Abstract
Cardiovascular complications remain the leading cause of mortality in adult human subjects with diabetes. Hyperglycemia has long been hypothesized to explain some of the effects of diabetes on cardiovascular complications caused by atherosclerosis, but a clear causative role for hyperglycemia has not been established. Recent studies in animal models indicate that glucose may play a role in diabetes-accelerated atherosclerosis by promoting pro-inflammatory responses in myeloid cells, which are key cell types in atherosclerosis. For example, monocytes and macrophages often take on a more pro-inflammatory phenotype in the setting of diabetes. Moreover, in-vitro studies demonstrate a connection between pro-inflammatory molecules and glucose metabolism in macrophages and dendritic cells. This review concerns the role of glucose metabolism in inflammatory macrophages, and their potential role in diabetic vascular disease. Further in-vivo studies, focusing on myeloid-specific effects of glucose metabolism as it relates to atherosclerosis, are needed to increase our understanding of the relationship between diabetes, myeloid cells, and cardiovascular disease.
| Abbreviations | ||
| AGE | = | advanced glycation end-product |
| AR | = | aldose reductase |
| FDG-PET | = | fluorine-18-labeled-2-deoxy-D-glucose positron emission tomography |
| FIH-1 | = | factor inhibiting HIF-1 |
| G6PD | = | glucose-6-phosphate dehydrogenase |
| GLUT1 (SLC2A1) | = | glucose transporter 1 |
| HbA1c | = | glycated hemoglobin A1c |
| HIF-1 | = | hypoxia-inducible factor-1 |
| IFN | = | interferon |
| IL | = | interleukin |
| LPS | = | lipopolysaccharide |
| PFK | = | phosphofructokinase |
| RAGE | = | receptor for AGE |
| TLR | = | Toll-like receptor |
Key message
Increased glucose metabolism in macrophages might contribute to the increased atherosclerosis associated with diabetes.
Clinical studies and animal studies suggest a role for hyperglycemia in diabetic vascular disease, but only under certain conditions
Many clinical studies demonstrate a correlation between suboptimal glycemic control and cardiovascular events in patients with type 1 diabetes (Citation1–3), although this relation is not consistently observed (Citation4–6). One issue, which has recently been brought to light, is that levels of glycated hemoglobin A1c (HbA1c), the current standard for analysis of glycemic control, have been shown to be regulated by genetic components, some of which are unrelated to elevated glucose levels (Citation7). Thus, levels of HbA1c might not always be a good measure of the extent of glycemic control. Another issue is that cardiovascular disease develops over long periods of time, and clinical intervention studies are rarely conducted over the time-span of several years or decades. Interestingly therefore, long-term follow-up studies provide the most compelling evidence for a beneficial effect of improved glycemic control on cardiovascular end-points (Citation2,Citation8). Furthermore, it is difficult to achieve optimal glycemic control in clinical practice. One interesting long-term study is the follow-up of the Diabetes Control and Complications Trial cohort. Strikingly, in this long-term follow-up, the effect of tight glycemic control early in the study led to significantly beneficial cardiovascular outcomes many years later in subjects with type 1 diabetes (Citation2).
Patients with type 2 diabetes often show increases in a number of cardiovascular risk factors, such as dyslipidemia, hypertension, and insulin resistance. Clinical intervention to improve either dyslipidemia or hypertension demonstrates the reduction of cardiovascular events, whereas the effect of glycemic control is controversial. Thus, hyperglycemia may play a less important role in patients with type 2 diabetes, as compared to patients with type 1 diabetes. Indeed, intensive blood glucose lowering in patients with type 2 diabetes has been shown to be either beneficial, to be without effect, or even to be detrimental to the development of cardiovascular events (Citation9–12). Although several other risk factors are likely to play a more important role than glucose in patients with established type 2 diabetes, epidemiological studies strongly indicate an association between blood glucose levels and the risk of cardiovascular disease (Citation13), and it cannot be ruled out that hyperglycemia plays a role, especially in patients with relatively normal lipids and few other risk factors.
Can we extrapolate information from animal studies to get a better understanding of the role of hyperglycemia in human cardiovascular disease? In mice, hyperglycemia is associated mainly with an increased formation of early macrophage-rich lesions of atherosclerosis (Citation14–16). It is therefore possible that the principal effect of hyperglycemia is to accelerate the first stages of lesion development (lesion initiation) and that this effect, perhaps together with other risk factors, could lead to the occurrence of cardiovascular events earlier in life in subjects with diabetes as compared with subjects without diabetes. The insight provided by long-term follow-up studies (Citation2,Citation8) may be consistent with these findings in mice, although several other explanations are possible. When thigh glycemic control was started early in the course of type 1 diabetes (Citation2), likely before the development of advanced lesions, the group receiving intense treatment to improve glucose control might have developed lesions at a slower rate, which resulted in fewer cardiovascular events after several years, even though at this time, blood glucose levels were similar between the groups. Another hypothesis for how elevated blood glucose levels might exert detrimental effects years later, even when glucose levels have been normalized, is that elevated glucose might induce long-term epigenetic changes in tissues susceptible to development of complications of diabetes (Citation17), or through increased formation of advanced glycation end-products.
In diabetic animals that are also severely dyslipidemic, a stimulatory effect of diabetes independent of that of dyslipidemia is often absent (Citation18–23). These animal models might therefore mimic some of the aspects of type 2 diabetes, in which dyslipidemia masks and overrides the stimulatory effects of hyperglycemia.
Thus, mouse models suggest that hyperglycemia may play a role in initiation of macrophage-rich atherosclerotic lesions, potentially leading to more advanced lesions later in life. Recent studies on new mouse models have begun to provide interesting evidence that glucose indeed does affect vascular cells in vivo.
How can direct effects of elevated glucose on vascular cells be evaluated in animal models?
How can the effect of glucose in different cell types within the atherosclerotic lesion be tested in vivo? One interesting approach was recently taken to address whether increased glucose uptake specifically in vascular smooth muscle cells would alter these cells’ properties in vivo (Citation24). In this study, the glucose transporter 1 (GLUT1; also known as SLC2A1) was over-expressed in smooth muscle cells in a transgenic mouse model. When mice with GLUT1 overexpressing smooth muscle cells were subjected to femoral artery injury, an increased accumulation of neutrophils was observed in the injured vessel (Citation24). Thus, increased glucose uptake in smooth muscle cells appears to enhance their pro-inflammatory potential. Although the vascular injury model used in this study should not be equated with atherosclerosis, these findings provide a first important view into the cell type-specific effects of glucose in vivo. It would be informative to evaluate atherosclerosis in mice with increased glucose uptake specifically in smooth muscle cells and other vascular cells involved in atherosclerosis.
Previous studies have used mouse models to investigate the role of advanced glycation end-products (AGEs) and the receptor of AGEs (RAGE) in atherosclerosis. These models did not provide cell type-specific data but demonstrated clearly that loss of RAGE prevents atherosclerosis in diabetic dyslipidemic mice (Citation21,Citation25). Subsequent studies demonstrated that RAGE deficiency also protects against atherosclerosis in non-diabetic mice and that RAGE binds several types of ligands (Citation26). Bone-marrow transplant studies suggested that the protective effects of RAGE deficiency in non-diabetic apoE–/– mice is due partially to RAGE expression in bone-marrow-derived cells (Citation27). Thus, RAGE activation does not occur only in diabetes, and RAGE does not specifically mediate effects of hyperglycemia but is likely to have a more wide-spanning importance in disease states associated with increased inflammation.
Another approach is to study enzymes in glucose metabolism believed to mediate the effects of glucose. An excellent example of this approach is the human aldose reductase (AR) transgenic mouse (Citation15). This mouse expresses levels of aldose reductase similar to those observed in humans, and exhibits increased atherosclerosis specifically in the setting of diabetes (Citation15). Thus, it is possible that hyperglycemia promotes atherosclerosis, at least in part, through increased flux through the polyol/sorbitol pathway (see ). It is not yet known what cell type might be primarily responsible for this effect.
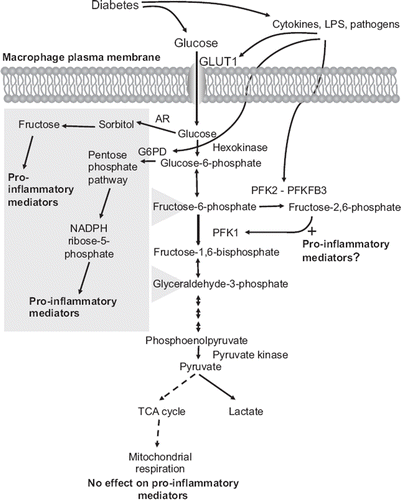
Figure 1. Schematic representation of the potential links between diabetes, glucose metabolites, and pro-inflammatory effects in macrophages. Macrophages rely heavily on glycolysis as an energy source, both under aerobic and anaerobic conditions. Diabetes is likely to increase glucose uptake in macrophages. Cytokines and pathogens also stimulate glycolysis by increasing expression of GLUT1, G6PD, and PFKFB3. Diabetes could therefore potentially stimulate glucose flux in macrophages through two mechanisms, by a direct effect through hyperglycemia, and by promoting a pro-inflammatory environment, which in turn further enhances glucose metabolism in a self-perpetuating cycle. Recent research shows that the pro-inflammatory phenotype of macrophages associated with diabetes might be due to increased glucose flux through the polyol/sorbitol pathway, the pentose phosphate pathway, and/or glycolysis, whereas mitochondrial respiration does not appear to affect the pro-inflammatory phenotype. The polyol/sorbitol pathway and pentose phosphate pathway both feed into glycolysis (as indicated by the gray box) and therefore could contribute to the increased flux through glycolysis.
These new mouse models, and other similar models, will provide much-needed insight into the potential role of glucose in different vascular cells in atherosclerosis in vivo.
High glycolysis in macrophages in atherosclerotic lesions
There is evidence that acutely elevated glucose levels promote leukocyte–endothelial interactions in vivo (Citation28), particularly in the presence of inflammatory mediators (Citation29). This effect of glucose could potentially provide an important first step in glucose-induced lesion formation. Myeloid cells—monocytes, macrophages, and dendritic cells—are major players in the immune system and rapidly accumulate at sites of inflammation, such as lesions of atherosclerosis (Citation30,Citation31). These sites are often characterized by poor vascularization and hypoxic conditions. Thus, in addition to lesions of atherosclerosis, high numbers of macrophages have been reported in joints affected by rheumatoid arthritis (Citation32) and in hypoxic areas of dermal wounds (Citation33). Macrophages must be able to move into a hypoxic environment during inflammation and to carry out their varied functions under such conditions, which requires substantial amounts of ATP.
Under normoxic conditions, most cells produce ATP from glucose via generation of pyruvate through glycolysis and subsequently use pyruvate to fuel the citric acid (TCA) cycle and oxidative phosphorylation. This is an efficient method of generating energy, since it theoretically results in 38 ATP molecules/glucose molecule used. However, under hypoxic conditions the cell's energy demand is met by glycolysis and conversion of pyruvate to lactate, rather than channeling into the citric acid cycle. This process produces only two ATP molecules from one glucose molecule, and is therefore an inefficient process, as compared to oxidative phosphorylation. Although macrophages and dendritic cells depend heavily on glycolysis for ATP production even under normal oxygen tension, these cells adapt to low oxygen tension by increasingly utilizing the anaerobic glycolytic pathway for ATP production (Citation34,Citation35). In isolated mouse peritoneal macrophages, glucose contributes to ATP production to a greater extent than does oleate (Citation36), and almost all of the utilized glucose is converted into lactate through glycolysis, whereas only ∼3% of glucose is oxidized under oxygenated conditions (Citation37). Furthermore, hypoxia stimulates glycolysis and prolongs cell survival in mouse bone-marrow-derived macrophages and human monocyte-derived macrophages (Citation38). These data suggest that glucose utilization and the glycolytic pathway might influence the activities of macrophages at sites of inflammation, such as atherosclerotic lesions (Citation39,Citation40).
Before a necrotic core has started to form, a large atherosclerotic lesion typically has a macrophage-rich core that has a high metabolic rate, and these macrophages are often restricted to anaerobic metabolism (Citation41). In fact, a high degree of anaerobic glycolysis was observed in rabbit lesions, in which high lactate production and high glucose consumption were seen in the core of advanced lesions (Citation42). The high glucose consumption is predicted to result in a reduction of glucose levels in the core of poorly vascularized large lesions, but absolute levels of glucose in macrophage-rich lesions in non-diabetic and diabetic settings are difficult to assess. Emerging imaging technology, using fluorine-18-labeled-2-deoxy-D-glucose positron emission tomography (FDG-PET) has also shed light onto the metabolic features of lesion macrophages. A number of studies have reported an increased uptake of FDG in atherosclerotic lesions (Citation43). There is a general agreement from these studies that the arterial FDG signal is proportional to the macrophage content of the lesion, although a few negative reports exist, presumably due to the non-specific nature of FDG-PET imaging (Citation43). High FDG uptake in atherosclerotic lesions has also been demonstrated in human subjects (Citation44). Generally, the degree of lesion FDG uptake increases with the number of cardiovascular risk factors, including diabetes (Citation43,Citation45).
Cellular glucose uptake is mediated by a family of facilitated diffusion glucose transporters, the GLUT or SLC2A family, and of these GLUT1 through 4 have specific and well established roles in glucose homeostasis (Citation46). Of these four GLUT isoforms, monocytes/macrophages from different species have been shown to express GLUT1 and GLUT3 (Citation47–49), with significant increases in GLUT1 expression during differentiation (Citation47,Citation48). GLUT4, the main insulin-stimulated glucose transporter (Citation46), is abundantly expressed in skeletal muscle and adipose tissue and is not observed in human monocyte/macrophages (Citation47). Glucose uptake in macrophages is therefore not critically dependent on insulin.
Once glucose has entered the cell, most of it is believed to be channeled into glycolysis. Glycolysis is regulated by three major enzymes: hexokinase, phosphofructokinase 1 (PFK1), and pyruvate kinase, which mediate irreversible steps in the pathway (). Hexokinase converts glucose to glucose-6-phosphate. Glucose-6-phosphate is then converted to fructose-6-phosphate by glucose-6-phosphate isomerase and then further to glucose-1,6-bisphosphate by PFK1. PFK1 is a rate-limiting enzyme in glycolysis and is regulated by several factors. Increased levels of ATP and citrate inhibit PFK1 activity, whereas ADP, AMP, and fructose-2,6-bisphosphate act as allosteric activators of PFK1. Importantly, fructose-2,6-bisphosphate, a strong activator of PFK1, is generated from fructose-6-phosphate by PFK2. PFK2 contains both kinase and phosphatase domains. Under low glucose conditions, the phosphatase is active, thus lowering fructose-2,6-bisphosphate levels, which in turn slows glycolysis. When glucose levels are high, the kinase domain is active, producing more fructose-2, 6-bisphosphate, which stimulates glycolysis. There are four subtypes of PFK2 reported in higher mammals, and each subtype has its own net activities and ratios of kinase/bisphosphatase activity (Citation50,Citation51). Therefore, the net activity of PFK2 is believed to influence largely the activity of PFK1 and, consequently, the activity of glycolysis.
Several of the glycolytic proteins are highly expressed or activated in macrophages. For example, the enzymatic activity of hexokinase in mouse peritoneal macrophages is reported to be high (Citation52). Hypoxia-inducible factor-1 (HIF-1) is a key regulator of the expression of genes necessary for adaptation to hypoxic conditions. Most of the glycolytic pathway-related genes are induced by HIF-1, including GLUT1, hexokinase, PFK, and phosphoglycerate kinase 1 (Citation53,Citation54). Loss of HIF-1α in macrophages by targeted disruption of the gene resulted in the reduction of glycolytic activity and ATP production to 20% of that observed within wild-type cells (Citation55). The activity of HIF-1 is regulated by factor inhibiting HIF-1 (FIH-1) and prolylhydroxylases. FIH-1 and prolylhydroxylases are oxygen sensors, and in most cells HIF activity is strongly suppressed by the action of these proteins under normoxic conditions (Citation56–59). However, macrophages and dendritic cells primarily use glycolysis for ATP production even under normoxic conditions. Recently it was demonstrated that in bone-marrow-derived macrophages, the adapter protein Mint3/APBA3 binds to FIH-1 and suppresses the activity of this protein, consequently enhancing the activity of HIF-1 (Citation60). Furthermore, membrane type 1 matrix metalloproteinase, a potent invasion-promoting protease expressed in macrophages, was shown to bind FIH-1 and lead to the inhibition of this protein even under normoxic conditions through a Mint3/APBA3-dependent pathway (Citation61).
Together, these studies demonstrate that macrophages utilize primarily glycolysis for energy production under both hypoxic and normoxic conditions.
There is a strong connection between glucose metabolism and a pro-inflammatory phenotype in macrophages
Recent research has revealed that pro-inflammatory activation of macrophages regulates their energy metabolism as well as inflammatory phenotype and that fuel utilization and inflammatory phenotype are closely related in these cells. Activation of macrophages has been classified into different states or populations in vitro, depending on the stimulus (Citation62,Citation63). The classical (M1) activation results in a highly pro-inflammatory macrophage phenotype, is a feature of microbicidal activity and pro-inflammatory cytokine production, and is mediated by like Toll-like receptor (TLR)-4 ligands and interferon-γ (IFN-γ). The alternative (M2) activation is a feature of a less inflammatory reaction, tissue repair and humoral immunity, and is thought to be mediated by IL-4 and/or IL-13. It was recently demonstrated that there is a clear difference in the flux of glucose metabolism between the classical activation and alternative activation (Citation64,Citation65). Interestingly, inflammatory classically activated macrophages are more dependent on glucose as a substrate, whereas the less inflammatory alternatively activated macrophage population is more dependent on fatty acid metabolism (Citation64). Although the glycolytic pathway gives a lower ATP yield than the TCA cycle/oxidative phosphorylation, glycolysis is believed to be able rapidly to provide ATP to macrophages, which need to meet the increased energy demand associated with defense against pathogens/infection (Citation64). Furthermore, pharmacological inhibitor studies have revealed that mitochondrial respiration is required for the alternatively activated macrophage phenotype, but not for the classically activated macrophage phenotype (Citation64). A similar increase in aerobic glycolysis has recently been observed in dendritic cells stimulated with TLR ligands (Citation35).
How do cytokines and pathogens promote glycolysis? It is possible that part of the effect is due to an increased expression of GLUT1 (Citation66), which would be likely to increase glucose uptake and subsequent glycolysis. Furthermore, an inducible form of PFK2 (PFKFB3) has caught attention because this enzyme is induced by hypoxia, inflammatory cytokines, and pathogens alike (Citation67). Classical activation of macrophages shifts the isoform expression of PFK2 from the liver type PFK2, which a has low net activity, to the more active ubiquitous PFK2 (PFKFB3), which keeps fructose-2,6-bisphosphate concentration at a high level due to its lower bisphosphatase activity, as compared to the liver type PFK2 (Citation68,Citation69). Consequently, this shift potentiates glycolytic ATP production to be utilized for pro-inflammatory responses in the classically activated macrophages. The PFKFB3 promoter contains putative HIF-binding sites, as well as an NF-κB site (Citation67). The effect of cytokines on induction of PFKFB3 does not appear to be due to HIF activation (Citation65) and is likely to be mediated by the NF-κB pathway. The increased glucose utilization in macrophages exposed to pro-inflammatory mediators could therefore be due in part to an increased PFKFB3 expression. The TLR4 ligand lipopolysaccharide (LPS) has also been shown to increase expression of glucose-6-phosphate dehydrogenase (G6PD) in macrophages (Citation70). G6PD therefore provides an additional enzyme induced by pro-inflammatory mediators, and its induction might increase glucose flux through the pentose phosphate pathway ().
The findings above demonstrate that pro-inflammatory molecules stimulate glucose metabolism/glycolysis in macrophages. Conversely, increased glucose uptake in macrophages can promote or contribute to a pro-inflammatory phenotype, suggesting that a pro-inflammatory environment and increased glucose levels might enhance each other in a vicious or self-perpetuating cycle. For example, recent studies suggest that down-regulation of PFKFB3 results in reduced expression of markers for the classically activated macrophage phenotype (Citation65), indicating that PFKFB3 activity promotes a pro-inflammatory response. However, PFKFB3 +/− mice exhibit increased insulin resistance, and macrophages isolated from the adipose tissue of these mice showed an increased production of cytokines (Citation71), possibly mediated by the pro-inflammatory in-vivo environment. The role of PFKFB3 induction in mediating increased inflammatory activation of macrophages therefore needs further study.
Consistent with the findings that glucose is poorly utilized in mitochondrial respiration in pro-inflammatory macrophages, pharmacological inhibition of mitochondrial respiration does not alter the macrophage's pro-inflammatory phenotype (Citation64), suggesting that glucose metabolism through mitochondrial respiration does not mediate the pro-inflammatory effects of glucose ().
In addition to the glycolytic pathway, increased glucose uptake might result in regulation of the macrophage's inflammatory phenotype, e.g. through the pentose phosphate pathway, the polyol/sorbitol pathway, or the hexosamine pathway (). Increased expression of G6PD increases glucose flux through the pentose phosphate pathway in macrophages, which associates with an enhanced inflammatory response in obesity (Citation72). LPS stimulation leads to G6PD up-regulation in macrophages, and over-expression of G6PD increases the expression of inflammatory markers, such as MCP-1, IL-6, and iNOS (Citation72). Mononuclear cells from G6PD-deficient human subjects produce reduced levels of cytokines (Citation73), further suggesting that this enzyme or downstream processes have a pro-inflammatory effect. G6PD-deficient mice have been shown to have reduced atherosclerosis, although the role of macrophages in this response is unknown (Citation74).
Likewise, the polyol/sorbitol pathway can mediate increased production of pro-inflammatory mediators in macrophages (Citation75). Aldose reductase, a key enzyme in this pathway, has been shown to promote LPS-induced pro-inflammatory responses in mouse macrophages. Inhibition or ablation of the activity of aldose reductase resulted in marked suppression of LPS-induced production of inflammatory cytokines (Citation75), whereas over-expression of aldose reductase resulted in increased expression of pro-inflammatory mediators in macrophages (Citation15). Furthermore, aldose reductase expression and activity are increased by foam cell formation induced by oxidized LDL in human monocyte-derived macrophages, and these effects are potentiated by high glucose conditions (Citation76). Importantly, both the polyol/sorbitol pathway and the pentose phosphate pathway feed into glycolysis, and these pathways are therefore closely related to glycolysis ().
There is a little information regarding the effect of the hexosamine biosynthesis pathway on macrophage inflammation, but one study concluded that this pathway does not significantly alter LPS-induced iNOS expression, a marker of classically activated macrophages (Citation77). The contribution of the hexosamine pathway in pro-inflammatory responses requires further study.
Together, these studies suggest that a glycolytic intermediate(s), perhaps with contribution from the polyol/sorbitol pathway and the pentose phosphate pathway, is the most likely mediator of the pro-inflammatory effects of glucose in macrophages (). The exact mechanisms whereby this might occur need further study.
Hyperglycemia may activate a pro-inflammatory program by mimicking the increased glycolysis induced by cytokines and pathogens in macrophages
Several human and animal studies show that monocytes/macrophages take on a pro-inflammatory phenotype in the setting of type 1 diabetes (Citation78–81), and this effect might be mediated, at least in part, by increased glucose levels (Citation80). It is tempting to speculate that diabetes, through hyperglycemia and subsequently increased macrophage glucose uptake, mimics the stimulatory effects of cytokines and pathogens on glycolysis in macrophages, thereby promoting a pro-inflammatory response in these cells, which would likely promote atherosclerosis. In addition, diabetes is often associated with a pro-inflammatory environment, which could in turn further promote macrophage glucose metabolism. Studies on new mouse models, for example models in which GLUT1 or glycolytic enzymes are modulated specifically in macrophages, and ultimately human studies, will be required to gain further understanding of the role of glucose in vascular cells and atherosclerosis in vivo.
Acknowledgements
This work was supported in part by NIH grants HL062887, HL092969 (Project 2), and HL097365 to KEB.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Poor glycemic control predicts coronary heart disease events in patients with type 1 diabetes without nephropathy. Arterioscler Thromb Vasc Biol. 1999;19:1014–9.
- Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, . Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–53.
- Eeg-Olofsson K, Cederholm J, Nilsson PM, Zethelius B, Svensson A-M, Gudbjörnsdóttir S, . Glycemic control and cardiovascular disease in 7,454 patients with type 1 diabetes. An observational study from the Swedish National Diabetes Register (NDR). Diabetes Care. 2010;33:1640–6.
- Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial (DCCT). Am J Cardiol. 1995;75:894–903.
- Orchard TJ, Olson JC, Erbey JR, Williams K, Forrest KY, Smithline Kinder L, . Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2003;26:1374–9.
- Prince CT, Becker DJ, Costacou T, Miller RG, Orchard TJ. Changes in glycaemic control and risk of coronary artery disease in type 1 diabetes mellitus: findings from the Pittsburgh Epidemiology of Diabetes Complications Study (EDC). Diabetologia. 2007;50:2280–8.
- Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J, . Common variants at 10 genomic loci influence hemoglobin A1C levels via glycemic and nonglycemic pathways. Diabetes. 2010;59:3229–39.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–53.
- Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, . Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, . Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72.
- Zoungas S, Patel A, Chalmers J, de Galan BE, Li Q, Billot L, . Severe hypoglycemia and risks of vascular events and death. N Engl J Med. 2010;363:1410–8.
- Kannel WB, McGee DL. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care. 1979;2:120–6.
- Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, . Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114: 659–68.
- Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, Tall AR, . Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest. 2005;115:2434–43.
- Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, . Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci USA. 2008;105:2082–7.
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, . Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–17.
- Duff GL, Payne TP. The effect of alloxan diabetes on experimental cholesterol atherosclerosis in the rabbit. III. The mechanism of the inhibition of experimental cholesterol atherosclerosis in alloxan-diabetic rabbits. J Exp Med. 1950; 92:299–317.
- Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001; 50:1654–65.
- Kanter JE, Johansson F, LeBoeuf RC, Bornfeldt KE. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ Res. 2007;100:769–81.
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr, Chow WS, . Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–31.
- Reaven P, Merat S, Casanada F, Sutphin M, Palinski W. Effect of streptozotocin-induced hyperglycemia on lipid profiles, formation of advanced glycation endproducts in lesions, and extent of atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 1997;17:2250–6.
- Dixon JL, Shen S, Vuchetich JP, Wysocka E, Sun GY, Sturek M. Increased atherosclerosis in diabetic dyslipidemic swine: protection by atorvastatin involves decreased VLDL triglycerides but minimal effects on the lipoprotein profile. J Lipid Res. 2002;43:1618–29.
- Adhikari N, Basi DL, Carlson M, Mariash A, Hong Z, Lehman U, . Increase in GLUT1 in smooth muscle alters vascular contractility and increases inflammation in response to vascular injury. Arterioscler Thromb Vasc Biol. 2011;31:86–94.
- Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, . Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–9.
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, . Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest. 2008;118:183–94.
- Preusch MR, Morris-Rosenfeld S, Bierhaus A, Albrecht C, Erwin Blessing E, Andrassy M, . Deletion of bone marrow derived receptor for advanced glycation end-products (RAGE) does not attenuate plaque formation but inhibits plaque progression in a mouse model of advanced atherosclerosis. Circulation. 2009;120:S1137–8, Abstract 5672.
- Booth G, Stalker TJ, Lefer AM, Scalia R. Elevated ambient glucose induces acute inflammatory events in the microvasculature: effects of insulin. Am J Physiol Endocrinol Metab. 2001;280:E848–56.
- Azcutia V, Abu-Taha M, Romacho T, Vázquez-Bella M, Matesanz N, Luscinskas FW, . Inflammation determines the pro-adhesive properties of high extracellular d-glucose in human endothelial cells in vitro and rat microvessels in vivo. PLoS One. 2010;5:e10091.
- Gerrity RG, Naito HK, Richardson M, Schwartz CJ. Dietary induced atherogenesis in swine. Morphology of the intima in prelesion stages. Am J Pathol. 1979;95:775–92.
- Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206:2141–9.
- Hansch A, Stiehl P, Geiler G. [Quantification of macrophages and granulocytes at the joint cartilage-pannus junction in rheumatoid arthritis]. Z Rheumatol. 1996;55: 401–9.
- Hunt TK, Knighton DR, Thakral KK, Goodson WH 3rd, Andrews WS. Studies on inflammation and wound healing: angiogenesis and collagen synthesis stimulated in vivo by resident and activated wound macrophages. Surgery. 1984;96:48–54.
- Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900.
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, . Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–9.
- Newsholme P, Newsholme EA. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem J. 1989;261:211–8.
- Newsholme P, Gordon S, Newsholme EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J. 1987;242: 631–6.
- Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, . Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol. 2009;182: 7974–81.
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–26.
- Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–15.
- Björnheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999;19:870–6.
- Leppänen O, Björnheden T, Evaldsson M, Borén J, Wiklund O, Levin M. ATP depletion in macrophages in the core of advanced rabbit atherosclerotic plaques in vivo. Atherosclerosis. 2006;188:323–30.
- Hiari N, Rudd JH. FDG PET imaging and cardiovascular inflammation. Curr Cardiol Rep. 2011;13:43–8.
- Chen W, Bural GG, Torigian DA, Rader DJ, Alavi A. Emerging role of FDG-PET/CT in assessing atherosclerosis in large arteries. Eur J Nucl Med Mol Imaging. 2009;36: 144–51.
- Kim TN, Kim S, Yang SJ, Yoo HJ, Seo JA, Kim SG, . Vascular inflammation in patients with impaired glucose tolerance and type 2 diabetes: analysis with 18F-fluorodeoxyglucose positron emission tomography. Circ Cardiovasc Imaging. 2010;3:142–8.
- Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141–5.
- Malide D, Davies-Hill TM, Levine M, Simpson IA. Distinct localization of GLUT-1, -3, and -5 in human monocyte-derived macrophages: effects of cell activation. Am J Physiol. 1998;274:E516–26.
- Fu Y, Maianu L, Melbert BR, Garvey WT. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: a role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol Dis. 2004;32:182–90.
- Chang M, Hamilton JA, Scholz GM, Masendycz P, Macaulay SL, Elsegood CL. Phosphatidylinostitol-3 kinase and phospholipase C enhance CSF-1-dependent macrophage survival by controlling glucose uptake. Cell Signal. 2009;21: 1361–9.
- Okar DA, Manzano A, Navarro-Sabatè A, Riera L, Bartrons R, Lange AJ. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci. 2001;26:30–5.
- Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. 2004;381:561–79.
- Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. 1986; 239:121–5.
- Aragonés J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9:11–22.
- Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13.
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, . HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57.
- Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1α. Mol Cell Biol. 2005;25: 6415–26.
- Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22:4082–90.
- Dann CE 3rd, Bruick RK, Deisenhofer J. Structure of factor-inhibiting hypoxia-inducible factor 1: an asparaginyl hydroxylase involved in the hypoxic response pathway. Proc Natl Acad Sci U S A. 2002;99:15351–6.
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71.
- Sakamoto T, Seiki M. Mint3 enhances the activity of hypoxia-inducible factor-1 (HIF-1) in macrophages by suppressing the activity of factor inhibiting HIF-1. J Biol Chem. 2009;284:30350–9.
- Sakamoto T, Seiki M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J Biol Chem. 2010; 285:29951–64.
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64.
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6.
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, . Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24.
- Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, . Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–14.
- Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun. 1996;64:108–12.
- Obach M, Navarro-Sabaté À, Caro J, Kong X, Duran J, Gómez M, . 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem. 2004;279:53562–70.
- Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg's effect. J Bioenerg Biomembr. 2007;39:223–9.
- Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, . Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277:6183–7.
- Spolarics Z. Endotoxin stimulates gene expression of ROS-eliminating pathways in rat hepatic endothelial and Kupffer cells. Am J Physiol. 1996;270:G660–6.
- Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, . Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J Biol Chem. 2010;285:3713–21.
- Ham M, Lee J-W, Choi AH, Masuzaki H, Kim JB. Increase of glucose-6-phosphate dehydrogenase in macrophage is associated with enhanced inflammatory responses in obesity. FASEB J. 2008;22:615.
- Sanna F, Bonatesta RR, Frongia B, Uda S, Banni S, Melis MP, . Production of inflammatory molecules in peripheral blood mononuclear cells from severely glucose-6-phosphate dehydrogenase-deficient subjects. J Vasc Res. 2007;44:253–63.
- Matsui R, Xu S, Maitland KA, Mastroianni R, Leopold JA, Handy DE, . Glucose-6-phosphate dehydrogenase deficiency decreases vascular superoxide and atherosclerotic lesions in apolipoprotein E(-/-) mice. Arterioscler Thromb Vasc Biol. 2006;26:910–6.
- Ramana KV, Fadl AA, Tammali R, Reddy ABM, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. 2006;281: 33019–29.
- Gleissner CA, Sanders JM, Nadler J, Ley K. Upregulation of aldose reductase during foam cell formation as possible link among diabetes, hyperlipidemia, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28: 1137–43.
- Woo HG, Jung YS, Baik EJ, Moon CH, Lee SH. The enhancement of endotoxin-induced nitric oxide production by elevation of glucose concentration in macrophage. Korean J Physiol Pharmacol. 1999;3:447–54.
- Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006; 55:774–9.
- Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, . Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol. 2009;183:4432–9.
- Wen Y, Gu J, Li SL, Reddy MA, Natarajan R, Nadler JL. Elevated glucose and diabetes promote interleukin-12 cytokine gene expression in mouse macrophages. Endocrinology. 2006;147:2518–25.
- Padmos RC, Schloot NC, Beyan H, Ruwhof C, Staal FJ, de Ridder D, . Distinct monocyte gene-expression profiles in autoimmune diabetes. Diabetes. 2008;57: 2768–73.