Abstract
Interleukin-23 (IL-23) is a pro-inflammatory cytokine composed of two subunits, p19 and p40. The p40 subunit is shared with IL-12. IL-23 and IL-12 have different receptors and different effects. Whereas IL-12 induces development of Th1 cells, which produce interferon-γ, IL-23 is involved in differentiation of Th17 cells in a pro-inflammatory context and especially in the presence of TGF-β and IL-6. Activated Th17 cells produce IL-17A, IL-17F, IL-6, IL-22, TNF-α, and GM-CSF. Inflammatory macrophages express IL-23R and are activated by IL-23 to produce IL-1, TNF-α, and IL-23 itself. These effects identify IL-23 as a central cytokine in autoimmunity and a highly promising treatment target for inflammatory diseases. IL-23 is found in the skin of patients with psoriasis, in the bowel wall of patients with chronic inflammatory bowel disease, and in synovial membrane of patients with rheumatoid arthritis. IL-23 is involved in osteoclastogenesis, independently from IL-17, via induction of RANKL expression. Debate continues to surround the role for IL-23 in the pathophysiology of inflammatory joint diseases (rheumatoid arthritis and spondyloarthritis). Ustekinumab, which inhibits IL-12 and IL-23 by blocking p40, has been found effective in cutaneous psoriasis and psoriatic arthritis, as well as in Crohn's disease. Treatments that specifically target IL-23 (antibodies to p19) are being developed.
| Abbreviations | ||
| ACR | = | American College of Rheumatology |
| AS | = | ankylosing spondylitis |
| BSA | = | bovine serum albumin |
| CIA | = | collagen-induced arthritis |
| DC | = | dendritic cell |
| EAE | = | experimental allergic encephalomyelitis |
| Ebi3 | = | Epstein–Barr virus-induced gene 3 |
| G-CSF | = | granulocyte colony-stimulating factor |
| gp130 | = | glycoprotein 130 |
| IBD | = | inflammatory bowel disease |
| IFN-γ | = | interferon-γ |
| IL-23 | = | interleukin-23 |
| IL-23R | = | interleukin-23 receptor |
| JAK | = | Janus kinase |
| MDP | = | muramyl dipeptide |
| NOD2 | = | nucleotide-binding oligomerization domain containing 2 |
| PASI | = | Psoriasis Area and Severity Index |
| RA | = | rheumatoid arthritis |
| Sca | = | stem cell antigen-1 |
| sCNTFR/CLC | = | soluble ciliary neurotrophic factor receptor cardiotrophin-like cytokine |
| SpA | = | spondyloarthropathy |
| STAT3 | = | signal transducer and activator of transcription 3 |
| TLR | = | Toll-like receptor |
| Treg | = | regulatory T cells |
| WNV | = | West Nile virus |
Key messages
Interleukin-23 plays a pivotal role in chronic inflammation.
It is a promising target for the development of treatment.
It is essential for treatments to be specific.
Description of interleukin-23 and its receptors
Interleukin-23 (IL-23) is a cytokine whose identification profoundly affected theories about chronic inflammation and autoimmunity. IL-23 belongs to the IL-12 family of cytokines, itself part of the IL-6 superfamily.
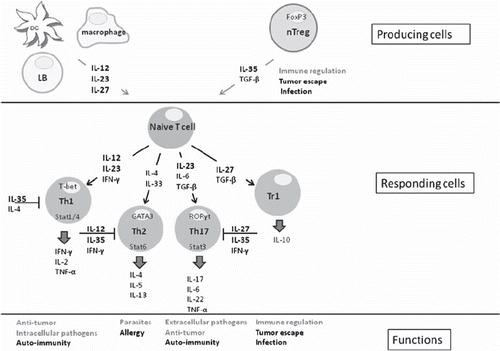
To date, the IL-12 family consists of four heterodimeric cytokines that share sequence homology. The first identified member was IL-12, composed of two subunits, p35 and p40. Substantial homology exists between p35 and IL-6 (Citation1), whereas p40 shares homology with the extracellular portion of the IL-6Rα chain and of sCNTFR (soluble ciliary neurotrophic factor receptor). The IL-12 receptor is composed of two chains, IL-12Rβ1 and IL-12Rβ2, which share homology with the IL-6 receptor (Citation2). Full signal transmission requires the phosphorylation of signal transducer and activator of transcription (STAT)-1, STAT3, STAT5, and, above all, STAT4. Long after IL-12 was identified, studies showed that p40 could combine with p19 to produce a new heterodimeric cytokine, IL-23 (Citation3). IL-23 shares some homology with IL-6 and granulocyte colony-stimulating factor (G-CSF). The IL-23 receptor (IL-23R) is composed of IL-12Rβ1 combined with a specific chain, IL-23R, which resembles IL-6gp130. The IL-23 signaling pathway is similar to that of IL-12, with a preponderant role for STAT3. The p35 subunit of IL-12, in addition to sharing homology with IL-6, is a component of the recently identified anti-inflammatory cytokine IL-35 (). IL-35 is a heterodimeric cytokine composed of the subunits p35 and Ebi3 (Epstein–Barr virus induced gene 3). Its receptor has not been identified, and its mechanism of action remains unclear. IL-35 is produced by regulatory T cells (Treg) and may contribute to inhibit T cell proliferation. Another member of the IL-12 family, IL-27, is composed of the subunit Ebi3, which is found also in IL-35 and interacts with IL-6gp130, and of p28, which interacts with IL-27Rα. IL-27 is produced by the dendritic cells (DCs), macrophages, and B cells (). It activates Th1 cells and inhibits Th2 cells (Citation4,Citation5).
Figure 1. The four cytokines of the IL-12 family and their respective receptors.
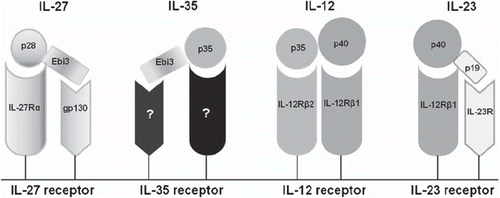
Figure 2. Mechanism of action of the cytokines from the IL-12 family. In grey their positive functions and in black their negative functions.
IL-23 is expressed chiefly by the macrophages and DCs. The IL-23R is found on memory T cells, NKT cells, macrophages, DCs, and naive T cells upon activation by TGF-β and IL-6 (Citation6). The main biological effects of IL-23 identified initially consist of stimulation of antigen presentation by DCs, T cell differentiation to Th17 cells, and production of interferon-γ (IFN-γ). IL-23 acts also as an end-stage effector cytokine through direct action on macrophages (Citation7). This can be in part interpreted as an autocrine loop of IL-23 on macrophages. In addition, intraperitoneal administration of recombinant IL-23 in mice induces expression of mRNA coding for IL-1 and TNF-α in peritoneal macrophages (Citation8).
IL-23 plays a major role in autoimmunity
Early research in a model of central nervous system autoimmunity suggested a crucial role for IL-23 in autoimmune disease. Indeed, IL-23 was found in the nervous system as well as in macrophages at the site of inflammation. Accordingly, it was demonstrated that its receptor, IL-23R, is expressed not only by T cells but also by myeloid cells. In a model of multiple sclerosis, the experimental allergic encephalomyelitis (EAE), it was shown that knock-out mice for the subunit p40 or for IL-12Rβ1 are protected against the illness. Protection against EAE was also observed in IL-23p19−/− mice, whereas EAE was exacerbated in IL-12p35 −/− mice. All these results, indicating a major role for IL-23, were also obtained in models of arthritis and chronic bowel inflammation (Citation9).
A key point is that IL-23 drives the development of T effector cells exhibiting a distinctive profile of inflammatory cytokine production. They produce IL-17A, IL-17F, IL-6, and TNF-α. Studies using the EAE model established that T cells stimulated with IL-23 were sufficient to transfer the disease to naive animals. Monoclonal antibodies to p19, specific of IL-23 and which fail to recognize IL-12, can prevent relapses in some EAE models and are associated with down-regulation of the expression of other inflammatory cytokines such as IFN-γ, IL-17, IL-6, and TNF-α. These results and a few others (Citation10) led to the classification of chronic inflammatory diseases into two groups, Th1 and Th17, although both profiles may exist concomitantly in some diseases (e.g. rheumatoid arthritis (RA)) (Citation11). Thus, T cell differentiation to Th17 cells, i.e. chiefly IL-17-producing T cells, requires IL-23. TGF-β and IL-6 are the major drivers for Th17 cell differentiation in mice and in humans (Citation12). A recent study showed that a combination of TGF-β, IL-1β, IL-6, IL-21, and IL-23 in serum-free conditions was necessary and sufficient to induce IL-17 expression in naive CD4+ T cells from cord blood (Citation13). On the other hand, a recent study demonstrated an alternative way for the differentiation of Th17 without TGF-β, but in presence of IL-23, IL-6, and IL-1β, in a model of EAE (Citation14). The specific transcription factor involved is RORγt, which has a central role in differentiation of Th17, but in addition RORα is required for complete Th17 cell differentiation.
Another role for IL-23 was demonstrated recently in the Tγδ subset of T cells. The IL-23R is expressed by many cells devoid of TCR αβ, and IL-23 can act independently from Tαβ (Th17) cells. In a colitis model, stimulation by IL-23 of lymphocytes present in the colon leads to the production of IFN-γ and IL-17 by lymphoid cells that express Thy1, stem cell antigen-1 (Sca-1), RORγt, and IL-23R (Citation15) ().
Figure 3. IL-23 is a central cytokine during inflammation. Most of the cells implicated in inflammation are regulated positively or negatively by IL-23.
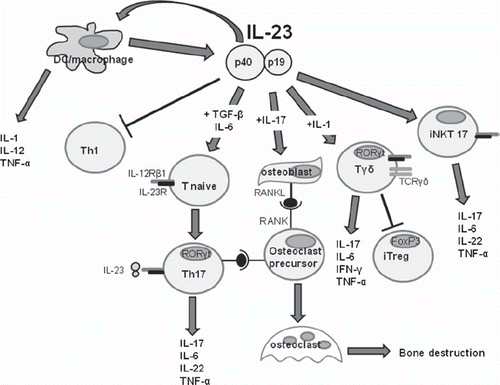
Tγδ cells exist in small numbers yet play a crucial role. When stimulated by IL-23, they produce IL-17, as shown in models of infection or autoimmunity. The result is Th17 cell differentiation under the combined influence of IL-23 and IL-1 (Citation16). In a model of collagen-induced arthritis (CIA) in which DBA/1 mice are injected with type II bovine collagen emulsified in complete Freund adjuvant to induce arthritis, the main source of IL-17 within the diseased joints is the Tγδ population induced by IL-23 and IL-1, which are expressed locally by the synovial cells. In contrast, Tγδ cells are virtually absent from the joints of human patients with RA (Citation17). In the model of antigen-induced arthritis with methyl bovine serum albumin (BSA), IL-23 is required for disease development. In IL-23p19−/− mice, the absence of IL-23 may limit the development of the joint disease and prevent progression to joint destruction. The development of synovitis is mediated by IL-23, which regulates the proportions of IFN-γ or IL-17-producing CD4+ T cells, as well as the expression of IL-17 and RORγt by Tγδ cells (Citation18). A recent study in the EAE model has established that Tγδ cells produce large amounts of IL-23R and exhibit a very early response to stimulation with IL-23. This response leads to the accumulation of IL-23R+ Tγδ cells, which produce IL-17, IL-21, and IL-22. The main role for these cells under pro-inflammatory conditions is to prevent Tαβ suppression by Treg cells and the differentiation of conventional T cells to Treg cells (Citation19).
In psoriasis, IL-23 expression is markedly up-regulated compared to normal skin. In patients with psoriasis, IL-23 acts through the chemokine receptor CCR6, causing inflammation and IL-22 production. IL-22 is a member of the IL-10 family produced by activated Th17 cells and plays a crucial role in mediating psoriasis-like modifications (Citation20).
IL-23 and chronic bowel inflammation
The bowel is the tissue that has the highest levels of IL-23 expression. It is also the site of greatest exposure to bacteria. The bowel contains about 1014 micro-organisms belonging to around 1,000 different species and having a total mass of about 1,000 grams (Citation21). One of the roles for IL-23 may be to counteract the suppressive effects of Treg cells in the bowel, thus allowing the induction of cellular immunity when an infection occurs. IL-23p19−/− mice are unable to develop an appropriate immune response specific to gut pathogens, perhaps because of an excessive Treg response due to the absence of IL-23. On the other hand, excessive IL-23 production may constitute a major precipitant of bowel inflammation in patients with conditions such as Crohn's disease (Citation22).
Interestingly, IL-12 was initially believed to play a pivotal role in the inflammatory process characteristic of Crohn's disease. Treatments targeting IL-12, most notably anti-IL-12p40 agents, were found effective in experimental models and subsequently yielded promising results in clinical studies (Citation23). However, the identification of IL-23 changed the hypothesized local hierarchy of cytokines in the gut. The efficacy of the IL-12p40 antibody has been ascribed to inhibition of the other cytokine having a p40 subunit, IL-23 (Citation22,Citation24), with IL-12 seeming to have a more ancillary role, perhaps because of the abundance in the bowel of Treg cells, which exert suppressive effects on IL-12-dependent Th1 cells.
Susceptibility to Crohn's disease and ulcerative colitis is influenced by the IL-23R polymorphism but not by polymorphisms in IL-12Rβ1 or IL-12p40 (Citation25). Recent studies have established that IL-23R is a risk factor for concomitant inflammatory bowel disease (IBD) and psoriasis (Citation26).
IL-23 may play a predominant role compared to IL-12 in all the mucous membranes of the body. Many pathogens found in the bowel induce marked IL-23 secretion, thereby stimulating innate and adaptive immune responses to cause ultimately the production of antimicrobial factors in the mucosa. Current data suggest that the primary role for IL-23 may be to drive a robust innate response that serves as the first line of defense against environmental pathogens not only in the gut but also in the lungs and skin. Experiments on infection by West Nile virus (WNV) showed that Toll-like receptor (TLR)-7 recognizes WNV and then promotes the immune cell homing to infected cells in an IL-12 and IL-23-dependent way (Citation27). It has been also been showed that muramyl dipeptide (MDP), a ligand of NOD2 (nucleotide-binding oligomerization domain containing 2), is responsible for stimulating the production of IL-23 and IL-1. In Crohn's disease, NOD2 is often mutated, there is a misregulation of IL-23, and the Th17 axis is implicated in the pathogenesis of the disease. Activation of DCs by MDP in combination with NOD2 leads to potent IL-23-stimulatory activity, whereas NOD2 mutant DCs from patients with Crohn's disease failed to activate Th17 cells even after stimulation by MDP and TLR ligand. These results suggest a pathway for priming of Th17 by IL-23 NOD2 (Citation28). The physiologic protective role may be overwhelmed under specific conditions, leading to pro-inflammatory effects of IL-23 (Citation29).
An association has been reported between chronic inflammation and the development of some types of cancer. For instance, the risk of colorectal cancer is increased in patients who have chronic IBD. The chronic inflammation in IBD is related to IL-23, which can act via two pathways, namely activation of IL-17-producing T cells, and induction of IL-6 and IL-1 production. Recent data indicate that bowel inflammation related to IL-23 and IL-6 influences the development of some types of colorectal cancer (Citation30). In addition, VEGF over-expression can increase the risk of colorectal cancer. A recent study investigated potential links among IL-23 levels, VEGF expression, and p53 expression in patients with colorectal cancer (Citation31). The results showed that IL-23 over-expression was correlated with VEGF levels, particularly in patients with high-grade colorectal cancer (Citation31).
IL-23 and joint disease: contribution of experimental models
Experimental models have helped to understand the role of IL-23 in autoimmune diseases, including chronic inflammatory joint disease. The first evidence that IL-23 played a key role in experimental arthritis was the complete resistance of IL-23p19−/− mice to CIA and the increased severity of CIA in IL-12p35−/− mice compared to wild-type controls (Citation32). Although IL-23p19−/− mice have no Th17 cells, they have lymphocytes capable of producing IFN-γ. In the presence of TGF-β and IL-6, IL-23 induces the co-expression of IL-17 and RANKL on CD4+T cells, leading to the production by endothelial and synovial cells of pro-inflammatory cytokines (IL-1, IL-6, IL-18, and TNF-α) and to osteoclast stimulation by RANKL via TRAF6 and transcription factors such as c-Fos. IL-23p19−/− mice and IL-17−/− mice are protected against inflammatory bone erosions, in contrast to mice with concomitant blockade of the IL-4 and IFN-γ pathways, illustrating the respective impacts of Th1 (IFN-γ), Th2 (IL-4), and Th17 (IL-17, IL-23). IFN-γ and the STAT1 pathway protect the bone in this setting. Consequently, IL-23 (or IL-17) is a promising treatment target in inflammatory joint disease associated with bone and joint damage.
The IL-23 activation pathway seems to play a pivotal role in lupus nephropathy. Thus, lpr/lpr mice bred on an IL-23R knock-out background were protected against the clinical and laboratory manifestations of lupus (including lupus nephropathy), compared to controls (Citation33). In addition, these animals had lower counts of CD3+CD4− CD8− lymphocytes and of IL-17A-producing lymphocytes (Citation33).
HLA-B27 transgenic rats, a model for spondyloarthritis, over-express IL-23 in the bowel. IL-23 over-expression is also found in these animals in the antigen-presenting cells and CD4+ T cells of the bowel (Citation34).
Data from humans with inflammatory joint disease
Ankylosing spondylitis (AS) is characterized by marked IL-23 over-expression in the bowel, with levels similar to those seen in Crohn's disease (Citation35). IL-23 is produced by Paneth cells, which are secretory resident epithelial cells found deep in the crypts of Lieberkühn in the small bowel. Paneth cells contribute to the innate immune response by secreting antimicrobial peptides. Normal individuals do not have Paneth cells producing large amounts of IL-23 (Citation36). In contrast to Crohn's disease, AS is not associated with IL-17 over-expression in the bowel, perhaps because of the low levels of IL-1β and IL-6, two cytokines that are involved synergistically in IL-17 production. Thus, IL-23/IL-23R axis dysregulation may lead to bowel inflammation and may constitute a marker for bowel inflammation in patients with AS.
Several studies compared the Th17 regulation in patients with RA or with spondyloarthropathies (SpA) other than psoriatic arthritis (Citation37). Even though it has been shown that Th17-related cytokines were expressed in joints from both RA and SpA patients, a correlation between disease activity (swollen joint count, C-reactive protein level, and DAS28) has only been demonstrated in patients with RA (Citation37). The studies showed that patients with RA had significantly more CCL20, a major Th17 cell chemoattractant (and therefore a factor responsible for secondary IL-23 production) in joint fluid than did patients with SpA (other than psoriatic arthritis). As well, patients with RA had significantly more IL-17A, IL-21, and IL-23 than did patients with SpA (Citation38). As other pro-inflammatory cytokines such as IL-1β, IL-2, IL-4, and even IL-12 are not different between SpA and RA patients, the studies demonstrated a different regulation of the IL-17/IL-23 pathway between RA and SpA. In RA, TNF-α antagonist therapy did not significantly influence IL-23 or IL-17 levels, suggesting independence of the TNF-α and IL-23/IL-17 systems and supporting a predominant position of TNF-α in the hierarchy of pro-inflammatory cytokines. This result is controversial. In the same study, IL-23 levels within joints were not significantly different between the two groups. IL-23 was found in established RA but not in very recent onset RA (Citation38). Previous work showed that IL-23 levels in joint fluid correlated with the presence of erosions (Citation39). A functional study of cultured synovial cells from patients with RA showed IL-23 production (Citation40). IL-23 blockade with IL-23R led to marked decreases in the production of TNF-α, IL-1β, and IL-6. A very interesting finding was that IL-17A blockade induced only modest decreases in the same pro-inflammatory cytokines, suggesting a hierarchy in the direct effects of IL-23 and IL-17, in keeping with current hypotheses. Infliximab therapy was found to diminish serum IL-23 levels in one study (Citation41) but not in another (Citation37).
Studies of cohorts of RA patients failed to identify relevant associations with the IL-23R haplotypes studied in patients with IBD. In contrast, the psoriasis-spondyloarthritis combination was associated with IL-23R haplotypes (Citation42,Citation43). In patients with lupus, no association was found with an IL-23R gene polymorphism (Citation44–46).
Is IL-23 a treatment target?
Therapeutic monoclonal antibodies
Ustekinumab (Stelara®, Centocor®, PA, USA) is a human monoclonal IgG1 kappa antibody against the p40 subunit of IL-12 and IL-23. Ustekinumab prevents IL-12 and IL-23 from interacting with the surface receptor IL-12Rβ1, thereby blocking the IL-12 and IL-23 cascade. This drug was developed for the treatment of psoriasis. The two pivotal studies are phase III randomized, double-blind, placebo-controlled trials, PHOENIX 1 and PHOENIX 2 (Citation47–49). In both studies, ustekinumab was promptly effective in patients with moderate-to-severe psoriasis. After 28 weeks of treatment (three injections), over 90% of patients had a significant response (as evaluated using the PASI-50). PASI (Psoriasis Area and Severity Index) is a score evaluating the severity of psoriasis. PASI-50 (respectively 75) response means an improvement of at least 50% (respectively 75%) of the base-line PASI score. The clinical improvements were preceded by decreases in the expression of mRNAs for IL-12/IL-23p40 and IL-23p19. In contrast, up-regulation of IL-12p35 has been reported (Citation50). The rates of adverse events, including infections, were not significantly different between the ustekinumab and placebo groups. A randomized trial (ACCEPT) compared ustekinumab (90 mg at weeks 0 and 4) to etanercept (50 mg twice weekly) for 12 weeks. The proportion of patients with a PASI 75 response was higher with ustekinumab (Citation51).
A clinical trial showed that ustekinumab was effective in patients with moderate-to-severe Crohn's disease (Citation52). In patients with psoriatic arthritis, a randomized, double-blind, placebo-controlled trial established that ustekinumab (90 mg at weeks 0, 1, 2, and 3) was effective. Thus, after 12 weeks, the proportion of patients with an ACR-20 response was 42.1% in the ustekinumab group and 14.3% in the placebo group (Citation53). American College of Rheumatology (ACR) criteria of response to a treatment are based on the improvement in tender or swollen joints and improvement in three of the following parameters: sedimentation rate, patient assessment, physician assessment, pain scale, and disability/functional questionnaire. Achievement of ACR-20 (respectively 50, 70) criteria means 20% (respectively 50%, 70%) improvement in tender or swollen joints counts, as well as 20% improvement of at least three of the five criteria. This phase II trial also produced evidence of efficacy in psoriasis. In contrast, the only published trial of ustekinumab in multiple sclerosis found no evidence of efficacy (Citation54,Citation55). However, the patients had advanced disease at base-line. The overall safety profile of ustekinumab was consistently good in clinical trials, with no significant differences in adverse event rates between the ustekinumab and placebo arms. Nevertheless, physicians must bear in mind at all times the possibility that ustekinumab might be associated with serious infections and malignancies (Citation56).
Other monoclonal antibodies to IL-12/IL-23 are being developed, including briakinumab (Citation57), a fully humanized recombinant IgG1 against p40. Phase II trials of briakinumab in patients with psoriasis are underway. The preliminary results indicate a significant treatment response. After 12 weeks, 90% of patients had a significant response (as assessed using the PASI-75). However, adverse events were recorded. More specifically, the infection rate was higher in the briakinumab arm than in the placebo arm (Citation58). Patients treated by briakinumab have higher risk to develop nasopharyngitis and upper respiratory tract infection followed by bronchitis and viral infection. To inhibit specifically IL-23 without inhibiting IL-12, the p19 subunit must be targeted. Monoclonal antibodies to p19 are being investigated.
IL-23 inhibition using drugs other than monoclonal antibodies
Our group has been working jointly with the Conservatoire National des Arts et Métiers (CNAM) Bioinformatics team to develop a novel IL-23 inhibition strategy. This approach consists in administering a vaccine composed of an IL-23p19 peptide coupled to Keyhole Limpet Hemocyanin (KLH). We obtained promising preliminary results in the CIA mouse model (Citation59).
Apilimod inhibits a transcription factor used for IL-12 and IL-23. Phase II clinical trials of apilimod are underway in patients with RA. An advantage of this agent is the possibility of oral administration. Promising results have also been obtained with triptolide, which is purified from a plant (Tripterygium wildordii hook F) used in traditional Chinese medicine to treat psoriasis, asthma, lupus, and RA. Triptolide inhibits the transcription of the p40 gene by acting on the promoter in the macrophages. Thus triptolide is a natural anti-IL-12/IL-23 agent (Citation60).
Conclusion
Interleukin-23 deserves careful attention both because it plays a pivotal role in chronic inflammation and because it can be inhibited effectively. It is important to bear in mind the manner in which IL-23 interacts with cells (including its heterodimeric structure) to interpret correctly the results obtained using inhibitors with various specificities. IL-23 is among the most promising treatment targets for chronic inflammatory joint disease.
Even though this review is focused on IL-23, other cytokines are involved in autoimmunity and deserve attention. Pro-inflammatory cytokines such as IL-1, IL-18, IL-17, IL-6, and TNF-α (as an unlimited list of players) induce both chronic and acute inflammatory response and play a role in autoimmunity by acting on innate or cognate phases of the process. All these cytokines are targets or potential targets for autoimmune diseases. From a cellular differentiation point of view, Th17 cells do not represent the only T cell population involved in autoimmunity and chronic inflammation. Th1 cells are in some cases major actors inducing autoimmunity, as recently described in EAE (Citation61). In another disease, such as lupus, IFN-γ and B cell activating factor (BAFF) are important mediators as shown by the two large phase III trials of belimumab which is a fully human antibody specific for BAFF (Citation62).
Acknowledgements
The authors wish to thank the following institutions for their funding in their research on IL-23: Arthritis-Fondation Courtin (France), Agence nationale de la recherche (ANR, France), Inserm (France), Société française de rhumatologie, University Paris 13, the DGA, and Andar.
Declaration of interest: The authors declare no conflict of interest.
References
- Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y. Role for interleukin-6 in structural joint damage and systemic bone loss in rheumatoid arthritis. Joint Bone Spine. 2010;77:201–5.
- Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202: 139–56.
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, . Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25.
- Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, . Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009;60:738–49.
- Collison LW, Vignali DA. Interleukin-35: odd one out or part of the family? Immunol Rev. 2008;226:248–62.
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, . A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708.
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, . Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
- Bastos KR, Marinho CR, Barboza R, Russo M, Alvarez JM, D'Imperio Lima MR. What kind of message does IL-12/IL-23 bring to macrophages and dendritic cells? Microbes Infect. 2004;6:630–6.
- Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42.
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, . Interleukin 17-producing CD4+effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
- Boissier MC. Cell and cytokine imbalance in rheumatoid arthritis. Joint Bone Spine. 2010. DOI: 10.1016/j.jbspin. 2010.08.017.
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89.
- Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9.
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, . Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010; 467:967–71.
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, . Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464: 1371–5.
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41.
- Ito Y, Usui T, Kobayashi S, Iguchi-Hashimoto M, Ito H, Yoshitomi H, . Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 2009;60:2294–303.
- Cornelissen F, Mus AM, Asmawidjaja PS, van Hamburg JP, Tocker J, Lubberts E. Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORgammat in gammadelta T cells. Arthritis Res Ther. 2009;11:R194.
- Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, . Gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33: 351–63.
- Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, Singh SP, . CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. 2009; 119:2317–29.
- Ehlers S, Kaufmann SH. Infection, inflammation, and chronic diseases: consequences of a modern lifestyle. Trends Immunol. 2010;31:184–90.
- Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, . Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18.
- Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, . Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–79.
- Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, . Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–70.
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, . A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
- Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, . Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007; 122:201–6.
- Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, . Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–53.
- Stetson DB, Medzhitov R. T helper 17 cells get the NOD. Immunity. 2007;27:546–8.
- Tato CM, Cua DJ. Reconciling id, ego, and superego within interleukin-23. Immunol Rev. 2008;226:103–11.
- Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets. 2008;9:375–80.
- Ljujic B, Radosavljevic G, Jovanovic I, Pavlovic S, Zdravkovic N, Milovanovic M, . Elevated serum level of IL-23 correlates with expression of VEGF in human colorectal carcinoma. Arch Med Res. 2010;41:182–9.
- Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, . Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82.
- Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–9.
- DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–43.
- Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, . Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009;60:955–65.
- Keshav S. Paneth cells: leukocyte-like mediators of innate immunity in the intestine. J Leukoc Biol. 2006;80:500–8.
- Melis L, Vandooren B, Kruithof E, Jacques P, De Vos M, Mielants H, . Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann Rheum Dis. 2010;69:618–23.
- Moura RA, Cascao R, Perpetuo I, Canhao H, Sousa E, Rosario AF, . Cytokine profile in serum and synovial fluid of established rheumatoid arthritis patients (Poster). 7th International Congress on Autoimmunity; Ljubljana, Slovenia, 5–9 May 2010. PS11.
- Kim HR, Kim HS, Park MK, Cho ML, Lee SH, Kim HY. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:259–64.
- Hillyer P, Larche MJ, Bowman EP, McClanahan TK, de Waal Malefyt R, Schewitz LP, . Investigating the role of the interleukin-23/-17A axis in rheumatoid arthritis. Rheumatology (Oxford). 2009;48:1581–9.
- Kageyama Y, Kobayashi H, Kato N. Infliximab treatment reduces the serum levels of interleukin-23 in patients with rheumatoid arthritis. Mod Rheumatol. 2009;19:657–62.
- Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, . The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2008;67:1451–4.
- Hollis-Moffatt JE, Merriman ME, Rodger RA, Rowley KA, Chapman PT, Dalbeth N, . Evidence for association of an interleukin 23 receptor variant independent of the R381Q variant with rheumatoid arthritis. Ann Rheum Dis. 2009; 68:1340–4.
- Safrany E, Hobor R, Jakab L, Tarr T, Csongei V, Jaromi L, . Interleukin-23 receptor gene variants in Hungarian systemic lupus erythematosus patients. Inflamm Res. 2010; 59:159–64.
- Kim HS, Kim I, Kim JO, Bae JS, Shin HD, Bae SC. No association between interleukin 23 receptor gene polymorphisms and systemic lupus erythematosus. Rheumatol Int. 2009;1:33–8.
- Li Y, Liang WB, Li C, Gao LB, Zhou B, Wang YY, . The association between interleukin-23 receptor gene polymorphisms and systemic lupus erythematosus. DNA Cell Biol. 2010;29:79–82.
- Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, . Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008; 371:1665–74.
- Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, . Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675–84.
- Krulig E, Gordon KB. Ustekinumab: an evidence-based review of its effectiveness in the treatment of psoriasis. Core Evid. 2010;5:11–22.
- Liu J, Cao S, Herman LM, Ma X. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J Exp Med. 2003;198:1265–76.
- Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, . Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362:118–28.
- Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, . A randomized trial of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn's disease. Gastroenterology. 2008;135:1130–41.
- Gottlieb A, Menter A, Mendelsohn A, Shen YK, Li S, Guzzo C, . Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009;373:633–40.
- Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804.
- Longbrake EE, Racke MK. Why did IL-12/IL-23 antibody therapy fail in multiple sclerosis? Expert Rev Neurother. 2009;9:319–21.
- Elliott M, Benson J, Blank M, Brodmerkel C, Baker D, Sharples KR, . Ustekinumab: lessons learned from targeting interleukin-12/23p40 in immune-mediated diseases. Ann N Y Acad Sci. 2009;1182:97–110.
- Lima XT, Abuabara K, Kimball AB, Lima HC. Briakinumab. Expert Opin Biol Ther. 2009;9:1107–13.
- Gandhi M, Alwawi E, Gordon KB. Anti-p40 antibodies ustekinumab and briakinumab: blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin Cutan Med Surg. 2010;29:48–52.
- Duvallet E, Ratsimendresy R, Assier E, Delavallée L, Bessis N, Zagury JF, . Active immunization against IL-23p19 improves collagen-induced-arthritis. Arthritis Rheum. 2009;10(Suppl):S1–2.
- Zhang Y, Ma X. Triptolide inhibits IL-12/IL-23 expression in APCs via CCAAT/enhancer-binding protein alpha. J Immunol. 2010;184:3866–77.
- Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, . T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–64.
- Looney RJ. B cell-targeted therapies for systemic lupus erythematosus: an update on clinical trial data. Drugs. 2010; 70:529–40.