH | = | thyroid hormone |
| THiRST | = | Thyroid Hormone Replacement Therapy in ST Elevation Myocardial Infarction |
| TR | = | thyroid receptor |
| TRH | = | thyrotropin-releasing hormone |
| TSH | = | thyroid-stimulating hormone |
Key messages
In the post-reperfusion era, molecular and genetic mechanisms of cardioprotection and regeneration represent new therapeutic challenges to limit infarct size and thus minimize post-ischemic remodeling after acute myocardial infarction.
Thyroid hormones (THs) participate in the regulation of molecular mechanisms of angiogenesis, cardioprotection, cardiac metabolism, and ultimately myocyte regeneration, changes that can improve left ventricular remodeling.
Treatment of cardiac patients with THs is largely discouraged. The origin of this belief is founded on weak or questionable scientific information. However, convincing experimental evidence may open the way to large-scale, controlled studies with synthetic TH use in patients with acute myocardial infarction.
Introduction
The term acute coronary syndrome is defined as any form of symptoms compatible with acute myocardial ischemia. Acute coronary syndrome covers all clinical conditions ranging from unstable angina to non-ST-segment elevation myocardial infarction to ST-segment elevation myocardial infarction. Each year in the United States, approximately 1.36 million hospitalizations are required for acute coronary syndrome, of which 0.81 million are for acute myocardial infarction (AMI) (Citation1). Myocardial infarction and its related short-term and long-term complications represent a major public health problem. While the pathophysiology of AMI has been sufficiently characterized, in particular during the last three decades, treatment goals and concomitant approaches have been markedly changing. This review is focused on the role of thyroid on AMI evolution and on the potential novel option of thyroid-related treatment of AMI. We will discuss in particular the pathophysiological basis of thyroid hormone (TH)-induced cardiac effects in the setting of AMI, as reported in experimental studies, and the potential therapeutic implications.
Acute myocardial infarction: from symptoms to molecular-based therapy and cell regeneration
When coronary care units were initially introduced in clinical practice, treatment was finalized to resolve the two main clinical consequences of AMI: life-threatening cardiac arrhythmias and acute cardiac insufficiency/cardiogenic shock. Later, with the development of coronary reperfusion, the goal of AMI treatment was to improve outcome for the leading cause of AMI, coronary occlusion due to atherosclerotic plaque rupture. The efficacy of reperfusion therapy was progressively enhanced with improved methods of intervention. These improvements were initially represented by intravenous thrombolytic agents and then by primary percutaneous transluminal coronary angioplasty and implantation of coronary stents. The current state-of-the art treatment of AMI is primary coronary angioplasty, defined as angioplasty and/or stenting without prior or concomitant fibrinolytic therapy (Citation2). The clinical impact of this therapeutic evolution was extraordinary when considering the drastic fall in in-hospital mortality. Before the development of coronary care units, in-hospital mortality was higher than 30% and is currently lower than 6% (Citation3). The direct effect of restoring myocardial perfusion is to limit infarct expansion by salvaging myocardium, to favor the contractile recovery of asynergic segments, and to improve cardiac performance, with each expected to improve long-term prognosis (Citation4,Citation5).
Both limitation of infarct size extent and recovery of dysfunctional contractile segments are key elements for preventing post-ischemic left ventricular (LV) dilatation and failure, a process defined as post-ischemic LV remodeling, that, despite improvements in reperfusion therapy, leads to almost 1 million new patients per year at risk for developing heart failure (Citation6). Post-ischemic LV remodeling is the final result of molecular, subcellular, cellular, and interstitial processes which involve changes in cardiomyocytes, extracellular matrix, and vasculature (microcirculation), affecting the infarct region, AMI border, and remote regions (Citation7). This is a dynamic process, lasting several months from the AMI phase, causing thinning of the infarct area, infarct expansion at the site of the necrotic border zone, and hypertrophy and fibrosis of the remote zone, likely occurring as a direct response to increased wall stress (Citation8). Increased wall stress is also the likely driving force for maladaptive remodeling of myocyte shape (e.g. increased myocyte length/width ratio and, consequently, increased chamber radius/wall thickness) in the remote zone (Citation9). The net final result of this process is progressive LV dilatation, its functional impairment, and the progression to heart failure (Citation10–12).
The 2020 World Health Organization projections view the high incidence of post-ischemic heart failure as the most important cause of mortality and morbidity. Consequently, the therapeutic challenge is to promote activation of mechanisms promoting myocyte self-protection and survival against acute and chronic ischemic bouts. Such approaches will minimize irreversible ischemic damage and favor functional recovery of the ischemic damaged myocardium, which should lead to reduced maladaptive remodeling. Ischemic preconditioning conferred by transient and repetitive episodes of ischemia, ischemic post-conditioning occurring from transient interruptions of reperfusion, and myocardial stunning and hibernation (two pathophysiological manifestations of dysfunctional but viable myocardium) are programs of cell survival in response to acute and chronic bouts of ischemia (Citation13–15). Genomic and molecular mechanisms favoring myocyte survival programs are complex and multifactorial and involve different and frequently cross-talking pathways. Improved cell survival includes mechanisms of cytoprotection through stimulation of heat shock proteins, activation of protein kinase C (PKC) and protein kinase B (AKT) pathways, inhibition of apoptosis, mechanisms of cell growth and angiogenesis including increased expression of the vascular endothelial growth factors, and mechanisms of metabolic adaptation such as the stimulation of glucose metabolism (Citation15,Citation16). Activation of cell survival mechanisms can be promoted by the administration of external drugs, stimulation of internal mechanisms, and genetic manipulation to delete or replace pathological genes or enhance gene expression.
Among internal cardiovascular regulatory mechanisms, THs may play a fundamental role. A physiological thyroid pattern plays an important role in tissue differentiation during the transition from fetal to postnatal growth. During this period, THs induce growth and transcriptional programming, leading to the typical gene expression profile of the adult heart. THs are also involved in the process of cell regeneration and/or differentiation and participate in the maintenance of cardiovascular homeostasis through direct genomic and non-genomic actions and other indirect actions (Citation17–20). Biologically active TH triiodothyronine (T3) mostly derives from deiodination of the prohormone thyroxine (T4) in peripheral tissues. Beside a normal hypothalamus–pituitary–thyroid axis, normal TH transport, and normal TH metabolism, normal receptor-mediated signaling into tissues is required to maintain a euthyroid condition. When one or more of the aforementioned and interrelated key elements of thyroid function homeostasis is altered, a dynamic disruption of homeostasis occurs and leads to altered TH action.
Experimental studies have shown that altered TH metabolism leads to abnormalities in cardiac morphology, gene expression and histology, and diastolic and systolic function, which favors altered post-ischemic remodeling (Citation21,Citation22). All these abnormalities are reversible when cardiac TH response is normalized (Citation20,Citation23). In the experimental setting, AMI reduces the expression of TH gene responders including a reduction in α-myosin heavy chain (MHC) and sarcoplasmic reticulum calcium-activated ATPase (SERCA2), whereas β-MHC and phospholamban (PLB) mRNA increase. These changes are part of the fetal recapitulation process consisting of reversal of genotype/phenotype to a more fetal-like cardiac state that has been documented in heart failure. Several experimental studies have clearly shown that THs participate in the regulation of molecular mechanisms of angiogenesis, cardioprotection, cardiac metabolism, and ultimately myocyte regeneration (Citation24), changes that can reverse LV remodeling by favorably improving myocyte shape and geometry of the left ventricular cavity, thus improving systolic and diastolic performance (Citation25,Citation26).
Thyroid function after AMI in the experimental setting
Thyroid hormone has a critical role in cardiovascular development and homeostasis in both physiological and pathological conditions. In experimental AMI, TH has been shown to affect cardiac contractility, LV function, and remodeling.
Serum TH level changes following AMI
In general, severe diseases produce down-regulation of thyroid function, which is called ‘euthyroid sick syndrome’ or ‘low T3 syndrome’. Ojamaa et al. (Citation20) have demonstrated that low T3 syndrome also occurs in an animal model of AMI, where serum T3 levels fell within 1 week of AMI and remained >40% lower than controls 4 weeks after AMI, while serum T4 levels remained unchanged. Olivares et al. (Citation27) further reported that serum thyrotropin-stimulating hormone (TSH) levels were elevated and T3 levels reduced until 8 weeks post-AMI. Serum T4 levels were low until 12 weeks after AMI despite increased thyroid stimulation 1 week after AMI. Meanwhile, type 1 iodothyronine deiodinase (D1) activity was decreased in the pituitary, thyroid, liver, and kidney. Increased type 3 iodothyronine deiodinase (D3) activity was observed in the infarcted myocardium. Decreased D1 activity leads to reduced conversion of T4 to T3, while increased D3 activity would enhance inactivation of T4 and T3, resulting in decreased serum TH levels after AMI. Both serum T3 and T4 levels were normalized 34 weeks after AMI (Citation28). Interestingly, the post-AMI cardiac D3 increased activity seems to be linked to ventricular remodeling, as recently shown in an AMI mouse model in which pathological ventricular remodeling leads to high and stable induction of D3 activity in cardiomyocytes and a local hypothyroid condition with reduced left ventricular tissue T3 concentrations (Citation29).
Cardiac thyroid receptor changes after AMI
The effects of TH can be mediated by genomic and non-genomic pathways. The genomic pathway starts when T3 enters the nucleus and binds to specific nuclear receptors (TRs). The T3–receptor complex then binds to thyroid hormone-response elements (TRE) in the promoter regions of specific target genes and modifies their expression (Citation30). There are several isoforms of TRs: TRα1 and TRβ1 (both bind T3) and TRα2 (which does not bind T3 but is able to bind to Thyroid Hormone Responsive Elements (TRE) and may exert a dominant negative effect on gene expression). The heart is a predominantly TRα1 organ, although TRβ1 is also expressed at a lower level. Pantos et al. (Citation31) reported that 8 weeks after AMI, (TRα1 and TRβ1) expression levels were decreased in the myocardium, as well as the expression levels of sarcoplasmic reticulum calcium-activated ATPase SERCA and Na+/H+exchanger, with increased expression of protein kinase C epsilon (PKCε) and tolerance of the post-infarcted heart to ischemia-reperfusion injury. In another study, these authors found that TRα1 and TRβ1 expressions remained unchanged at 2 weeks after AMI, while a 2-fold increase in TRα1 expression and a 2.2-fold decrease in TRβ1 expression were observed after 13 weeks in the non-infarcted area (Citation32). Actually, reduced expression of TRβ1 receptors is a consistent finding in the course of post-ischemic remodeling, while TRα1 expression is variable depending on the time-course of myocardial remodeling development. Accordingly, recent results show that TRα1 has a distinct pattern of expression in relation to different stages of disease progression, being up-regulated in rat hearts with compensated chronic post-AMI heart failure and markedly decreased in decompensated post-AMI heart failure (Citation28).
TH and LV function in AMI models
In AMI, 3-day treatment of T3 initiated immediately after AMI has been shown to increase+dp/dt (Citation33). Long-term (13 weeks) TH treatment initiated 24 hours after AMI increased LV ejection fraction (EF), LV-developed pressure,+dp/dt, and −dp/dt (Citation34). When treatment was initiated 3 weeks after AMI, Gay et al. found that thyroxine (T4; 1.5 μg/100 g body weight) increased+dp/dt and decreased LV end-diastolic pressure (Citation35,Citation36). Pantos et al. (Citation37) showed that a higher dose of TH starting 13 weeks after AMI and continued for 4 weeks significantly improved LV ejection fraction, LV-developed pressure, +dp/dt, and −dp/dt. Although the TH doses used for these studies were relatively high, Henderson et al. (Citation38) found that physiological replacement of T3 1 week after AMI for 8 weeks significantly improved LV +dp/dt and tended to improve diastolic function. In a diabetic AMI model, beneficial effects of TH were also observed by Kalofoutis et al. (Citation39), where TH administration for 2 weeks resulted in increased LV ejection fraction, normalized wall stress, and increased systolic radial strain in all myocardial segments after acute AMI.
Anti-apoptotic and mitochondrial effects of TH
Preservation of cell survival pathways resulting in inhibition of myocyte apoptosis is one of the major therapeutic objectives after AMI. We have previously shown that T3 can activate AKT signaling and protect cultured neonatal rat cardiomyocytes against serum starvation-induced apoptosis (Citation40). We also found that T3 treatment for 3 days after acute MI reduced myocyte apoptosis in the border area, possibly via AKT signaling (Citation33). The anti-apoptotic effect of T3 was also reported by Pantos et al. in an ischemia/reperfusion model with decreased p38 mitogen-activated protein kinase (MAPK) activation (Citation41). Over the past decade, convincing evidence has shown that the nuclear transcription factors mt-TFA and HIF1-α have a critical role in the maintenance of cell survival during myocardial ischemia (Citation42) and their cardiac over-expression attenuates LV remodeling and preserves cardiac performance after myocardial infarction (Citation37,Citation43). Early and sustained L-T3 supplementation enhances the expression of mt-TFA and HIF1-α in the peri-infarcted myocardium compared to control animals. Moreover, the mitochondrial respiratory defects are significantly attenuated in the border zone of L-T3-treated hearts in the presence of preserved expression of nuclear transcriptor factor PPAR-gamma co-activator (PGC)-1α, which is a powerful promoter of mitochondrial biogenesis (Citation44). L-T3 supplementation after AMI may even have a critical role in modulating the mitochondrial permeability transition opening, which is the primary cause of necrotic cell death in ischemic myocardium (Citation45).
TH and LV remodeling and myocyte gene expression changes
LV remodeling after MI is characterized by LV geometric, structural, and functional changes. At 2 weeks after MI, Pantos et al. (Citation23) showed that there was a decrease in LV function, LV posterior wall thickness/LV internal dimension ratio, a decreased expression of α-myosin heavy chain (α-MHC) and an increased expression of β-myosin heavy chain (β-MHC), with no change in the expression of SERCA and phospholamban (PLB) in the non-infarcted myocardium. TH treatment after MI resulted in normalization of LV posterior wall thickness/LV internal dimension ratio with increased expression of α-MHC and decreased expression of β-MHC and a 2-fold increase in the ratio of SERCA/PLB. Preliminary data from our lab indicate that post-MI LV hypertrophy in rats induced by TH can increase myocyte cross-sectional area without further myocyte lengthening (Citation46). Similarly, Ojamaa et al. (Citation20) reported that the expression levels of several T3-responsive genes were altered in the hypertrophied LV after MI, including significant decreases in α-MHC, SERCA2, and Kv1.5 (a K+ channel gene) mRNA, whereas β-MHC and PLB mRNA were significantly increased. Treatment of the MI rats with either replacement or elevated doses of T3 significantly increased LV ejection fraction. However, normalization of serum T3 did not restore expression of all T3-regulated genes, indicating altered T3 responsiveness in the post-infarcted myocardium. Although β-MHC and Kv1.5 mRNA content was returned to control levels, α-MHC and SERCA were unresponsive to T3 at replacement doses. Only at higher doses of T3 was α-MHC mRNA returned to control values.
Long-term (13 weeks) TH treatment resulted in ellipsoidal reshaping of the LV chamber with a shift from β-MHC to α-MHC, as well as increased ratio of SERCA/PLB to normal values and increased heat shock protein (HSP) 70 myocardial content (Citation34). In addition, late treatment with TH also provided LV remodeling benefits after MI. Pantos et al. (Citation37) reported that 4 weeks of treatment with a higher dose of TH starting at 13 weeks after MI significantly improved cardiac geometry, leading to a less spherical shape, with increased α-MHC, and decreased β-MHC expression. A physiological dose of T3 therapy starting 1 week after MI and continuing for 8 weeks also significantly increased α- and reduced β-MHC expression (Citation38).
TH and angiogenesis
TH-induced cardiac hypertrophy has been shown to be accompanied by increased capillary density in both young and old rats, with capillary proliferation actually preceding the development of cardiac hypertrophy (Citation47–49). In a porcine model, Briesch et al. found that both capillary and arteriolar beds experienced growth paralleled to myocyte growth during TH-induced cardiac hypertrophy (Citation50). The pro-angiogenic effect of TH can be mediated by both non-genomic and genomic pathways. TH binds to a cell surface receptor for TH, the integrin αVβ3, via activation of MAPK signaling to promote the release of fibroblast growth factor, leading to new vessel formation (Citation51,Citation52). TRβ in coronary endothelial cell is associated with angiogenesis during pathological cardiac hypertrophy (Citation43).
TH and LV function in ischemia/reperfusion models
Pre-ischemic treatment with T3 for 10 minutes has been shown to improve post-ischemic LV functional recovery with no changes in oxygen consumption and coronary flow (Citation53). This was associated with enhanced β-adrenergic responses in the stunned myocytes (Citation54). A number of studies have shown that T3 administration upon reperfusion can facilitate recovery of stunned myocardium and LV function (Citation41,Citation55–62). These beneficial effects might be due to improvement in local and global contractile function, in ventriculoarterial coupling, and in energy efficiency (Citation55,Citation56). T3 treatment after reperfusion also led to decreased coronary arterial resistance and increased coronary blood flow (Citation60). T3 has been shown to increase myocardial adenosine triphosphate levels and decrease lactate levels, indicating an improvement of mitochondrial function and increased aerobic metabolism (Citation63), and improved glucose oxidation (Citation62) without increasing myocardial oxygen consumption (Citation61,Citation62) and oxygen wasting (Citation60). Although no changes in post-ischemic high-energy phosphate concentrations were reported in T3 treatment of severe LV ischemic injury models (Citation57–59) and there was no change in myocardial infarct size (Citation61), T3 treatment improved overall survival in animals suffering from ischemia/reperfusion injury (Citation64).
Pantos et al. (Citation42,Citation65) showed that pre-ischemic, 14-day T4 administration increased HSP70 expression and decreased p38 MAPK activation in response to ischemia, resembling ischemic preconditioning. The same treatment also led to increased basal expression and phosphorylation of HSP27, and earlier and sustained redistribution of HSP27 from the S (Triton soluble, cytosol membrane) to P (Triton insoluble, cytoskeleton-nucleus) fraction (Citation66). Such changes might help to protect myocardium against ischemic insult, resulting in the improvement of post-ischemic functional recovery. Furthermore, the expression of parathyroid hormone-related peptide and the related type 1 receptor throughout the ischemia and reperfusion phase were enhanced with prior 2 weeks’ administration of T4, which can also be cardioprotective (Citation67).
While evidence for the cardioprotective effects of THs is robust (Citation18), contrary data are limited. Venditti et al. have shown that hyperthyroidism can sensitize the heart to reperfusion injury, resulting in low functional recovery (Citation68–70). Hyperthyroid hearts displayed an increase in mitochondrial permeability (Citation70) with mitochondrial dysfunction. The mitochondria showed a decrease in the rate of O2 consumption, anti-oxidant concentration and the capacity to remove H2O2, and an increase in the rate of H2O2 release and susceptibility to Ca2+-induced swelling. The increased oxidative stress by ischemia-reperfusion and T3 resulted in increased mitochondrial damage with increased hydroperoxide levels and protein-bound carbonyl (Citation68). Hyperthyroid hearts also showed an increase in both lipid peroxidation and susceptibility to oxidative stress, with over-production of nitric oxide (Citation69). Clearly, long-term hyperthyroidism is a different condition and results in cardiac physiological/histological changes that are not seen with therapeutic TH treatment (replacement dose or slightly higher doses) which are often reversible. Moreover, in Venditti's studies, the authors used an isolated rat heart model of ischemia reperfusion without maintaining constant flow and heart rate (Citation68). As a result, hyperthyroid hearts during reperfusion had significantly lower coronary flow and nearly double the heart rate of normal hearts. Despite these unfavorable conditions, hyperthyroid hearts had only a slightly lower recovery of function.
TH expression and AMI in humans: an old question without a good answer
As observed in animal studies with AMI, a more frequent change in TH metabolism in patients is a significant fall in circulating T3 levels and a corresponding increase in reverse T3 (rT3), the inactive T3 metabolite. These changes occur rapidly within 12 hours from the onset of symptoms, reaching the nadir by 72 hours. Greater T3 down-regulation has been observed in patients with LV dysfunction and intense pro-inflammatory and stress response, as documented by the correlation between interleukin 6 and T3 and rT3, and cortisol and T3 and rT3 (Citation71,Citation72). A negative correlation was also found between plasma free T3 concentrations and the extent of myocardial damage as assessed by asynergic area and wall motion score index in repeated measurements throughout a 14-day period after AMI (Citation73). Notably, most data on the TH time-course after AMI were collected prior to the introduction of reperfusion (Citation74) or no information on revascularization procedures were given in the remaining studies (Citation71). The occurrence of a low T3 syndrome does not represent a peculiar pattern of AMI because it has been documented in several additional cardiac and non-cardiac illnesses. Independently of type (i.e. acute or chronic), severity, and time-course of disease, a low T3 state has been generally interpreted as a merely adaptive mechanism finalized to reduce catabolic processes of illness and thus having beneficial effects through the reduction in metabolic demand (Citation75). This thesis has been rebutted in the last few years, at least in cardiac disease, by clinical evidence showing the prognostic negative impact of altered TH metabolism such as low T3 syndrome or pre-existing subclinical (mild) primary hypothyroidism being associated with a higher risk of short-term and long-term adverse outcomes, including mortality (Citation76–78). At present, there is no doubt that thyroid hypofunction is associated with an increased risk of cardiovascular disease. Importantly, a recent large-scale analysis based on individual data from 55,287 participants from 11 prospective cohorts showed that subclinical hypothyroidism is associated with an increased risk of coronary heart disease events and mortality, particularly in those with a TSH concentration of 10 mIU/L or greater (Citation79). The combination of increased arterial stiffness, impaired endothelial function, increased atherosclerosis, and altered coagulability together with a direct low action of TH at the cardiovascular level may have a pathogenetic role in the development of coronary artery disease in patients with subclinical hypothyroidism. Subclinical hypothyroidism, independently of other major factors, doubled the relative risk of AMI in females (Citation80). Moreover, levels of TSH in the upper part of the normal range were associated with multivessel disease (Citation81), and relatively low T3 levels were predictive of both single- and multivessel disease (Citation82), thus suggesting that even minimal alterations of TH state may contribute to coronary disease progression. Contextually, in 331 patients with AMI, TH abnormalities showed a strong prognostic power, with increased levels of rT3 resulting as an independent predictor of short-term and long-term mortality (Citation83). However, association does not imply cause/effect relationship, and only well designed large-scale interventional trials can clarify the true pathogenetic role of TH dysfunction in the progression of ischemic heart disease.
The dangerous “dogma” of thyroid drugs’adverse effect in cardiac patients
For a long time, treatment of cardiac patients with THs or TH analogs has been largely discouraged. The origin of this belief, however, is founded on weak or questionable information. For example, at the beginning of the twentieth century, surgical hypothyroidism was actually proposed as an extracardiac approach to treat ischemic heart disease, based on the observation that a patient with angina became asymptomatic after thyroidectomy (Citation84). This approach, mostly based on the concept of reducing myocardial demand after ischemic insult, has not continued over time due to inadequate evidence. This concept contrasted with the previous hypothesis of a causal relationship between hypothyroidism and atherosclerosis raised by Greenfield at the end of the nineteenth century (Citation85). At the same time, it is recognized that ischemic heart disease is a common complication of thyrotoxicosis in the presence of pre-existing coronary artery disease as a result of increased oxygen demand. Angina can also occur in the presence of normal coronary arteries. The exact mechanisms have not been well defined, although it has been postulated that coronary arteries may be sensitive to spasm in the presence of thyroid hormone excess (Citation86). Based on these apparently negative premises it is not surprising that the current, common belief of physicians is that exogenous TH therapy in cardiac patients represents in any case an injudicious and dangerous approach with concomitant risks of arrhythmia, myocardial ischemia/infarction, and worsening of congestive heart failure. Unfortunately, some additional support for this ‘dogma’ was based on a large-scale interventional trial named the Coronary Drug Project (CDP). This problematic study was done in the early 1970s (Citation87). The CDP demonstrated adverse outcomes, particularly with respect to the pro-arrhythmic effects of D-T4. D-T4 is the inactive form of thyroxine; L-T4 is the active form. First of all, the relevancy of this trial with regard to clinical outcomes from active L-T4 treatment is not clear since inactive D-T4 was the administered agent. The original assumption was that D-T4 was a safe analog of L-T4 that possessed lipid-lowering effects without adverse effects on cardiac function or metabolism. Patients were given 6 mg/day D-T4 which is equivalent to 225 μg of L-T4 and corresponds to more than double the endogenous production of T4 which is ∼90–100 μg/day (Citation88). It was later found that the D-T4 preparation (Choloxin; Flint Laboratories) used in the CDP was contaminated with a high level of active L-T4 (Citation89). Therefore, the cumulative dose of the D-T4 and L-T4 being administered was equivalent to several times the L-T4 dose that would be given to a patient to correct overt hypothyroidism. Notably, the potential toxic effect of D-T4 in patients with coronary artery disease was already reported before the start of the CDP (Citation90). For this reason, the CDP did not provide helpful information regarding the therapeutic use of TH supplementation in the treatment of cardiac patients.
Only convincing experimental evidence can open the way to large-scale, controlled studies with TH use in cardiac patients. On the other hand, all pilot interventional studies with the use of synthetic THs conducted in humans to this point were, paradoxically, done in cardiac disorders such as coronary bypass and chronic heart failure or cardiogenic shock, for which good animal models have not been clearly identified (Citation91–93). To our knowledge, no studies evaluating the safety and efficacy of TH supplementation started after AMI have been completed despite good evidence in favor of treatment in animals. In any case, and independently of TH type, timing, and dosage employed, the initial clinical experience of supplementation with synthetic TH has suggested its safety (Citation92) and provides some evidence for beneficial effects, mainly in patients undergoing cardiac surgery in adults and infants as well as in those with heart failure (Citation91,Citation92). In particular, an improvement in overall cardiac performance characterized by increased stroke volume was documented in T3-treated patients with chronic and clinically stable dilated cardiomyopathy and low T3 syndrome (Citation93).
In the perspective of treatment in patients with AMI, several questions need to be addressed to optimize treatment and avoid potential side-effects. Also, at variance with coronary artery bypass and heart failure (HF), additional potential targets of treatment of AMI with THs include positive effects on cardiac tissue remodeling by combined actions on cardiomyocyte interstitium and vasculature in addition to functional cardiac improvement. An additional fascinating new perspective is also the use of THs to facilitate differentiation toward an adult cardiac phenotype of embryonic stem cells via classical genomic pathways, thus enhancing maturation of electrophysiologic, as well as calcium homeostasis, properties of embryonic stem cell-derived cardiomyocytes (Citation24).
The understood goal of TH treatment after AMI should be to restore and maintain euthyroidism in patients with altered peripheral TH metabolism. Many issues, however, still remain largely undefined, in particular: 1) the type of hormone to be administered, i.e. T3, T4, and/or thyrotropin-releasing hormone (TRH); 2) the dosage to be administered and TH/TSH circulating levels to be maintained; and 3) timing of administration. Moreover, it should be taken into account that acute MI in humans may differ in several aspects from experimentally induced AMI in animals. Likewise there are differences between humans and animals in TH-induced gene regulation and acute actions of TH.
Regarding the type of TH to administer, the rationale for treating patients with T3 instead of T4 is based on the notion that the physiological pattern of peripheral conversion from T4 into T3 is impaired after AMI. The administration of T3 could then directly bypass this critical point through application of the biologically active TH. Due to impaired T4-to-T3 conversion from altered deiodinase activity, T4 treatment may lead to a delay in the TH response, with reduced production of active hormone. This delay in the initial phase of the disease could be critical. An alternative promising approach could be the concomitant infusion of both TRH and growth hormone (GH). Some pilot studies have shown that such a combination was able to sustain both hypothalamic–pituitary–thyroid feedback and stimulate D1 deiodinase and thus T3 peripheral production (Citation94). However, the short half-life, in particular of TRH, could make administration difficult.
The TH dosage is a key point considering that the therapeutic objective is to normalize overall TH metabolism and tissue response to TH. Regarding this point, evidence suggests that a hypothyroid tissue state can be present independently of the normal circulating levels of TH (Citation27,Citation28,Citation95). Therefore, the normalization of TH circulating levels does not automatically mean normalization of the tissue response to TH. However, in the absence of a good biomarker for a hypothyroid-like tissue state, a more logical initial approach is to normalize the circulating levels of conventionally used indices of thyroid function, i.e. T3, T4, and TSH. This approach is supported by the notion that almost all intracardiac T3 arises from circulating hormone and that serum TH levels from critically ill patients strictly correlates with intratissue TH levels, with D3 activity inversely correlated and D1 directly correlated with intratissue T3/rT3 ratio. So, it is likely that when low circulating T3 occurs, a vicious circle is activated with a low intratissue T3 favoring its progressive reduction (Citation96) ().
Figure 1. The vicious cycle induced by low T3 circulating levels leads to further reduction in T3 cardiac tissue availability as shown on the left side. The potential effects of systemic T3 replacement therapy on cardiac T3 metabolism and TR receptors in AMI are shown on the right.
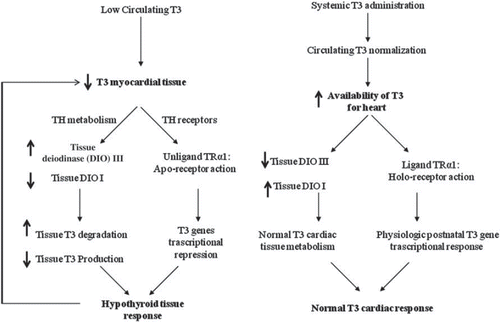
On the other hand, experimental data clearly documented that high doses of T3 were only initially associated with physiological myocardial hypertrophy and improved cardiac performance, but after 1 month of treatment a decline of cardiac performance was observed (Citation97). Indirect proof can also be derived from results of the CDP study and the more recent study using the TH analog 3,5-diiodothyropropionic acid (DITPA) in patients with HF, with both clearly indicating the negative impact in terms of prognosis and quality of life of induction of a thyrotoxic state (Citation98). Perhaps the best analogy for a hyperthyroid stimulus would be continuous exercise. While exercise and hyperthyroidism produce a physiologic type of stimulus with increased heart rate, cardiac output, and peripheral vasodilatation, non-stop exercise would clearly have adverse effects. Indeed, our group showed that over-exercising Spontaneously Hypertensive Heart Failure rats had accelerated cardiac pathology and worsening of heart function (Citation99).
When considering treatment of acute MI with THs, we should fairly consider possible pros and cons of THs treatment. Thyroid hormones are indeed a very powerful tool to affect not only heart function but also its susceptibility to atrial and ventricular arrhythmias by both genomic and non-genomic mechanisms (Citation100). This issue is particularly important due to the evidence that acute MI itself is known to be associated with an increased risk of malignant arrhythmias secondary to the development of a pro-arrhythmic substrate (Citation101). TH action may resemble the actions of catecholamines such as positive inotropic, lusitropic, chronotropic, and bathmotropic effects, while, on the other hand, a hypothyroid condition is associated with an increase in circulating catecholamines (Citation102). Thus, maintenance of thyroid homeostasis is likely the more important target in light of the potential pro-arrhythmic effects of any kind of dysthyroidism. In fact, while the pro-arrhythmic effects of hyperthyroidism are well known, hypothyroidism may favor cardiac arrhythmia, by determining QT prolongation and reduced activity of SERCA2. Moreover, it should be taken into account that acute MI in humans may differ in several aspects from experimentally induced AMI in animals. Likewise there are differences between humans and animals in TH-induced gene regulation and acute actions of THs, and co-expression of myosin heavy chain (MHC) isoform V1. Intracellular Ca2+ overload is the key event triggering ventricular fibrillation and preventing its reversal. Hearts with low levels of both SERCA2a and V1-MHC, as in the case of a hypothyroid state, can only defibrillate following administration of compounds that increase SERCA2a activity (Citation103).
The timing of starting TH treatment strictly depends on whether and when an altered TH pattern is adaptive or maladaptive. In fact, the drop in T3 may represent a genetically determined, beneficial adaptive response for a brief time that has been optimized through millennia by natural selection and evolution (Citation104). For instance, in the case of acute MI there is an open question whether the MI-related occurrence of low T3 syndrome may be beneficial by preserving energy to maintain its function as well as to be more electrically stable. On the other hand, if the initial insult or concurrent illness is not corrected, then ‘allostatic overload’ occurs, and pathological effects can be detected (Citation104). Therefore low T3 syndrome may be both physiological and beneficial early after the insult and pathological and maladaptive at a later time point. If this interpretative model is sound, better understanding of signals involved in the shift from an adaptive to a maladaptive response represents a key element in the evolution of the disease and in implementing a tailored TH therapy able to achieve the goal and to prevent detrimental consequences.
Taking into account all these premises, our group recently started a pilot clinical trial investigating the effects of T3 treatment in patients with AMI. The Thyroid Hormone Replacement therapy in ST elevation myocardial infarction (THiRST) trial is a phase II, randomized, double blind, placebo-controlled study. Patients admitted to the coronary care unit for chest pain and subsequently proven STEMI, undergoing standard medical treatment and myocardial revascularization of the culprit lesion, with border-line/reduced free T3 plasma levels (FT3 <2.2 pg/mL, or reduction of FT3 plasma levels more that 20% with respect to entry levels after a 72-hour period of hospitalization) are being randomly allocated with a 1:1 ratio to receive TH replacement therapy (maximum daily dosage 15 μg/m2 body surface) with Liotir (Liothyronine Sodium, oral T3 in drop form, IBSA, Institute Biochimicue SA, Lugano Swtzerland) or placebo. Primary end-points are: 1) evaluation of safety and feasibility of treatment with TH, after MI; and 2) the effects of therapy on post-ischemic remodeling and left ventricular function before starting therapy and at the end of the study. The treatment regimen utilized is able to restore early and to maintain over time a ‘physiological’ thyroid condition as documented by normal circulating values of both T4 and T3 and TSH.
Concluding remarks
In the post-reperfusion era, molecular and genetic mechanisms of cardioprotection and regeneration represent new therapeutic challenges to limit infarct size and thus minimize post-ischemic remodeling. THs may play an important homeostatic and regulatory role due to their different effects on these mechanisms and their homeostatic role on the cardiovascular system. The liaison between heart and thyroid is, however, an old and still unresolved question. The possibility of investigating this liaison from different perspectives, and not only from the angle of the functional role of TH on heart performance, is today possible by the development of research technologies and knowledge of the fine biomolecular and genetic mechanisms regulating the cardiovascular system. As wisely stated by Professor Attilio Maseri, ‘major breakthroughs in our knowledge often occur by looking at problems from different perspectives, which not infrequently reveals that some long-held and broadly accepted notions are not universally true, such that new, unexpected avenues for research, diagnosis, and treatment are suddenly opened’ (Citation105).
Acknowledgements
The THiRST Trial is partly funded by on-going European Community ITC-STREP FP7 PONTE Research Project (Project number 247945) ‘Efficient Patient Recruitment for Innovative Clinical Trials of Existing Drugs to Other Indications’.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Lloyed-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, . Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee.
- Van de Werf F, Bax J, Betriu A, Blomstrom-Lundqvist C, Crea F, Falk V, . ESC Committee for Practice Guidelines (CPG). Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation. Eur Heart J. 2008;29:2909–45.
- Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D Jr, O'Neill WW, . Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA. 2007;297:43–51.
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20.
- Araskiewicz A, Grajek S, Lesiak M, Prech M, Pyda M, Janus M, . Effect of impaired myocardial reperfusion on left remodelling in patients with anterior wall acute myocardial infarction treated with primary coronary intervention. Am J Cardiol. 2006;98:725–8.
- Krumholz HM, Anderson JL, Bachelder BL, Fesmire FM, Fihn SD, Foody JM, . ACC/AHA 2008 performance measures for adults with ST-elevation and non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (writing Committee to Develop Performance Measures for ST-elevation and Non ST-elevation Myocardial Infarction). J Am Coll Cardiol. 2008; 52:2046–99.
- Dixon JA, Spinale FG. Pathophysiology of myocardial injury and remodelling: implications for molecular imaging. J Nucl Med. 2010;51:102S–6S.
- Pfeffer MA. Left ventricular remodelling after acute myocardial infarction. Ann Rev Med. 1995;46:455–66.
- Gerdes AM, Kellerman SE, Moore JA, Muffly KE, Clark LC, Reaves PY, . Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation. 1992;86:426–30.
- Bolognese L, Neskovic AN, Parodi G, Cerisano G, Buonamici P, Santoro GM, . Left venticular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long term prognosis. Circulation. 2002; 106: 2351–7.
- St John Sutton M, Pfeffer MA, Moye L, Plappert T, Rouleau JL, Lamas G, . Cardiovascular death and left ventricular remodelling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 1997;96: 3294–9.
- Wong SP, French JK, Lydon AM, Manda SO, Gao W, Ashton NG, . Relation of left ventricular sphericity to 10-year survival after acute myocardial infarction. Am J Cardiol. 2004;94:1270–5.
- Depre C, Vatner SF. Cardioprotection in stunned and hibernating myocardium. Heart Fail Rev. 2007;12:307–17.
- Kloner RA, Rezkalla SH. Preconditioning, postconditioning and their application to clinical cardiology. Cardiovasc Res. 2006;70:297–307.
- Kitakaze M. How to mediate cardioprotection in ischemic hearts—accumulated evidence of basic research should translate to clinical medicine. Cardiovasc Drugs Ther. 2010; 24:217–23.
- Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, . Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–12.
- Mai W, Janier MF, Allioli N, Quignodon L, Chuzel T, Flamant F, . Thyroid hormone receptor α is a molecular switch of cardiac function between fetal and postnatal life. Proc Natl Acad Sci U S A. 2004;101:10332–7.
- Pantos C, Mourouzis I, Cokkinos DV. New insights into the role of thyroid hormone in cardiac remodeling: time to reconsider? Heart Fail Rev. 2011;16:79–96.
- Pantos C, Mourouzis I, Xinaris C, Papadopoulou-Daifoti Z, Cokkinos D. Thyroid hormone and ‘cardiac metamorphosis’: potential therapeutic implications. Pharmacol Ther. 2008; 118:277–94.
- Ojamaa K, Kenessey A, Shenoy R, Klein I. Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am J Physiol Endocrinol Metab. 2000;279:E1319−24.
- Chen YF, Redetzke RA, Said S, Beyer AJ, Gerdes AM. Changes in left ventricular function and remodeling after myocardial infarction in hypothyroid rats. Am J Physiol Heart Circ Physiol. 2010;298:H259–62.
- Tang YD, Kuzman JA, Said S, Anderson BE, Wang X, Gerdes AM. Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation. 2005;112:3122–30.
- Pantos C, Mourouzis I, Markakis K, Dimopoulos A, Xinaris C, Kokkinos AD, . Thyroid hormone attenuates cardiac remodeling and improves hemodynamics early after acute myocardial infarction in rats. Eur J Cardiothorac Surg. 2007;32:333–9.
- Lee YK, Ng KM, Chan YC, Lai WH, Au KW, Ho CY, . Triiodothyronine promotes cardiac differentiation and maturation of embryonic stem cells via the classical genomic pathway. Mol Endocrinol. 2010;24:1728–36.
- Campbell SE, Gerdes AM. Regional changes in myocyte size during the reversal of thyroid-induced cardiac hypertrophy. J Mol Cell Cardiol. 1988;20:379–87.
- Thomas TA, Kuzman JA, Anderson BE, Andersen SM, Schlenker EH, Holder MS, . Thyroid hormones induce unique and potentially beneficial changes in cardiac myocyte shape in hypertensive rats near heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2118–22.
- Olivares EL, Marassi MP, Fortunato RS, da Silva AC, Costa-e-Sousa RH, Ara jo IG, . Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology. 2007;148:4786–92.
- Pantos C, Mourouzis I, Galanopoulos G, Gavra M, Perimenis P, Spanou D, . Thyroid hormone receptor alpha1 downregulation in postischemic heart failure progression: the potential role of tissue hypothyroidism. Horm Metab Res. 2010;42:718–24.
- Pol CJ, Muller A, Zuidwijk MJ, van Deel ED, Kaptein E, Saba A, . Left-ventricular remodeling after myocardial infarction is associated with a cardiomyocyte-specific hypothyroid condition. Endocrinology. 2011;152:669–79.
- Brent GA. The molecular basis of thyroid hormone action. N Engl J Med. 1994;331:847–53.
- Pantos C, Mourouzis I, Saranteas T, Paizis I, Xinaris C, Malliopoulou V, . Thyroid hormone receptors alpha1 and beta1 are downregulated in the post-infarcted rat heart: consequences on the response to ischaemia-reperfusion. Basic Res Cardiol. 2005;100:422–32.
- Pantos C, Mourouzis I, Xinaris C, Kokkinos AD, Markakis K, Dimopoulos A, . Time-dependent changes in the expression of thyroid hormone receptor alpha 1 in the myocardium after acute myocardial infarction: possible implications in cardiac remodelling. Eur J Endocrinol. 2007;156:415–24.
- Chen YF, Kobayashi S, Chen J, Redetzke RA, Said S, Liang Q, . Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol. 2008;44: 180–7.
- Pantos C, Mourouzis I, Markakis K, Tsagoulis N, Panagiotou M, Cokkinos DV. Long-term thyroid hormone administration reshapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic Res Cardiol. 2008;103:308–18.
- Gay R, Gustafson TA, Goldman S, Morkin E. Effects of L-thyroxine in rats with chronic heart failure after myocardial infarction. Am J Physiol. 1987;253(2 Pt 2):H341–6.
- Gay RG, Graham S, Aguirre M, Goldman S, Morkin E. Effects of 10- to 12-day treatment with L-thyroxine in rats with myocardial infarction. Am J Physiol. 1988;255(4 Pt 2): H801–6.
- Pantos C, Mourouzis I, Tsagoulis N, Markakis K, Galanopoulos G, Roukounakis N, . Thyroid hormone at supra-physiological dose optimizes cardiac geometry and improves cardiac function in rats with old myocardial infarction. J Physiol Pharmacol. 2009;60:49–56.
- Henderson KK, Danzi S, Paul JT, Leya G, Klein I, Samarel AM. Physiological replacement of T3 improves left ventricular function in an animal model of myocardial infarction-induced congestive heart failure. Circ Heart Fail. 2009;2:243–52.
- Kalofoutis C, Mourouzis I, Galanopoulos G, Dimopoulos A, Perimenis P, Spanou D, . Thyroid hormone can favorably remodel the diabetic myocardium after acute myocardial infarction. Mol Cell Biochem. 2010;345:161–9.
- Kuzman JA, Gerdes AM, Kobayashi S, Liang Q. Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:841–4.
- Pantos C, Mourouzis I, Saranteas T, Clav G, Ligeret H, Noack-Fraissignes P, . Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol. 2009; 104:69–77.
- Eckle T, Köhler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008 118:166–75.
- Makino A, Suarez J, Wang H, Belke DD, Scott BT, Dillmann WH. Thyroid hormone receptor-beta is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinology. 2009;150:2008–15.
- Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC1alpha. Cardiovasc Res. 2008;79:208–17.
- Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial transition pore opening during myocardial reperfusion: a target for cardioprotection. Cardiovasc Res. 2004;61:372–85.
- Chen YF, Bathia S, Redetzke RA, Coburn T, Gerdes AM. Thyroid hormone improves left ventricular myocyte remodeling after myocardial infarction in rats. J Card Fail. 2009; 15:S73.
- Chilian WM, Wangler RD, Peters KG, Tomanek RJ, Marcus ML. Thyroxine-induced left ventricular hypertrophy in the rat. Anatomical and physiological evidence for angiogenesis. Circ Res. 1985;57:591–8.
- Tomanek RJ, Connell PM, Butters CA, Torry RJ. Compensated coronary microvascular growth in senescent rats with thyroxine-induced cardiac hypertrophy. Am J Physiol. 1995;268(1 Pt 2):H419–25.
- Tomanek RJ, Busch TL. Coordinated capillary and myocardial growth in response to thyroxine treatment. Anat Rec. 1998;251:44–9.
- Breisch EA, White FC, Hammond HK, Flynn S, Bloor CM. Myocardial characteristics of thyroxine stimulated hypertrophy. A structural and functional study. Basic Res Cardiol. 1989;84:345–58.
- Davis FB, Mousa SA, O'Connor L, Mohamed S, Lin HY, Cao HJ, . Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ Res. 2004;94:1500–6.
- Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, . Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146:2864–71.
- Kazmierczak P, Polak A, Mussur M. Influence of preischemic short-term triiodothyronine administration on hemodynamic function and metabolism of reperfused isolated rat heart. Med Sci Monit. 2004;10:BR381–7.
- Tse J, Gandhi A, Yan L, He YQ, Weiss HR. Effects of triiodothyronine pretreatment on beta-adrenergic responses in stunned cardiac myocytes. J Cardiothorac Vasc Anesth. 2003;17:486–90.
- Novitzky D, Matthews N, Shawley D, Cooper DK, Zuhdi N. Triiodothyronine in the recovery of stunned myocardium in dogs. Ann Thorac Surg. 1991;51:10–6; discussion 16–7.
- Yokoyama Y, Novitzky D, Deal MT, Snow TR. Facilitated recovery of cardiac performance by triiodothyronine following a transient ischemic insult. Cardiology. 1992;81:34–45.
- Holland FW 2nd, Brown PS Jr, Clark RE. Acute severe postischemic myocardial depression reversed by triiodothyronine. Ann Thorac Surg. 1992;54:301–5.
- Wechsler AS, Kadletz M, Ding M, Abd-Elfattah A, Dyke C. Effects of triiodothyronine on stunned myocardium. J Card Surg. 1993;8(2 Suppl):338–41.
- Dyke CM, Ding M, Abd-Elfattah AS, Loesser K, Dignan RJ, Wechsler AS, . Effects of triiodothyronine supplementation after myocardial ischemia. Ann Thorac Surg. 1993;56: 215–22.
- Klemperer JD, Zelano J, Helm RE, Berman K, Ojamaa K, Klein I, . Triiodothyronine improves left ventricular function without oxygen wasting effects after global hypothermic ischemia. J Thorac Cardiovasc Surg. 1995;109:457–65.
- Hsu RB, Huang TS, Chen YS, Chu SH. Effect of triiodothyronine administration in experimental myocardial injury. J Endocrinol Invest. 1995;18:702–9.
- Liu Q, Clanachan AS, Lopaschuk GD. Acute effects of triiodothyronine on glucose and fatty acid metabolism during reperfusion of ischemic rat hearts. Am J Physiol. 1998;275 (3 Pt 1):E392–9.
- Novitzky D, Human PA, Cooper DK. Effect of triiodothyronine (T3) on myocardial high energy phosphates and lactate after ischemia and cardiopulmonary bypass. An experimental study in baboons. J Thorac Cardiovasc Surg. 1988;96:600–7.
- Novitzky D, Human PA, Cooper DK. Inotropic effect of triiodothyronine following myocardial ischemia and cardiopulmonary bypass: an experimental study in pigs. Ann Thorac Surg. 1988;45:50–5.
- Pantos C, Malliopoulou V, Paizis I, Moraitis P, Mourouzis I, Tzeis S, . Thyroid hormone and cardioprotection: study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Mol Cell Biochem. 2003; 242:173–80.
- Pantos C, Malliopoulou V, Mourouzis I, Karamanoli E, Moraitis P, Tzeis S, . Thyroxine pretreatment increases basal myocardial heat-shock protein 27 expression and accelerates translocation and phosphorylation of this protein upon ischaemia. Eur J Pharmacol. 2003;478:53–60.
- Halapas A, Lembessis P, Mourouzis I, Pantos C, Cokkinos DV, Sourla A, . Experimental hyperthyroidism increases expression of parathyroid hormone-related peptide and type-1 parathyroid hormone receptor in rat ventricular myocardium of the Langendorff ischaemia-reperfusion model. Exp Physiol. 2008;93:237–46.
- Venditti P, Agnisola C, Di Meo S. Effect of ischemia-reperfusion on heart mitochondria from hyperthyroid rats. Cardiovasc Res. 2002;56:76–85.
- Masullo P, Venditti P, Agnisola C, Di Meo S. Role of nitric oxide in the reperfusion induced injury in hyperthyroid rat hearts. Free Radic Res. 2000;32:411–21.
- Pavon N, Aranda A, Garcia N, Hernandez-Esquivel L, Chavez E. In hyperthyroid rats octylguanidine protects the heart from reperfusion damage. Endocrine. 2009;35:158–65.
- Friberg L, Werner S, Eggertsen G, Ahnve S. Rapid down-regulation of thyroid hormones in acute myocardial infarction. Is it cardioprotective in patients with angina? Arch Int Med. 2002;162:1388–94.
- Kimur T, Kotajima N, Kanda T, Kuwabara A, Fukumura Y, Kobayashi I. Correlation of circulating interleukin-10 with thyroid hormone in acute myocardial infarction. Res Commun Mol Pharmacol. 2001;110:63–58.
- Ceremuzynski L, Czerwosz L, Chamiec T, Bartoszewicz Z, Herbaczynska-Cedro K. Low serum triiodotrhyronine in acute myocardial infarction indicates major heart injury. Kardiol Pol. 2004;60:468–80.
- Wiersinga WM, Lie KI, Touber JL. Thyroid hormones in acute myocardial infarction. Clin Endocrinol. 1981;14:367–74.
- De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999; 84:151–64.
- Iervasi G, Pingitore A, Landi P, Raciti M, Ripoli A, Scarlattini M, . Low-T3 syndrome: a strong prognostic predictor of death in patients with heart disease. Circulation. 2003;107:708–13.
- Iervasi G, Molinaro S, Landi P, Taddei MC, Galli E, Mariani F, . Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526–32.
- Pingitore A, Landi P, Taddei MC, Ripoli A, L'Abbate A, Iervasi G, . Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am J Med. 2005;118: 132–6.
- Rodondi N, den Elzen WP, Bauer DC, Cappola AR, Razvi S, Walsh JP, .; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010;304:1365–74.
- Hak AE, Pols HAP, Visser TJ, Drexhage HA, Hofman A, Witteman JCM. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: The Rotterdam Study. Ann Intern Med. 2000;132:70–8.
- Yun KH, Jeong MH, Lee EM, Lee J, Rhee SJ, Yoo NJ, . Relationship of thyroid stimulating hormone with coronary athrosclerosis in angina patients. Int J Cardiol. 2007;122:56–60.
- Coceani M, Iervasi G, Pingitore A, Carpeggiani C, L'Abbate A. Thyroid hormone and coronary artery disease: from clinical correlations to prognostic implications. Clin Cardiol. 2009;32:380–5.
- Friberg L, Drvota V, Bjelak AH, Eggertsen G, Ahnve S. Association between increased levels of reverse T3 and mortality after acute myocardial infarction. Am J Med. 2001; 111: 699–703.
- Kocher A. Ueber morbus Basedowi. Mitt Grenzgeb Med Chir. 1901;1:1–13.
- Greenfield WS. Autopsy findings in a 58 year old woman with myxoedema. Published as an appendix to Ord WM. Med Chir Trans. 1878;61:57.
- Somerville W, Levine SA. Angina pectoris and thyrotoxicosis. Br Heart J. 1950;12:245–57.
- The coronary drug project. Findings leading to further modifications of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA. 1972;220:996–1008.
- Pilo A, Iervasi G, Vitek F, Ferdeghini M, Cazzuola F, Bianchi R. Thyroidal and peripheral production of 3,5,3'-triiodothyronine in humans by multicompartmental analysis. Am J Physiol. 1990;258:E715–26.
- Young WF Jr, Gorman CA, Jiang NS, Machacek D, Hay ID. L-thyroxine contamination of pharmaceutical D-thyroxine: probable cause of therapeutic effect. Clin Pharmacol Ther. 1984;36:781–7.
- Jones RJ, Cohen L. Sodium dextro-thyrxine in coronary disease and hypercholesteremia. Circulation. 1961;24;164–70.
- Ranasinghe AM, Quinn DW, Pagano D, Edwards N, Faroqui M, Graham TR, . Glucose-insulin-potassium and tri-iodothyronine individually improve hemodynamic performance and are associated with reduced troponin I release after on-pump coronary artery bypass grafting. Circulation. 2006;114:I245–50.
- Portman MA, Slee A, Olson AK, Cohen G, Karl T, Tong E, . TRICC Investigators. Triiodothyronine Supplementation in Infants and Children Undergoing Cardiopulmonary Bypass (TRICC): a multicenter placebo-controlled randomized trial: age analysis. Circulation. 2010;122:S224–33.
- Pingitore A, Galli E, Barison A, Iervasi A, Scarlattini M, Nucci D, . Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized, placebo-controlled study. J Clin Endocrinol Metab. 2008;93:1351–8.
- Mebis L, van den Berghe G. The hypothalamus-pituitary-thyroid axis in critical illness. Neth J Med. 2009;67: 332–40.
- Liu Y, Redetzke RA, Said S, Pottala JV, de Escobar GM, Gerdes AM. Serum thyroid hormone levels may not accurately reflect thyroid tissue levels and cardiac function in mild hypothyroidism. Am J Physiol Heart Circ Physiol. 2008;294:H2137–43.
- Peeters RP, van der Geyten S, Wouters PJ, Darras VM, van Toor H, Kaptein E, . Tissue thyroid hormone levels in critical illness. J Clin Endocrinol Metab. 2005;90:6498–507.
- De K, Ghosh G, Datta M, Konar A, Bandyopadhyay J, Bandyopadhyay D, . Analysis of differentially expressed genes in hyperthyroid-induced hypertrophied heart by cDNA microarray. J Endocrinol. 2004;182:303–14.
- Goldman S, McCarren M, Morkin E, Ladenson PW, Edson R, Warren S, . DITPA (3,5-diiodothyropropionic acid), a thyroid hormone analog to treat heart failure: phase II trial veterans affairs cooperative study. Circulation. 2009;119:3093–100.
- Schultz RL, Swallow JC, Waters RP, Kuzman JA, Redetzke RA, Said S, . Effects of excessive long-term exercise on cardiac function and myocyte remodeling in hypertensive heart failure rats. Hypertension. 2007;50: 410–6.
- Tribulova N, Knezl V, Shainberg A, Seki S, Soukup T. Thyroid hormones and cardiac arrhythmias. Vascul Pharmacol. 2010;52:102–12.
- Betsuyaku T, Kanno S, Lerner DL, Schuessler RB, Saffitz JE, Yamada KA. Spontaneous and inducible ventricular arrhythmias after myocardial infarction in mice. Cardiovasc Pathol. 2004;13:156–64.
- Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and the heart. Circulation. 1993;87;1435–41.
- Manoach M, Tribulova N, Vogelezang D, Thomas S, Podzuweit T. Transient ventricular fibrillation and myosin heavy chain isoform profile. J Cell Mol Med. 2007;11:171–4.
- Cini G, Carpi A, Mechanick J, Cini L, Camici M, Galetta F, . Thyroid hormones and the cardiovascular system: Pathophysiology and interventions. Biomed Pharmacother. 2009;63:742–53.
- Maseri A. Forewords. Iervasi G, Pingitore A. Thyroid and heart failure: from pathophysiology to clinics. Milan and New York: Springer; 2009.