Abstract
Hemostasis is traditionally defined as a physiological response to blood vessel injury and bleeding, which entails a co-ordinated process involving the blood vessel, platelets, and blood clotting proteins (i.e. coagulation factors). Hemostasis can be divided into primary and secondary components. The former rapidly initiates after endothelial damage and is characterized by vascular contraction, platelet adhesion, and formation of a soft aggregate plug. The latter is initiated following the release of tissue factor and involves a complex sequence of events known as the blood coagulation cascade, encompassing serial steps where each coagulation factor activates another in a chain reaction that culminates in the conversion of fibrinogen to fibrin. Patients carrying abnormalities of the coagulation cascade (i.e. deficiencies of coagulation factors) have an increased bleeding tendency, where the clinical severity is mostly dependent upon the type and the plasma level of the factor affected. These disorders also impose a heavy medical and economic burden on individual patients and society in general. The aim of this article is to provide a general overview on the pathophysiology, clinics, diagnostics, and therapy of inherited disorders of coagulation factors.
Key words::
Key messages
Inherited bleeding disorders can be classified as acquired or inherited, and as affecting primary or secondary hemostasis.
Inherited disorders of the coagulation system typically comprehend von Willebrand disease, hemophilia A, hemophilia B, hemophilia C, and rare bleeding disorders.
Patients carrying abnormalities of the coagulation system (i.e. quantitative or qualitative abnormalities of coagulation factors) have an increased bleeding tendency, where the clinical severity is mostly dependent upon the type and the plasma level of the factor affected.
Introduction
Hemostasis is traditionally defined as a physiological response to blood vessel injury and bleeding, which involves the co-ordinated activity of vascular, platelet, and plasma factors. It is an accepted convention to divide this process into primary and secondary components. The former rapidly initiates after endothelial damage and is characterized by vascular contraction, platelet adhesion, and formation of a soft aggregate plug. The latter is initiated following the release of tissue factor (TF) and involves a complex sequence of events known as the blood coagulation cascade. This process encompasses serial steps where each coagulation factor (F) activates another in a chain reaction that culminates in the conversion of fibrinogen to fibrin. Primary hemostasis is short-lived, and the immediate post-injury vascular constriction abates quickly; when the blood-flow is allowed to increase, the soft plug might be rapidly sheared from the injured surface. Thus, the main goal of secondary hemostasis is to stabilize the soft plug and therefore facilitate the arrest of the hemorrhage.
Overview of blood coagulation
Although blood coagulation has been classically classified into two pathways, sometimes referred to as intrinsic and extrinsic, important biological advances over the past decades have helped to clarify the precise mechanisms involved in the initiation and propagation of the coagulation cascade, including the involvement of cellular elements.
The initial and essential event is the release of TF from the injured endothelium, which combines with FVII to form the TF–FVIIa (activated FVII) complex (). This complex rapidly catalyzes the activation of both FX and FIX. The former (activated) factor (FXa) combines with activated FV (FVa) to form a complex also known as ‘prothrombinase’, while the latter (FIXa) combines with activated FVIII (FVIIIa) to form a complex named ‘tenase’. Both complexes actively convert prothrombin (FII) into thrombin (FIIa), which in turn finally converts fibrinogen into fibrin.
Figure 1. Overview of the in-vivo blood coagulation cascade.
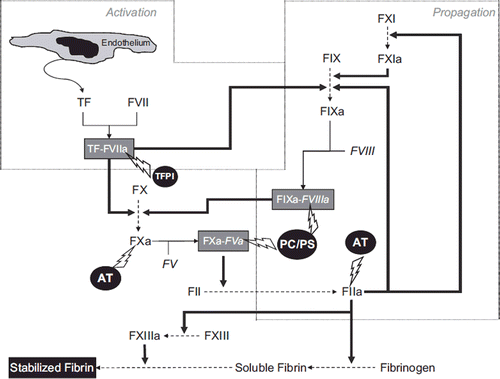
The amount of thrombin generated by this initial activation step is, however, ineffective (∼2% of the total amount required) to produce a sufficient amount of fibrin to stabilize the soft plug. The main role of the TF pathway is therefore to generate the so-called ‘thrombin burst’, a process by which thrombin initiates a feedback process by activating several components of the coagulation cascade, including FV and FVIII (which activates FXI, which in turn activates FIX). This process also facilitates the ‘propagation’ of the coagulation cascade.
FVII might also be activated by thrombin, FXIa, and FXa. Fibrin monomers are initially held together by non-covalent interactions, but the catalysis of covalent cross-links by FXIII definitively stabilizes the clot. According to this model, the older concept of the coagulation cascade encompassing two different (intrinsic/extrinsic) activation pathways is therefore no longer legitimate, and the role of FXII (i.e. the initiator of the ‘intrinsic pathway’) is virtually insignificant. A delicate balance exists between coagulation and fibrinolysis for determining the true stability of the fibrin clot. Importantly, thrombin is not only involved in generation of the clot but stabilizes it through activation of the thrombin activatable fibrinolysis inhibitor (TAFI), which exerts an essential function for protecting the fibrin clot against lysis (Citation1).
The entire complex mechanism of activation and propagation of blood coagulation is further regulated at several steps by different inhibitors, including antithrombin (AT), the co-ordinated action of protein C (PC) and protein S (PS), and the tissue factor pathway inhibitor (TFPI) (Citation2,Citation3).
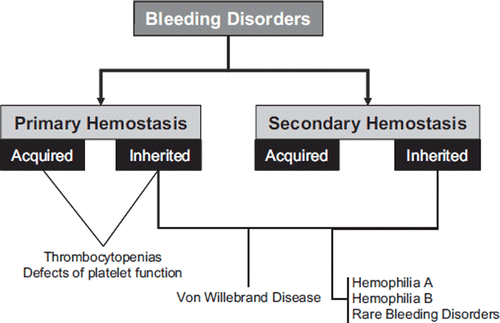
According to the World Federation of Hemophilia (WFH), the general term ‘bleeding disorder’ defines a broad range of medical problems that lead to poor blood clotting and thus continuous or uncontrollable bleeding (Citation4). Bleeding disorders can be classified as acquired or inherited, and as affecting primary or secondary hemostasis ().
Figure 2. Classification of bleeding disorders. Von Willebrand disease (VWD) is primarily a disorder of ‘primary hemostasis’ (affecting platelet adhesion and aggregation), but as von Willebrand factor (VWF) can also contribute to ‘secondary hemostasis’ by providing factor VIII to sites of injury, VWD can also express clinical symptoms associated with failure of the coagulation cascade.
The aim of this article is to provide a general overview on the pathophysiology, clinics, diagnostics, and therapy of inherited disorders of procoagulant factors (summarized in ). Although the plasma protein von Willebrand factor (VWF) is per se not involved in the coagulation pathway, one of its roles is to protect and stabilize FVIII and thus provide this procoagulant protein at sites of thrombus formation. Moreover, VWF deficiencies and defects give rise to clinical bleeding that may sometimes be difficult to differentiate from those arising from deficiencies in the clotting factors. Accordingly, this review will cover von Willebrand disease (VWD), hemophilia A, hemophilia B, hemophilia C, and rare bleeding disorders. Whilst the latter comprise a diverse group of relatively rare congenital bleeding disorders, education and information related to these have not been a priority of the medical community according to the WFH (Citation4), leading to their relatively poor diagnosis and management.
Table I. The coagulation factors.
Platelet disorders
Platelet disorders, which are an additional cause of symptomatic bleeding, will not be covered in this review. Nevertheless a brief overview is useful to facilitate their differential diagnosis from inherited disorders of secondary hemostasis. Platelet disorders define a primary hemostasis disturbance that can arise due to abnormalities of platelet number (i.e. thrombocytopenias) or function (i.e. disorders of platelet function), or both, and can be further classified as acquired or congenital (). These pathologies are mainly characterized by easy bruising, epitasis, menorrhagia, and excessive bleeding from trauma and dental or surgical interventions, whereas defects in secondary hemostasis, as previously noted, exhibit delayed deep bleeding (e.g. muscles and joints), and the characteristic physical examination finding is hemarthrosis, which is instead absent in primary hemostatic disorders. As regards laboratory testing, the hall-mark of platelet disorders is the normality of routine coagulation tests such as prothrombin time (PT) and activated partial thromboplastin time (APTT). Conversely, the platelet count is typically reduced in thrombocytopenias, while disorders of platelet function generate abnormal results of bleeding time and other primary hemostasis tests (e.g. platelet function testing by aggregometry or by the PFA-100).
Table II. Platelet disorders.
Von Willebrand disease
VWD is reportedly the most common of the inherited factor-related bleeding disorders, with reported prevalence of mild disease ranging from 1:100 to 1:10,000, depending on the study setting (Citation5). VWD arises due to defects or deficiencies in VWF, which is encoded by chromosome 12. However, genetic testing is generally not required or useful for the diagnosis of VWD, which is instead based on clinical observations and laboratory phenotypic testing. Indeed, the complexity and heterogeneity of VWD will often lead to failure in the identification of any genetic mutation in VWF (Citation6). VWD may be autosomal dominant or autosomal recessive depending on the type of defect. VWF has two primary functions: 1) facilitating the binding of platelets to each other and to subendothelium (in order to promote the formation of the platelet plug—i.e. primary hemostasis), and 2) binding and protecting FVIII from proteolysis (and thus promoting FVIII activity at sites of injury to facilitate secondary hemostasis). Clinically, VWD patients therefore present with a range of symptoms but primarily mucocutaneous symptoms such as bruising, epistaxis, menorrhagia, and gastric hemorrhages, but also post-surgical bleeding. Hemophilia-like symptoms may also be present when FVIII levels are also low (Citation7).
VWD patients are classified into one of six groups or types, with type 1 defining a partial quantitative deficiency of VWF, type 3 defining a total deficiency of VWF, and type 2 defining qualitative defects comprising: 1) type 2A (representing a loss of high-molecular-weight (HMW) VWF), 2) type 2B (representing a hyper-adhesive VWF defect classically also leading to clearance of HMW VWF and VWF-bound platelets), 3) type 2N (representing a defect of FVIII binding), and 4) type 2M (representing a heterogeneous group of other inherent VWF defects) (Citation8).
The complexity and heterogeneity of VWD typically mandates a comprehensive laboratory work-up that includes a broad range of assays to characterize appropriately both the level and activity of VWF. This includes assessment of FVIII activity, VWF protein (antigen; VWF:Ag), ristocetin co-factor (VWF:RCo), and preferably also collagen binding (VWF:CB) (Citation9). Testing must often be repeated on a new sample to confirm initial findings and, depending on the pattern of initial findings, also extended to include additional tests such as ristocetin-induced platelet agglutination (RIPA), VWF multimers, and VWF-FVIII binding (Citation10).
The differential diagnosis and typing of a patient's VWD depends on both the level of VWF and the functional defect (if any) determined. In brief, if VWF is not identified by any test, then type 3 VWD (the most severe form of VWD) would be identified. These patients also present with very low levels (<10%) of FVIII, so clinical symptoms of classical hemophilia are also evident. If VWF is low but similar levels of VWF are identified with all VWF assays, then the patient has type 1 VWD, and clinical symptoms are usually associated to the level of VWF (the lower the VWF level, the more severe the clinical symptoms). Patients in whom a discordance is observed between the VWF:Ag and activity assays (such that lower levels are detected by the latter) are classified as type 2 VWD, with the specific type (2A, 2B, 2N, or 2M) identified according to the test patterns determined (). An additional rare disorder called platelet type (PT-) VWD can give phenotypic test patterns similar to type 2B VWD but is caused by a platelet defect (Citation11).
Table III. Von Willebrand disease diagnosis and treatment.
In developed countries, mild type 1 VWD represents the majority of cases of VWD (>70%), with type 3 VWD representing <5% of cases, and type 2 VWD representing the rest. Within type 2 VWD, types 2A and 2M each represent the most common forms (about 30%–40% each), and types 2B and 2N are relatively rare forms (about 10%–15% each). In developing countries the situation is somewhat reversed, and types 3 and 2 tend to dominate, either because of consanguinity or because only the most severe forms of VWD are identified (Citation12).
Treatment of VWD depends on type and severity but generally consists of desmopressin (effective in mild type 1 VWD and some forms of type 2 VWD) or VWF replacement therapy (VWD types and/or treatment settings where desmopressin is not effective) (Citation13) (see also ). Anti-fibrinolytic therapy using tranexamic acid is also useful in some cases or in some settings, and hormonal replacement therapy may help control menorrhagia in some cases. VWF replacement therapy currently comprises plasma-derived VWF concentrates. Although a recombinant VWF is in development and has progressed to phase I human trials, this product is not yet available or licensed for use (Citation14).
Hemophilia A and B
Hemophilia A and B comprise inherited X-linked bleeding disorders respectively resulting from a deficiency of clotting factors VIII (FVIII) or IX (FIX). Hemophilia A occurs in 1 out of 10,000 male births, while hemophilia B occurs in 1 out of 30,000 male births (Citation15,Citation16). A deficiency of either FVIII or FIX results in the absence of a functioning intrinsic tenase complex leading to diminished thrombin generation and an inability to form and maintain a stable clot (Citation17).
The 186 kb FVIII gene, located on the Xq28 chromosome, encodes a heterotrimer protein composed of the A1, A2, A3, -C1 and -C2 domains. Multiple mutations leading to hemophilia A have been described on the FVIII gene, 30% of them occurring de novo. One of the most common, accounting for 40%–50% of patients, is a combined gene inversion and crossing-over, which completely disrupts the gene (Citation18). Patients with an inversion or a deletion, which leads to an absence of FVIII levels, are more susceptible to the development of anti-FVIII antibodies.
The FIX gene, located on chromosome Xq27, is 33 kb in length and encodes a vitamin K-dependent, single-chain glycoprotein consisting of 415 amino acids. Multiple mutations have been described that characterize the hemophilia B phenotype, such as point mutations, frameshifts, deletions, and other abnormalities that cause structural or functional change in the FIX protein. Over 33% of mutations arise de novo, and over 30% of mutations occur at CG dinucleotides, which involve critical arginine amino acids (Citation18).
Excluding familial disorders, the diagnosis of hemophilia A or B may first be suspected following an isolated prolongation of the routine coagulation test called the APTT (although different commercial reagents used for this test are characterized by a heterogeneous sensitivity to deficiencies of factors of the intrinsic pathway deficiency, so that sometimes mild deficiencies may be missed) and then be confirmed following performance of the specific factor assay. In familial disorders where the defect is already established, other family members are typically directly assessed by specific factor assays. The bleeding tendency in both hemophilia A and B is related to the concentration of the factor present in plasma and is typically classified as mild (5%–40% of normal), moderate (1%–5% of normal), or severe (<1% of normal) (Citation19).
Hemophilia A and B are clinically indistinguishable from each other, with both manifesting spontaneous hemarthrosis, soft-tissue hematomas, retroperitoneal bleeding, intracerebral hemorrhage, and delayed post-surgical bleeding. Over time, complications from recurrent hemarthrosis and soft-tissue hematomas include severe arthropathy, joint contractures, and pseudotumors, leading to chronic pain and disability. Around one-third of hemophilia carriers have low levels of FVIII or FIX and may experience bleeding symptoms similar to those described in people with mild hemophilia.
The introduction of virus-inactivated, plasma-derived coagulation factors first followed by recombinant products has revolutionized the care of people with hemophilia (Citation20). Indeed, these therapeutic weapons have improved the quality of life of hemophilia A and B patients and their families and have permitted regular infusion of factor concentrate replacement therapy to prevent bleeding and resultant joint damage (i.e. primary prophylaxis), home treatment, and ultimately a near normal life-style and life expectancy (Citation21–27).
Accordingly, in the era of the wide-spread diffusion (at least in developed countries) of recombinant products and of prophylaxis to prevent arthropathy, the most serious and challenging complication of replacement therapy has now become the development of inhibitors against coagulation FVIII or FIX. Inhibitory alloantibodies develop in approximately 20%–30% and 3%–5% of patients with severe hemophilia A and B, respectively, making replacement therapy ineffective in these patients, thereby precluding their access to a safe and effective standard of care and predisposing them to an unacceptably high risk of morbidity and mortality (Citation28,Citation29).
As regards inhibitors in hemophilia A, other genetic factors have been documented to influence inhibitor formation together with FVIII gene mutations, such as polymorphisms in genes involved in the regulation of the immune system (polymorphisms in the tumor necrosis factor and interleukin-10 genes), differences in the major histocompatibility complex, and ethnicity (Citation29,Citation30). Moreover, the role of environmental or treatment-related factors, such as an early age at first factor concentrate exposure, the intensive exposure to FVIII, the immunologic challenge (i.e. infection, surgery, vaccination), or the choice of FVIII replacement therapy (i.e. recombinant or plasma-derived concentrates), in the likelihood of developing inhibitors has been explored by a number of studies but with inconclusive results (Citation31–34).
Similarly to hemophilia A, the inhibitor development in hemophilia B is influenced by the severity and localization of the FIX gene defect (Citation29,Citation35). In particular, there are data indicating that inhibitors in hemophilia B are especially associated with large deletions in the FIX gene, and that these patients are at higher risk of severe anaphylactic reactions to FIX concentrate therapy (Citation36,Citation37).
The laboratory investigation of inhibitors to FVIII or FIX is problematic. Testing is based on so-called ‘Bethesda’ assays preferably with Nijmegen modifications, but results are compromised by poor relative consistency in test results (Citation38–40). This high assay variability compromises therapy since this will be differentially applied depending on whether inhibitors are low or high titer (see below); thus, patients with false low or high titers may be inappropriately managed.
The main short-term objective of the treatment of alloantibodies against FVIII and FIX is the management of bleeding diathesis (Citation41,Citation42). Acute bleeding events may be controlled with high doses of FVIII or FIX concentrates in hemophiliacs with low-titer factor inhibitors (<5 Bethesda units (BU)), while bypassing agents (i.e. activated prothrombin complex concentrates and recombinant activated factor VII) are necessary for patients with high-titer inhibitors (>5 BU) (Citation43–46). The eradication of the alloantibody through immune tolerance induction (ITI) in order to restore normal FVIII or FIX pharmacokinetics is the main long-term goal of the treatment of hemophiliacs with high-responding (anamnestic) inhibitors of recent onset (Citation47). Finally, disappointing clinical results have been obtained to date with gene therapy (based on genetically modified cells or direct in-vivo gene delivery using viral or plasmid vectors) for hemophilia (Citation48). Nevertheless, as genetic research advances and new results become available, clinicians and regulators need to be open to reconsider that some inherent risks of gene therapy might be outweighed by potential benefits, which include the use of effective, long-lasting, and safer treatments (Citation49).
Rare coagulation factor deficiencies
Rare coagulation factor deficiencies are autosomal recessive bleeding disorders in which one or more of the clotting factors other than FVIII or FIX (i.e. factors I, II, V, V+VIII, VII, X, XI, or XIII) is missing or not working properly (Citation50). Less information is available about these disorders (most data have been reported by the European Network of Rare Bleeding Disorders (EN-RBD) and the European Hemophilia Safety Surveillance (EUHASS)), because many of them have only been discovered in the last 40 years; they are reportedly less frequent than hemophilia or VWD and therefore more challenging to diagnose (Citation51).
Basically, the estimated prevalence in the general population varies between 1 in 500,000 and 1 in 2 million (3% to 5% of all the inherited coagulation deficiencies), and the clinical manifestations range from mild to severe (Citation51). According to the most recent WFH report, describing data on people with hemophilia, VWD, and other rare factor deficiencies throughout the world, the number of people with bleeding disorders other than hemophilia A and B is 18,762 out of 213,904 subjects with identified bleeding disorders (8.8%) (Citation52), more than 70% of which is attributed to rare factor deficiencies.
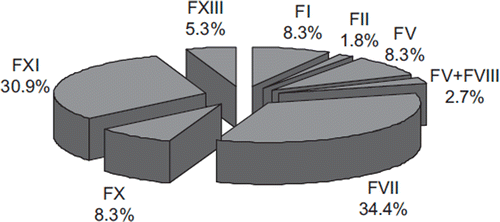
The relative frequency of the different rare factor deficiencies is synthesized in . These figures confirm that deficiencies of FVII and FXI are the most frequent, whereas that of FII and the combined FV + FVIII deficiencies are the most rare. Typically, none of the bleeding manifestations that affect patients with rare coagulation factor deficiencies can be considered specific for any specific defect, so that the precise diagnosis of the defect can only be made following laboratory investigation. Patients affected are habitually characterized by a wide spectrum of clinical presentations that range from a mild or moderate bleeding tendency, to potentially serious or life-threatening hemorrhages. As such, physicians are frequently challenged by individuals with mild hemorrhagic symptoms, particularly occurring after a surgical intervention, and the leading diagnostic problem is therefore to establish whether these subjects have a hemorrhagic disorder requiring special investigations and treatments.
Figure 3. Relative frequency of rare inherited bleeding disorders. Official data of the World Federation of Hemophilia (Citation51).
Particular attention should be paid to women affected by rare bleeding disorders, as they may experience excessive menstrual bleeding or menorrhagia and bleeding during pregnancy and child-birth. Affected women suffer also from the secondary consequences of such events as chronic iron deficiency anemia and a significantly reduced quality of life, due mainly to a complicated reproductive life but also due to limitations in social activities and work. Post-partum bleeding often occurs if replacement therapy is not administered after delivery. Pregnancy is not contraindicated in patients with RBDs but requires a multidisciplinary approach: the best management of pregnancy should be decided through the co-ordinated action of a team composed of pediatricians, hematologists, and obstetricians.
Factor I (fibrinogen) deficiency
Inherited fibrinogen deficiency has an estimated prevalence of 1 in 1,000,000 and may manifest as quantitative (afibrinogenemia and hypofibrinogenemia) or qualitative (dysfibrinogenemia) fibrinogen abnormalities, or both (hypodysfibrinogenemia) (Citation53). Afibrinogenemia is the most common inherited fibrinogen disorder and is characterized by virtually immeasurable plasma fibrinogen. Hypofibrinogenemia is rarer and is characterized by fibrinogen levels lower than 1.5 g/L. Around 400 cases of dysfibrinogenemia have been characterized to date, and the affected patients have fibrinogen protein levels typically in the normal range (1.5–3.5 g/L), so that its presence can only be suspected (and diagnosed) by a discrepancy between functional (clottable) and immunoreactive fibrinogen.
Patients carrying inherited fibrinogen disorders typically suffer from a bleeding diathesis but might paradoxically be exposed to the risk of thromboembolic complications as well as pregnancy loss (Citation54). The vast majority of patients with afibrinogenemia manifest the first symptoms during the neonatal period, especially with umbilical cord bleeding. Bleeding from other sites is also frequent, including skin, gastrointestinal and genitourinary tracts, and central nervous system (intracranial hemorrhage is the leading cause of mortality). Other hemorrhages that characterize hemophilia (e.g. hemarthrosis and muscular bleeding) are relatively infrequent. Since fibrin is capable of binding thrombin (thereby down-regulating its activity), afibrinogenemia (at variance with hemophilia) is characterized by conserved thrombin generation and platelet aggregation, which would both explain the paradoxical development of arterial and venous thrombosis in these patients. Patients with hypofibrinogenemia are usually asymptomatic, and serious hemorrhages are only observed after surgery, traumas, or in association with other hemostatic abnormalities. Importantly, liver disease can be occasionally observed in patients with hypofibrinogenemia due to the accumulation of abnormal fibrinogen molecules aggregated in the hepatocytes. Although most patients with inherited dysfibrinogenemia are asymptomatic, some experience bleeding (∼25%), thromboembolic complications (∼20%), or both. The hemorrhagic complications usually onset only after surgery or traumas, or post-partum.
The main laboratory findings are represented by variable prolongations of both PT and APTT (), associated with decreased levels of both clottable and immunoreactive fibrinogen (afibrinogenemia and hypofibrinogenemia) or decreased levels of clottable fibrinogen in the presence of normal immunological values (dysfibrinogenemia). A definitive diagnosis may require characterization of the underlying molecular defect. The mainstay of therapy of the most severe forms of inherited fibrinogen disorders is based on the use of fibrinogen concentrates. Nevertheless, no standard protocol is available since therapy is individualized according to the nature and severity of the deficit, as well as to the thrombotic risk of the patient, to balance and prevent potential thromboembolic complications associated with therapy. Nevertheless, plasma fibrinogen levels above 0.5 g/L are recommended for effective long-term secondary prophylaxis, or above 1.0 g/L in the case of active bleeding (Citation55).
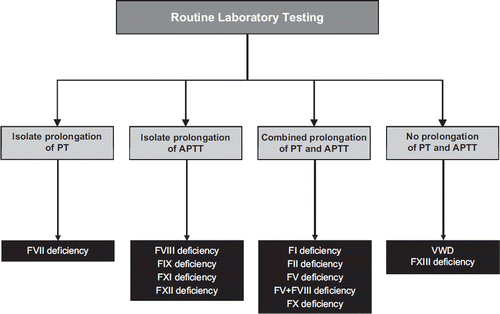
Figure 4. Abnormalities of routine coagulations tests in patients with inherited coagulation factor (F) disorders. Some cases of VWD may also present with isolated prolongation of APTT (when low FVIII levels are also evident). Owren-based PT results may be normal in FV deficiency. Primary hemostasis defects such as thrombocytopenias and platelet function disorders should yield normal PT and APTT values.
Factor II (prothrombin) deficiency
FII (prothrombin) deficiency has an overall prevalence in the general population of 1 in 2,000,000 (Citation56) and can be classified in two main phenotypes: type I (hypoprothrombinemia, which is characterized by decreased levels of both FII activity and antigen) and type II (dysprothrombinemia, which is instead characterized by the normal or near-normal levels of FII antigen but decreased activity due to the synthesis of a dysfunctional protein). Rarely, hypoprothrombinemia and dysprothrombinemia have both been reported in compound heterozygotes for FII deficiency. Interestingly, the complete deficiency of this protein is seemingly incompatible with life since it has never been described and experimental inactivation of the FII gene in the animal model is characterized by embryonic or neonatal lethality.
Patients affected by FII deficiency in the heterozygote form and typically displaying plasma levels of the protein ranging between 40% and 60% are usually asymptomatic and might only manifest a hemorrhagic tendency after traumas, surgery, or other invasive procedures (e.g. tooth extractions, tonsillectomy, endoscopy). More severe deficits, i.e. patients with FII plasma levels <10%, are instead characterized by a severe bleeding syndrome. Lancellotti and De Cristofaro have recently reviewed 32 cases of severe FII deficiency, reporting serious hemorrhages in 12% to 60% of the cases, mainly represented by intracerebral bleeding, intracranial hemorrhages, spontaneous hematomas, bruising, hemarthrosis, menorrhagia, and bleeding after tooth extraction (Citation56). The routine laboratory tests (i.e. PT and APTT) are variably prolonged (), and specific one-stage clotting assays using tissue thromboplastin and prothrombin-depleted plasma are necessary to diagnose FII deficiency. As for other defects, a discrepancy between functional (clotting) and antigenic values is suggestive for the present of a dysfunctional protein (dysprothrombinemia).
The care of patients carrying FII efficiency is determined by the severity of the disease. Replacement therapy is usually needed in homozygous patients only (i.e. FII levels <10%), in the presence of active bleeding or for prophylaxis prior to invasive procedures. Unfortunately, no purified prothrombin concentrates have been developed so far, so that fresh-frozen plasma (FFP) and prothrombin complex concentrates (PCCs) are the only therapeutic choice. Although the definitive plasma FII level required for efficient hemostasis has not been determined, a minimum level of 10%–15% may be sufficient for preventing minor hemorrhages, while levels between 20% and 40% might be necessary in the presence of trauma or surgery (Citation56).
Factor V deficiency
Congenital FV deficiency has a prevalence of 1 in 1,000,000 in the general population (Citation57). Analogously to other similar disorders, type I is characterized by concordantly low or immeasurable antigen and functional plasma levels (quantitative defect), while type II deficiency is characterized by normal or mildly reduced antigen plasma levels associated with a reduced coagulant activity (qualitative defect). Homozygotes or compound heterozygotes usually display FV plasma levels <10%, while the heterozygous state in the mild or moderate form is associated with FV plasma levels between 20% and 30%. The relatively low platelet content of FV is important, however, because patients with very low to undetectable plasma FV may still possess functional FV in their platelets which, in combination with low tissue factor pathway inhibitor (TFPI) levels, allows sufficient thrombin generation to rescue these patients from fatal bleeding.
The bleeding tendency of FV-deficient patients is highly dependent upon the residual plasma FV level. In severe patients, the first bleeding episodes occur in childhood and typically comprise skin and mucosal tract hemorrhages (e.g. epistaxis and menorrhagia). Other severe hemorrhages, like hematomas, hemarthroses, bleeding in the central nervous system and in the gastrointestinal tract, are less frequent or very rare (i.e. <20%) (Citation57). The FV deficiency is characterized by a concomitant prolongation of both PT (using the Quick but not the Owren assay) and APTT (), which can be corrected, as in other inherited clotting factor deficiencies, by mixing the patient plasma with a plasma pool. The deficiency needs to be confirmed and thus diagnosed by performing a specific FV assay. However, it is reasonable in suspected FV-deficient patients to assess also FVIII in order to rule out a combined deficiency of FV and FVIII (Citation58). The standard of care is mainly based on replacement therapy with FFP because FV concentrates are unavailable and FV is not contained in cryoprecipitate or prothrombin complex concentrates. The minimum recommended FV plasma level in case of severe bleeding or as a prophylaxis before surgery is between 20% and 25% (Citation57).
Factor V and VIII deficiency
The combined deficiency of FV and FVIII, also known as F5F8D or FV + FVIII, is a rare autosomal recessive bleeding disorder (1:1,000,000) attributable in up to 70% of the cases to null mutations in the ERGIC-53 gene, also known as LMAN1 (lectin mannose binding protein), which encodes an endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) marker protein. Less frequently, in nearly 15% of cases, the pathology can also be determined by mutations in another gene, the MCFD2 (multiple coagulation factor deficiency 2), which encodes a co-factor of LMAN1 (Citation58). Both proteins are involved in the intracellular transport of FV and FVIII. At variance with other rare bleeding disorders, F5F8D is thereby determined by ineffective processing of the proteins rather than by specific mutations in the FV and FVIII genes.
Phenotypically, F5F8D is characterized by concomitantly low levels (i.e. 5% to 20%) of the two separate clotting factors and is associated with a mild to moderate bleeding tendency, but the residual plasma levels of both FV and FVIII are usually sufficient to prevent severe hemorrhages. The most frequent symptoms include bruising, epistaxis, gum bleeding, menorrhagia, hemorrhages after surgery, dental extraction, and trauma (Citation58). The main laboratory findings include a combined prolongation of both PT and APTT, while demonstration of reduced coagulant activity of both FV and FVIII is necessary for diagnosis.
The differential diagnosis of F5F8D with a FV deficiency or mild hemophilia A or even a FV deficiency associated with mild hemophilia A is assisted by assessment of the genetic transmission (hemophilia A is X-linked), the type and severity of bleeding, and relative levels of each factor. As for the therapeutic approach, the bleeding in F5F8D patients is usually treated according to the nature of the hemorrhage and the plasma levels of the coagulation factors, and the disorder does not require regular prophylaxis. Replacement therapy is that already described individually for mild hemophilia A and FV deficiency (Citation58).
Factor VII deficiency
FVII deficiency is reportedly the most common among the rare congenital coagulation disorders (1 case per 500,000 in the general population; relative prevalence among rare bleeding disorders: 34.4%) and is typically transmitted as an autosomal recessive disorder (Citation59). Type 1 deficiency results from decreased biosynthesis or accelerated clearance, while type 2 abnormalities are characterized by the presence of a dysfunctional molecule in plasma. The clinical phenotypes of FVII deficiency are rather heterogeneous, ranging from asymptomatic to severe disease. To investigate the relationship between clinical phenotype, clotting activity (FVIIc), and FVII genotype, Mariani et al. carried out a multicenter study of FVII congenital deficiency with centralized genotyping and specific functional assays, finally proposing a severity classification for this disorder (Citation60). Basically, clinical phenotypes include asymptomatic conditions (also including homozygotes and double heterozygote carriers), miscellaneous minor bleedings, and patients with a severe disease characterized by life-threatening and disabling symptoms (important surgical bleeding, hemarthrosis, central nervous system and gastrointestinal hemorrhages).
The final classification is based on the nature and number of the symptoms included, with severely affected patients having a median FVIIc concentration of 1.4% (interquartile range (IQR) 0.9%–3.8%), those moderately affected displaying a median FVIIc concentration of 3% (IQR 1%–21.7%), and those considered ‘mild bleeders’ having a median FVIIc concentration of 14% (IQR 3%–31%). This classification was further supported by a significantly different prevalence of homozygote or double heterozygote carriers among the different classes of severity (severe: 98%, moderate: 84%, mild bleeders: 56%). Most interestingly, homozygote carriers for an identical mutation and with nearly overlapping FVIIc and FXa generation levels were characterized by substantially discordant clinical phenotypes, so that it was concluded that additional environmental and/or inherited factors might modulate the expressivity of the FVII deficiency (Citation59,Citation60).
Importantly, an excess of symptomatic women (experiencing menorrhagia, easy bruising, and gum bleeding) among carriers of inherited FVII deficiency has been reported in the International Registry on Congenital Factor VII Deficiency as compared with men (Citation61).
The laboratory features of patients with FVII deficiency typically show a variable prolongation of PT in the presence of normal values of the APTT (). The final diagnosis is made by demonstration of isolated reduction of FVIIc in plasma. However, different thromboplastins used for the FVIIc clotting assay as well as the residual levels of FVII in the substrate used for the assay might influence the diagnostic performances (i.e. the sensitivity) of the assay (Citation62).
The differential diagnosis between inherited and acquired FVII deficiency (e.g. due to vitamin K deficiency, vitamin K antagonist therapy, or liver disease), is usually based on the presence of an isolated reduction of FVII with normal levels of the other vitamin K-dependent factors (FII, FIX, and FX) (Citation63,Citation64). The clinical management of patients with inherited FVII deficiency is particularly challenging due to uncertainty regarding minimal FVII levels needed to avoid bleeding in different clinical situations, the lack of a significant correlation between bleeding phenotype and FVIIc clotting levels, the frequent onset of surgery-related bleeding, and the more severe presentation in affected women as compared with men (Citation61). Accordingly, although several treatment options are available for this disorder, including plasma-derived FVII concentrates and recombinant activated FVII (rFVIIa), prophylaxis and treatment (i.e. optimal dosages and administration times) of bleeding episodes still have to be clearly defined, and the clinical management must be individualized for the single patient. It is, however, generally accepted that unlike the hemophilias, prophylaxis might not be routinely administered to FVII-deficient patients, and it must be limited to unweaned infants prone to severe and frequent bleedings, in patients with recurrent hemarthrosis, and in women with severe menorrhagia with concomitant iron deficiency anemia (Citation61). Excessive menstrual bleeding in women with FVII deficiency may also be controlled with hormonal contraceptives (birth control pills) or anti-fibrinolytic drugs (Citation50).
Factor X deficiency
Factor X (FX) deficiency has an estimated prevalence of 1 in 1,000,000 in the general population (Citation65). A concomitant reduction of functional and antigen levels typically indicates a type I deficiency caused by defective synthesis or ineffective protein secretion, whereas a discrepancy between functional (low) and antigen (normal or only mildly reduced) levels is suggestive for a type II deficiency, which globally resembles the effect of the new direct activated FX inhibitors (DFXaI).
Although patients with FX deficiency have a highly variable hemorrhagic phenotype, the bleeding symptoms are usually more accentuated than in other rare inherited coagulation disorders, due to the central role of FX in hemostasis (). Thus, severely affected patients (i.e. FX plasma levels <1%) can manifest early with life-threatening hemorrhages such as those involving the umbilical cord or the central nervous system. Hemarthrosis, hematomas, gastrointestinal bleeding, menorrhagia, and hematuria might also frequently occur.
Conversely, patients with mild to moderate FX deficiency (i.e. FX plasma levels between 5% and 20%) bleed characteristically after hemostatic challenges such as trauma or surgery (Citation65). From a laboratory perspective, combined prolongation of PT and APTT in association with a reduced FX clotting activity (but normal levels of other vitamin K-dependent clotting factors) might be sufficient for a correct diagnosis of FX deficiency.
There is no specific FX concentrate available, either derived or recombinant, so that the treatment of patients with FX deficiency is still based on the administration of FFP or prothrombin complex concentrates (PCCs) containing FX, although this option should be carefully considered for the risk of thromboembolic complication due to the high concentrations of FII, FVII, and FIX in these preparations. Although the minimum FX levels necessary for efficient hemostasis as well as the dose and the frequency of administration of replacement therapy remain to be established, a residual plasma FX level of 5% might be sufficient to achieve 50% of thrombin generation and prevent serious bleeding (Citation65).
Factor XI deficiency
Hereditary FXI deficiency is an autosomal bleeding disorder with a prevalence of 1 in 1,000,000 in the general population, although its frequency is remarkably higher among Ashkenazi Jews (1 every 450 individuals) (Citation66). As for other inherited disorders, the concordance or discordance between FXI antigen and activity levels determines the classification, which can be type I (activity and antigen levels both reduced) or type II (dysfunctional, normal to moderately reduced activity levels in the presence of a normal antigen concentration).
The disorder is also known as hemophilia C, since it was discovered in 1953 by Rosenthal and colleagues as the third hemorrhagic disease after hemophilia A and B. The disease is less clinically severe compared with the other forms of hemophilia, and the onset of hemorrhages is usually related to trauma or invasive procedures. Nevertheless, the bleeding tendency in patients affected by FXI deficiency is very subjective and thereby highly unpredictable. Notably, the bleeding phenotype is correlated with the site of injury and not with the genotype.
The sites most affected by bleeding are mainly those with local high fibrinolytic potential (e.g. urogenital tract, oral cavity after dental extraction or tonsillectomy), where hemorrhages might be relatively frequent (50% to 70%) as compared with other sites (1% to 40%) (Citation66). This peculiarity has been explained by the fact that FXI is strongly involved in the activation of TAFI, a protein necessary for attenuating fibrinolysis and thereby stabilizing the clot. As mirrored by the biological function of FXI in blood coagulation, the typical laboratory finding is a prolonged APTT associated with a normal PT.
The final diagnosis requires the determination of FXI plasma levels, which is also helpful for establishing the severity of the disease (i.e. severe deficiency is defined by FXI levels <15%–20%). The assessment of the plasma antigen concentration might be necessary to distinguish between quantitative and qualitative defects (Citation66).
Patients with severe forms of FXI deficiency are usually managed with fresh-frozen plasma to obtain FXI plasma activity levels of 40%. FXI concentrates are also available, but their use has been questioned especially in the elderly and in patients with cardiovascular disease or surgery due to the intrinsic thrombotic potential. Due to the biological interplay between FXI and fibrinolysis, patients with FXI deficiency undergoing surgery might also benefit from the administration of anti-fibrinolytic agents such as tranexamic acid or epsilon aminocaproic acid (Citation66).
Factor XII deficiency
Although FXII is a coagulation protein that is essential for surface-activated blood coagulation tests (e.g. APTT), its deficiency is not associated with a bleeding tendency (Citation67). Nevertheless, FXII deficiency is still worthwhile mentioning here because it represents the most frequent cause (up to one-third) of prolonged APTT in daily laboratory practice (Citation68). As such, the recognition of this coagulation disorder might be advantageous for the clinical management of these patients, in that it would prevent additional analyses and prolonged hospitalization in order to troubleshoot the cause of a prolonged APTT during hospital admissions, as well as the inadvertent treatment of such patients to correct their prolonged APTTs.
Although the incidence of FXII deficiency is estimated at 1 in 1,000,000 this should be confirmed given the high laboratory detection rate. FXII deficiency is inherited as an autosomal recessive condition. Even patients with virtually undetectable plasma FXII levels are completely asymptomatic and, in most cases, are diagnosed by chance or during pre-screening blood tests for surgery. The typical laboratory features are represented by a prolonged APTT in the presence of a normal PT. The quantification of residual FXII clotting activity in plasma is necessary to confirm the initial diagnosis. Being a ‘benign’ condition, treatment of carriers, even in the homozygous state, is unnecessary. There are some suggestions that FXII deficiency might instead predispose to thromboembolic complications, but this has not been definitely established.
Factor XIII deficiency
The general frequency of FXIII deficiency is very low, being estimated at 1 in 2,000,000 (Citation69). The clinical features and bleeding tendency vary widely according to the plasma FXIII levels, although patients affected by severe deficit (i.e. plasma FXIII levels <3%) might experience serious and frequent hemorrhages, including umbilical bleeding (80%), superficial bruising (60%), subcutaneous hematoma (55%), mouth and gums hemorrhages (30%), intracranial hemorrhage (30%), and/or hemarthrosis (24%). Other forms of bleeding are rarer (<15%), as are perioperative hemorrhages. Nevertheless, severe bleeding complications have been reported in patients with severe deficiency undergoing abdominal, gynecologic, plastic, urologic, and neurologic surgery.
Due to the important role of FXIII in wound healing, a typical feature of this disease is represented by abnormal scar formation, which might be present in up to one-third of affected patients (Citation69). The diagnosis of FXIII deficiency represents a major challenge for the hemostasis laboratory, because both the PT and APTT are normal in these patients (). Accordingly, it is the clinics that should primarily guide the diagnostic reasoning (e.g. abnormal bleeding in a patient with normal routine coagulation tests, exclusion of other defects of both primary and secondary hemostasis, presence of abnormal scars). The demonstration of reduced FXIII plasma levels using a quantitative FXIII activity assay might be sufficient for the diagnosis, although this assay is not routinely performed by most laboratories.
The characterization of the disorder might also require further complicated testing, such as the measurement of FXIII-A2B2 antigen concentration in plasma, measurement of FXIII-A and FXIII-B in plasma and FXIII-A in platelet lysate, and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of cross-linked fibrin (Citation69). Heterozygous carriers of FXIII deficiency (i.e. those with FXIII plasma levels ∼50%) do not usually require therapy. Conversely, due to the high risk of spontaneous hemorrhages, life-long prophylaxis might be recommended in patients severely affected (i.e. those with FXIII plasma levels <3%). Whole blood, FFP, and cryoprecipitate have been classically used as adequate sources of FXIII for the management and treatment of this deficiency. Nevertheless, the availability of plasma FXIII concentrate containing FXIII-A2B2 is now recommended as the treatment of choice for reasons of both efficacy and safety. A new recombinant FXIII-A2 concentrate (rFXIII-A2) has also become available and holds promise as a suitable alternative to FXIII concentrate (Citation69).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bouma BN, Mosnier LO. Thrombin activatable fibrinolysis inhibitor (TAFI)—how does thrombin regulate fibrinolysis? Ann Med. 2006;38:378–88.
- Lippi G, Franchini M. Pathogenesis of venous thromboembolism: when the cup runneth over. Semin Thromb Hemost. 2008;34:747–61.
- Laterre PF, Wittebole X, Collienne C. Pharmacological inhibition of tissue factor. Semin Thromb Hemost. 2006; 32: 71–6.
- World Federation of Hemophilia. What is a bleeding disorder? Available at: http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=26&contentid=5&rptnam=bleeding (accessed 23 July 2010).
- Franchini M, Targher G, Lippi G. Prophylaxis in von Willebrand disease. Ann Hematol. 2007;86:699–704.
- Favaloro EJ. Genetic testing for von Willebrand disease: the case against. J Thromb Haemost. 2010;8:6–12.
- Franchini M, Mannucci PM. Von Willebrand factor: another janus-faced hemostasis protein. Semin Thromb Hemost. 2008;34:663–9.
- Sadler JE, Budde U, Eikenboom JCJ, Favaloro EJ, Hill FGH, Holmberg L, . Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4: 2103–14.
- Favaloro EJ. Laboratory identification of von Willebrand disease: technical and scientific perspectives. Semin Thromb Hemost. 2006;32:456–71.
- Favaloro EJ. Towards a new paradigm for the identification and functional characterisation of von Willebrand disease. Semin Thromb Hemost. 2009;35:60–75.
- Favaloro EJ. Phenotypic identification of platelet-type von Willebrand disease and its discrimination from type 2B von Willebrand disease: a question of 2B or not 2B? A story of nonidentical twins? Or two sides of a multidenominational or multifaceted primary-hemostasis coin? Semin Thromb Hemost. 2008;34:113–27.
- Favaloro EJ, Mohammed S, Koutts J. Identification and prevalence of von Willebrand Disease type 2N (Normandy) in Australia. Blood Coag Fibrinolysis. 2009;20:706–14.
- Federici AB, Mannucci PM. Management of inherited von Willebrand disease in 2007. Ann Med. 2007;39:346–58.
- Turecek PL, Schrenk G, Rottensteiner H, Varadi K, Bevers E, Lenting P, . Structure and function of a recombinant von Willebrand factor (VWF) drug candidate. Semin Thromb Hemost. 2010;36:510–21.
- Bolton-Maggs PH, Pasi KJ. Haemophilia A and B. Lancet. 2003;361:1801–9.
- Mannucci PM, Tuddenham EG. The hemophilias—from royal genes to gene therapy. N Engl J Med. 2001;344: 1773–9.
- Broze GJ, Higuchi D. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88: 3815–23.
- Oldenburg J, Schwaab R. Molecular biology of blood coagulation. Semin Thromb Hemost. 2001;27:313–24.
- White GC II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia: recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85:560.
- Franchini M, Coppola A, Molinari AC, Santoro C, Schinco P, Speciale V, . Forum on: the role of recombinant factor VIII in children with severe haemophilia A. Haemophilia. 2009;15:578–86.
- Franchini M, Lippi G. Recombinant factor VIII concentrates. Semin Thromb Hemost. 2010;36:493–7.
- Monahan PE, Di Paola J. Recombinant factor IX for clinical and research use. Semin Thromb Hemost. 2010;36:498–509.
- Coppola A, Franchini M, Tagliaferri A. Prophylaxis in people with hemophilia. Thromb Haemost. 2009;101:674–81.
- Tagliaferri A, Rivolta GF, Iorio A, Oliovecchio E, Mancuso ME, Morfini M, . for the Italian Association of Hemophilia Centers. Mortality and causes of death in Italian persons with haemophilia, 1990–2007. Haemophilia. 2010;16:437–46.
- Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. 2008;14 (Suppl 3): 10–18.
- Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, . Possible transmission of variant Creutzfeldt–Jakob disease by blood transfusion. Lancet. 2004;363: 417–21.
- Pipe SW. Recombinant clotting factors. Thromb Haemost. 2008;99:840–50.
- Gringeri A, Mantovani LG, Scalone L, Mannucci PM. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS Study Group. Blood. 2003;102:2358–63.
- Gouw SC, van den Berg HM. The multifactorial etiology of inhibitor development in hemophilia: genetics and environment. Semin Thromb Hemost. 2009;35:723–34.
- Oldenburg J, Schröder J, Brackmann HH, Müller-Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41(Suppl 1):82–8.
- Santagostino E, Mancuso ME, Rocino A, Mancuso G, Mazzucconi MG, Tagliaferri A, . Environmental risk factors for inhibitor development in children with haemophilia A: a case-control study. Br J Haematol. 2005;130:422–7.
- Gouw SC, van der Bom JG, Auerswald G, Ettinghausen CE, Tedgård U, van den Berg HM. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007;109:4693–7.
- Goudemand J, Rothschild C, Demiguel V, Vinciguerrat C, Lambert T, Chambost H, . FVIII-LFB and Recombinant FVIII study groups. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006;107:46–51.
- Chalmers EA, Brown SA, Keeling D, Liesner R, Richards M, Stirling D, . Paediatric Working Party of UKHCDO. Early factor VIII exposure and subsequent inhibitor development in children with severe haemophilia A. Haemophilia. 2007;13:149–55.
- Berntorp E, Shapiro A, Astermark J, Blanchette VS, Collins PW, Dimichele D, . Inhibitor treatment in haemophilias A and B: summary statement for the 2006 international consensus conference. Haemophilia. 2006;12 (Suppl 6):1–7.
- Jadhav M, Warrier I. Anaphylaxis in patients with hemophilia. Semin Thromb Hemost. 2000;26:205–8.
- Franchini M, Lippi G, Montagnana M, Targher G, Zaffanello M, Salvagno GL, . Anaphylaxis in patients with congenital bleeding disorders and inhibitors. Blood Coagul Fibrinolysis. 2009;20:225–9.
- Verbruggen B, van Heerde WL, Laros-van Gorkom BA. Improvements in factor VIII inhibitor detection: From Bethesda to Nijmegen. Semin Thromb Hemost. 2009;35: 752–9.
- Kershaw G, Jayakodi D, Dunkley S. Laboratory identification of factor inhibitors: the perspective of a large tertiary hemophilia center. Semin Thromb Hemost. 2009;35:760–8.
- Favaloro EJ, Bonar R, Kershaw G, Duncan E, Sioufi J, Marsden K. Investigations from external quality assurance programs reveal a high degree of variation in the laboratory identification of coagulation factor inhibitors. Semin Thromb Hemost. 2009;35:794–805.
- Franchini M, Lippi G. Recent improvements in the clinical treatment of coagulation factor inhibitors. Semin Thromb Hemost. 2009;35:806–13.
- Mathews V, Nair SC, David S, Viswabandya A, Srivastava A. Management of hemophilia in patients with inhibitors: the perspective from developing countries. Semin Thromb Hemost. 2009;35:820–6.
- Astermark J. Treatment of the bleeding inhibitor patient. Semin Thromb Hemost. 2003;29:77–85.
- Von Depka M. Managing acute bleeds in the patient with haemopilia and inhibitors: options, efficacy and safety. Haemophilia. 2005;11(Suppl 1):18–23.
- Franchini M, Manzato F, Salvagno GL, Montagnana M, Zaffanello M, Lippi G. Prophylaxis in congenital hemophilia with inhibitors: the role of recombinant activated factor VII. Semin Thromb Hemost. 2009;35:814–9.
- Ananyeva NM, Lee TK, Jain N, Shima M, Saenko EL. Inhibitors in hemophilia A: advances in elucidation of inhibitory mechanisms and in inhibitor management with bypassing agents. Semin Thromb Hemost. 2009;35:735–51.
- DiMichele D, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recommendations. Haemophilia. 2007;13(Suppl 1):1–22.
- Viala NO, Larsen SR, Rasko JE. Gene therapy for hemophilia: clinical trials and technical tribulations. Semin Thromb Hemost. 2009;35:81–92.
- Lippi G, Franchini M, Saenko EL. Gene therapy for hemophilia A. Friend or foe? Blood Coagul Fibrinolysis. 2009;20: 395–9.
- World Federation of Hemophilia. What are rare clotting factor deficiencies? World Federation of Hemophilia; 2009.
- Peyvandi F, Palla R, Menegatti M, Mannucci PM. Introduction. Rare bleeding disorders: general aspects of clinical features, diagnosis, and management. Semin Thromb Hemost. 2009;35:349–55.
- World Federation of Hemophilia. WFH report on the Annual Global Survey 2007. Available at: http://www.wfh.org/2/docs/Publications/Statistics/2007-Survey-Report.pdf (accessed 23 July 2010).
- de Moerloose P, Neerman-Arbez M. Congenital fibrinogen disorders. Semin Thromb Hemost. 2009;35:356–66.
- de Moerloose P, Boehlen F, Neerman-Arbez M. Fibrinogen and the risk of thrombosis. Semin Thromb Hemost. 2010;36: 7–17.
- Bolton-Maggs PHB, Perry DJ, Chalmers EA, Parapia LA, Wilde JT, Williams MD, . The rare coagulation disorders—review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004;10:593–628.
- Lancellotti S, De Cristofaro R. Congenital prothrombin deficiency. Semin Thromb Hemost. 2009;35:367–81.
- Asselta R, Peyvandi F. Factor V deficiency. Semin Thromb Hemost. 2009;35:382–9.
- Spreafico M, Peyvandi F. Combined factor V and factor VIII deficiency. Semin Thromb Hemost. 2009;35:390–9.
- Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost. 2009;35:400–6.
- Mariani G, Herrmann FH, Dolce A, Batorova A, Etro D, Peyvandi F, . International Factor VII Deficiency Study Group. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005; 93:481–7.
- Lapecorella M, Mariani G; International Registry on Congenital Factor VII Deficiency. Factor VII deficiency: defining the clinical picture and optimizing therapeutic options. Haemophilia. 2008;14:1170–5.
- Girolami A, Scandellari R, Lombardi AM. The use of tissue thromboplastins of different origin is a fundamental tool in the initial characterization of FVII defects on ‘factor VII deficiency (Semin Thromb Hemost 2009;35(4):400–406)’. Semin Thromb Hemost. 2010;36:123–4; author reply 125–7.
- Brenner B, Kuperman AA, Watzka M, Oldenburg J. Vitamin K-dependent coagulation factors deficiency. Semin Thromb Hemost. 2009;35:439–46.
- Wada H, Usui M, Sakuragawa N. Hemostatic abnormalities and liver diseases. Semin Thromb Hemost. 2008;34:772–8.
- Menegatti M, Peyvandi F. Factor X deficiency. Semin Thromb Hemost. 2009;35:407–15.
- Duga S, Salomon O. Factor XI deficiency. Semin Thromb Hemost. 2009;35:416–25.
- Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res. 2010;125:210–5.
- Lippi G, Franchini M, Brazzarola P, Manzato F. Preoperative screening: the rationale of measuring APTT in risk assessment. Haematologica. 2001;86:328.
- Karimi M, Bereczky Z, Cohan N, Muszbek L. Factor XIII deficiency. Semin Thromb Hemost. 2009;35:426–38.