Abstract
Mitochondria are essential organelles with multiple functions, the most well known being the production of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). The mitochondrial diseases are defined by impairment of OXPHOS. They are a diverse group of diseases that can present in virtually any tissue in either adults or children. Here we review the main molecular mechanisms of mitochondrial diseases, as presently known. A number of disease-causing genetic defects, either in the nuclear genome or in the mitochondria's own genome, mitochondrial DNA (mtDNA), have been identified. The most classical genetic defect causing mitochondrial disease is a mutation in a gene encoding a structural OXPHOS subunit. However, mitochondrial diseases can also arise through impaired mtDNA maintenance, defects in mitochondrial translation factors, and various more indirect mechanisms. The putative consequences of mitochondrial dysfunction on a cellular level are discussed.
| Abbreviations | ||
| ad | = | autosomal-dominant |
| ar | = | autosomal-recessive |
| AS | = | Alpers syndrome |
| ATP | = | adenosine triphosphate |
| CI | = | complex I |
| CII | = | complex II |
| CIII | = | complex III |
| CIV | = | complex IV |
| CV | = | complex V |
| CoQ10 | = | coenzyme Q |
| COX | = | cytochrome c oxidase |
| dATP | = | deoxyadenosine triphosphate |
| dCTP | = | deoxycytidine triphosphate |
| dGK | = | deoxyguanosine kinase |
| dGTP | = | deoxyguanosine triphosphate |
| dNTP | = | deoxynucleoside triphosphate |
| dTTP | = | thymidine triphosphate |
| GRACILE | = | growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death |
| IMM | = | inner mitochondrial membrane |
| IMS | = | inter-membrane space |
| IOSCA | = | Infantile-onset spinocerebellar ataxia |
| KSS | = | Kearns–Sayre syndrome |
| LHON | = | Leber's hereditary optic neuropathy |
| LS | = | Leigh syndrome |
| LSBL | = | leukoencephalopathy with brain-stem and spinal cord involvement and lactate elevation |
| MDS | = | mitochondrial DNA depletion syndrome |
| MELAS | = | mitochondrial encephalopathy with lactic acidosis and stroke-like episodes |
| MERRF | = | myoclonic epilepsy with ragged-red fibers |
| MIRAS | = | mitochondrial recessive ataxia syndrome |
| MLASA | = | mitochondrial myopathy, lactic acidosis, and sideroblastic anemia |
| MNGIE | = | mitochondrial neurogastrointestinal encephalopathy |
| MRP | = | mitochondrial ribosomal protein |
| mtDNA | = | mitochondrial DNA |
| NARP | = | neurogenic weakness, ataxia, retinitis pigmentosa |
| nDNA | = | nuclear DNA |
| OMM | = | outer mitochondrial membrane |
| OXPHOS | = | oxidative phosphorylation |
| PEO | = | progressive external ophthalmoplegia |
| PS | = | Pearson's syndrome |
| Q | = | ubiquinone (oxidized) |
| QH2 | = | ubiquinone (reduced) |
| RC | = | respiratory chain |
| RNR | = | ribonucleotide reductase |
| ROS | = | reactive oxygen species |
| SANDO | = | sensory ataxic neuropathy, dysarthria, and ophthalmoparesis |
| SCA-E | = | spinocerebellar ataxia with epilepsy |
| TK2 | = | thymidine kinase 2 |
| TP | = | thymidine phosphorylase |
Key messages
Mitochondrial diseases can manifest in any organ at any age. They are caused by mutations in the genes of mitochondrial DNA or nuclear DNA, and their inheritance can be autosomal-dominant or recessive, X-linked, or maternal.
Many of the genes that are mutated in mitochondrial diseases encode proteins that are structural components or assembly factors of the OXPHOS complexes. Others encode proteins involved in mtDNA maintenance or mitochondrial protein synthesis. In addition, OXPHOS dysfunction can arise through indirect mechanisms.
The metabolic consequences of mitochondrial dysfunction are incompletely understood, both on the level of the cell and the level of the organism, and cannot be explained by mere ATP deficiency.
Mitochondrial diseases are among the most common of the inherited disorders of metabolism (reviewed in (Citation1)). For the diseases caused by mutations in mitochondrial DNA (mtDNA), a population prevalence of at least 9.2 per 100,000 people was measured in the working-age population of north-east England. In addition, 16.2 per 100,000 people were identified as asymptomatic mtDNA mutation carriers who are at risk of developing mitochondrial disease (Citation2). These figures did not include mitochondrial diseases caused by nuclear gene defects, and thus the total prevalence of mitochondrial diseases is thought to be higher, at least in the order of 1 in 5,000 people.
The mitochondrial diseases may cause symptoms in any organ and present at any age (reviewed in (Citation3,Citation4)). A central function of mitochondria is production of the cellular energy currency adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). OXPHOS is carried out by five enzyme complexes in the inner mitochondrial membrane (IMM) (). Apart from energy conversion, mitochondria take part in a number of other processes including heme biosynthesis, steroid hormone biosynthesis, calcium homeostasis, and apoptosis (reviewed in (Citation5)). Mitochondrial diseases are generally understood as diseases where one or more of the OXPHOS complexes are dysfunctional. This review focuses on the various molecular mechanisms behind these diseases.
Figure 1. This figure shows human mtDNA represented by a circle. The positions of each of the protein-coding genes as well as the 12S and 16S rRNA genes and the tRNA genes are indicated. The OXPHOS system is shown above in simplified form. The protein-coding genes are color-coded according to the complex to which their product belongs. The non-coding D-loop region is also shown. The two strands of mtDNA are divided into a heavy strand and a light strand. The regions known as origins of heavy and light strand replication (OH and OL, respectively) are indicated, as are the transcription promoters for heavy and light strands (HSP and LSP, respectively). In mitochondrial energy conversion, acetyl-coenzyme A (CoA), which can be derived from pyruvate produced in cytoplasmic glycolysis or from mitochondrial fatty acid oxidation, conveys the carbon atoms of the acetyl group into the tricarboxylic acid (TCA) cycle. The enzymes of the TCA cycle oxidize these carbons and generate high-energy electrons that are transferred to the electron carriers nicotinamide adenine dinucleotide (NADH (reduced), NAD+ (oxidized)), or flavin adenine dinucleotide (FADH2 (reduced), FAD (oxidized)). The OXPHOS system comprises five large enzyme complexes, labeled complex I-V. All OXPHOS complexes are embedded in the inner mitochondrial membrane (IMM). Complexes I–IV together constitute the respiratory chain (RC), which uses energy released during electron transfer to pump protons from the matrix into the inter-membrane space (IMS). Complex I (CI) is a NADH oxidase, and complex II (CII) is a succinate dehydrogenase. CII is also a TCA cycle enzyme that oxidizes succinate and uses FADH2 as the electron carrier. It is the only RC complex that does not pump protons and that does not contain mtDNA-encoded subunits. Both CI and CII transfer electrons onward to the lipid-soluble electron carrier ubiquinone (coenzyme Q10, CoQ10, or Q (oxidized), QH2 (reduced)) inside the IMM. QH2 is oxidized by complex III (CIII), and electrons are transferred to cytochrome c (Cyt c), which is a water-soluble electron carrier in the IMS. Complex IV (CIV) catalyzes the final transfer of electrons to molecular oxygen to generate water. The combined action of the RC generates an electrochemical potential difference across the IMM. This is utilized by ATP synthase or complex V (CV) to catalyze the phosphorylation of ADP to ATP.
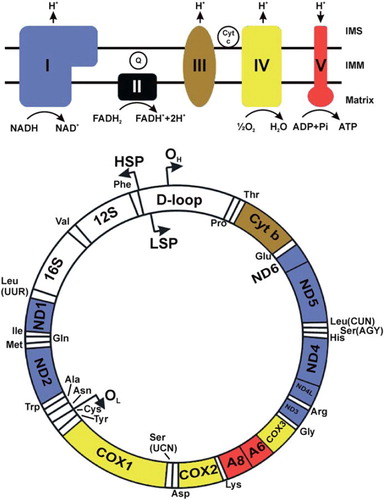
Mitochondria are surrounded by two membranes. The outer mitochondrial membrane (OMM) contains large amounts of porin (also known as voltage-dependent anion channel or VDAC), which allows passage of small hydrophilic molecules such as sugars, ions, and amino acids. The IMM is impermeable to most solutes. It has numerous folds called cristae, and it encloses the mitochondrial matrix. Mitochondria are dynamic organelles that can move, fuse, and divide according to the needs of the cell. In fact, in many cell types, it is often more appropriate to regard the mitochondria as a dynamic network rather than multiple discrete organelles (reviewed in (Citation5)).
Genetics of mitochondrial disease
Mitochondrial diseases can be inherited by maternal, autosomal, or X-chromosomal transmission. This exceptional genetic variability is due to the unique genetic control of the organelle, which depends on two separate genomes. Mitochondria contain their own genome, called mtDNA. Human mtDNA is an intron-less multi-copy, circular 16,569 bp molecule that is exclusively inherited via the maternal line (Citation6). Mitochondrial DNA encodes 13 proteins, each of which is an essential subunit of one of the OXPHOS complexes (). In addition, mtDNA encodes 22 tRNAs and 2 rRNAs required for the translation of these proteins (Citation6). Primary or secondary defects of mtDNA are an important cause of mitochondrial disease, often leading to one of several classical mtDNA syndromes (). The mtDNA syndromes are sporadic or maternally inherited, when the primary defect is a mutation or deletion in mtDNA, and autosomally inherited when the mtDNA defect is secondary to a mutation in a nuclear gene that is required for mtDNA maintenance. However, most of the mitochondrial proteins, including the majority of OXPHOS complex subunits and assembly factors, are encoded by the nuclear DNA (nDNA) (Citation7). Therefore, the majority of the mitochondrial diseases are caused by defects in nuclear genes and are inherited according to their chromosomal location. A mitochondrial disease may involve isolated dysfunction of only one OXPHOS complex or combined dysfunction of several complexes. The latter results for instance from defects in mtDNA maintenance, mitochondrial protein synthesis, or mitochondrial protein importation.
Table I. This table lists the common mitochondrial syndromes and the main organs that are affected in each case. The syndromes are grouped according to their typical molecular etiology. MELAS, MERRF, NARP, and LHON are caused by mtDNA point mutations. KSS, PS, and PEO are caused by single mtDNA deletions that are either sporadic or maternally inherited; ad/ar PEO, MDS, MNGIE, MIRAS, and AS are caused by defective mtDNA due to mutations in nuclear genes responsible for mtDNA maintenance. LS is typically caused by mutations in mitochondrial or nuclear genes encoding structural proteins or assembly factors of the OXPHOS complexes. The inheritance of LS may thus be maternal, autosomal, or X-linked. Adapted from reference (Citation4).
Mitochondrial DNA genetics
Inheritance of mtDNA mutations is different from that of nDNA mutations because mtDNA is a multi-copy genome with hundreds or thousands of copies per cell, and because mtDNA is only inherited from the mother. In most cases, all of the mtDNA molecules within the cells of an individual are identical. This situation is known as homoplasmy. However, in mtDNA diseases, the patients are often heteroplasmic, meaning that they have two different mtDNA populations. In general, disease severity correlates with the relative proportion of mutated to intact mtDNA. A heteroplasmic person will only develop the disease if the mutant load exceeds a certain threshold. The threshold may be different for different mutations and different tissues.
The uniparental inheritance pattern of mtDNA means that it is in theory possible that mutations accumulate slowly over successive generations until, eventually, a threshold mutant level is reached that manifests as a disease (reviewed in (Citation8)). Therefore, a mechanism is required to prevent the transmission of defective mtDNA to offspring. This protection has been attributed to a genetic bottle-neck of mtDNA in developing primordial germ cells (reviewed in (Citation8)), meaning that only a small subset of the mtDNAs of the original oocyte populate the germ cells that will become the next generation. The bottle-neck may result from a strong reduction of mtDNA copy number in primordial germ cells, followed by amplification and random segregation of mtDNA species as the germ cells divide (Citation9). Alternatively, the bottle-neck may be caused by replication of only a subset of mtDNAs in primary oocytes that undergo folliculogenesis (Citation10). Because of the bottle-neck, heteroplasmy levels can shift rapidly between generations. Therefore, members of the same family may exhibit striking clinical variability. Evidence from mouse models of mtDNA disease suggests that severely defective mtDNA molecules can be selected against during germ cell or oocyte development (Citation11,Citation12). This purifying selection may explain why moderately pathogenic mtDNA mutations that do not cause complete inhibition of OXPHOS and lead to late-onset disease are more common than highly deleterious mtDNA mutations (Citation11).
Mitochondrial DNA mutations are also known to occur sporadically in somatic tissues. The mutations may undergo clonal expansion, eventually reaching the threshold level and causing mitochondrial dysfunction at a later age. Accumulating mtDNA mutations, and subsequent progressive mitochondrial dysfunction, are thought to contribute to the normal aging process (reviewed in (Citation13)).
Defects of structural OXPHOS subunits and assembly factors
The OXPHOS complexes are made up of at least 89 structural protein subunits encoded by either mtDNA or nDNA. In addition, a number of nDNA-encoded assembly factors are needed for the intricate assembly processes of the complexes. Defects in a number of structural and assembly proteins are known to cause mitochondrial disease (reviewed in (Citation14)). The diseases present with either isolated or combined complex deficiencies, and their clinical manifestations are diverse ().
Table II. This table lists the disease-associated genes that encode either structural or assembly factors of the OXPHOS complexes. Note that structural proteins are encoded by either nDNA or mtDNA, whereas assembly factors, i.e. proteins needed to make the complex but that are not part of the final complex, are only encoded by nDNA.
Complex I deficiency
Isolated complex I deficiency is among the most common forms of mitochondrial disease. Complex I, also known as NADH dehydrogenase or NADH:quinone reductase, is a ∼980 kDa structure composed of 45 subunits, of which 7 are encoded by mtDNA and 38 by nDNA (Citation15). The complex is L-shaped, with a hydrophobic arm embedded in the IMM and a perpendicular hydrophilic peripheral arm that protrudes into the matrix. The conserved structures of both subunits from bacteria and yeast have been characterized recently (Citation16–19). CI has three functional modules: a dehydrogenase module that oxidizes NADH, a hydrogenase module that reduces ubiquinone, and a proton-transferring module that pumps protons. The latter module constitutes much of the membrane arm and also contains all of the mtDNA-encoded subunits. Electron transfer in the hydrophilic arm induces conformational changes that push an α-helix that is positioned in a parallel orientation within the membrane arm, and whose movement tilts other helices in the membrane arm to allow the passage of protons from the matrix side to the inter-membrane space (IMS) (Citation17,Citation18).
CI deficiency is associated with various phenotypes both in children and adults. Many patients have Leigh syndrome (LS), which is characterized clinically by a severe symptom constellation including developmental delay and failure to thrive, and radiologically or histologically by typical necrotic lesions in the basal ganglia and brain-stem (reviewed in (Citation4)). Others present with fatal infantile lactic acidosis, neonatal cardiomyopathy, leukoencephalopathy, myopathy, combined tubulopathy and hepatopathy, or less well defined phenotypes (reviewed in (Citation20)). Another classical CI-related disorder is Leber's hereditary optic neuropathy (LHON), which is an adult-onset disease affecting almost exclusively the optic nerve. The first disease-causing mtDNA mutations were described in LHON patients, in the gene encoding the ND4 subunit of CI (Citation21). Children with CI-related disease tend to have normal prenatal development, which may reflect a relatively low demand for mitochondrial function during embryonic life, and up-regulation of OXPHOS following birth (reviewed in (Citation20)).
Pathogenic mutations have been reported in all of the mtDNA-encoded CI subunits and a number of nDNA-encoded subunits (). Some genetic defects interfere with the insertion of the polypeptide into the complex, while others allow complex assembly but impair the enzymatic activity of the final complex (Citation22). In the former, complex assembly intermediates accumulate depending on which stage of the assembly process the defective subunit is normally inserted in (Citation23).
Two different models of human CI assembly have been suggested. In the first model, the membrane arm and peripheral arms are first assembled individually and then attached to each other. This is reminiscent of the well studied CI assembly process in the fungus Neurospora crassa (Citation23). In the second model, the subunits from both arms form subcomplexes that come together already before the process is fully finished. The second model is supported by the accumulated assembly intermediates that have been described in patients (Citation14,Citation22,Citation24). Proteins that are needed for the assembly of CI, but that are not themselves components of the final structure, are referred to as CI assembly factors. Several such proteins exist, and disease-causing mutations have so far been found in the genes encoding six of them (). Further, in many cases where CI fails to assemble, the responsible gene defects have not been found. Therefore, it is expected that more assembly factor mutations will be found in the future. Also, more information will be needed on the precise mechanisms of CI assembly.
The tissue-selectivity of clinical manifestations in CI deficiency may depend on differences in the residual level of CI activity in different tissues. The cases of CI deficiency that are caused by mtDNA mutations exhibit greater variation in severity, which can be partially accounted for by the relative load of mutant mtDNA (Citation20). Finally, CI deficiency may be important in the pathogenesis of ataxia, having been found in the mitochondrial ataxia syndromes (described under disorders of mtDNA maintenance), and a CI-deficient mouse model (Citation25).
Complex II deficiency
Succinate-coenzyme Q reductase, or CII, is different from the other OXPHOS complexes since all of its four subunits, SDH-A, SDH-B, SDH-C, and SDH-D, are encoded by nDNA. Furthermore, CII provides a physical link between OXPHOS and the citric acid cycle, as it catalyzes the transfer of electrons from the citric acid cycle intermediate succinate to the OXPHOS electron carrier ubiquinone. Homozygous or compound heterozygous mutations in the SDH-A gene cause LS (Citation26). Mutations in the other three genes predispose to tumors, specifically paragangliomas of the carotid bodies and pheochromocytomas (Citation27–29). Tumor predisposition is inherited in an autosomal-dominant manner. The mechanisms by which citric acid cycle or OXPHOS defects lead to tumor formation are not known. One possibility is that changes in the cell's metabolic state are sensed as hypoxia, leading to the activation of growth-promoting transcriptional pathways (reviewed in (Citation30)).
Complex III deficiency
Isolated CIII defects are among the least common respiratory chain (RC) disorders (reviewed in (Citation31)). The full name of complex III is coenzyme Q:cytochrome c oxidoreductase. It consists of 11 subunits, of which only cytochrome b is encoded by mtDNA. Electron transfer from QH2 to cytochrome c is achieved by a stepwise process known as the Q cycle (reviewed in (Citation32)). The Q cycle involves a partially oxidized semi-quinone intermediate that is prone to auto-oxidation, i.e. transfer of one electron to oxygen to make superoxide. Therefore, CIII is a major source of reactive oxygen species (ROS), which can damage many cellular macromolecules. Increased ROS may thus be an important feature in diseases caused by CIII deficiency (reviewed in (Citation31)).
Almost all of the known CIII subunit mutations are in the gene encoding cytochrome b. Most of the pathogenic amino acid changes are located outside of the transmembrane domains of the protein, and they affect the catalytic activity and/or assembly of CIII (reviewed in (Citation31)). The clinical consequences of defective cytochrome b are diverse: predominant symptoms may arise in optic nerve, heart, brain, or muscle. Some cytochrome b amino acid changes lead to CI deficiency, which supports the notion that CI and CIII function as a supercomplex and that supercomplex formation is dependent on intact cytochrome b (Citation33). Furthermore, supercomplex stabilization appears to depend on cardiolipin, which is a phospholipid that is unique to the IMM. Deficiency of cardiolipin causes Barth syndrome (cardioskeletal myopathy, neutropenia, and 3-methylglutaconic aciduria), probably through destabilization of respiratory supercomplexes (Citation34).
Disease-causing mutations have been identified in two nDNA genes encoding structural CIII subunits and in the BCS1L gene, which encodes an essential CIII assembly factor (). At least 20 BCS1L mutations have been reported, and they underlie a remarkably wide range of phenotypes ranging from the GRACILE syndrome (growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death) (Citation35) to the Björnstad syndrome that includes sensorineural hearing loss and hair changes known as pili torti (Citation36). The function of the BCS1L gene product is to insert the Rieske iron-sulfur protein during the late stages of complex assembly (Citation31). Accordingly, BCS1L gene mutations have been found to cause the accumulation of an almost fully assembled CIII that is severely lacking in enzymatic activity. Mutations causing more severe phenotypes have been found to induce greater ROS production, which may account for variable disease severity and organ involvement (Citation36).
Complex IV deficiency
Complex IV, or cytochrome c oxidase (COX), has 13 subunits, of which the 3 large core subunits (COX1, COX2, and COX3) that are responsible for electron transfer are mtDNA-encoded. The 10 nDNA-encoded subunits are not directly involved in catalysis but are apparently needed for stability and regulation of the complex (reviewed in (Citation14)).
Similar to the other RC complexes, COX-deficiency is responsible for a diverse number of clinical presentations. Mutations in the COX1-3 genes of mtDNA cause single-organ or multi-organ manifestations, such as myopathy or MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes), respectively. The mutations have been found to alter the assembly or stability of the complex. The regulation of COX activity is complex, involving tissue-specific isozymes, phosphorylation, and allosteric regulation. Therefore, tissue variability of regulatory factors is a potential explanation for the observed clinical diversity (reviewed in (Citation37)).
The assembly of CIV is a stepwise process that is estimated to require more than 20 nuclear-encoded factors that are not part of the final holoenzyme (reviewed in (Citation14)). Defects in a number of these factors are associated with variable diseases including LS or Leigh-like diseases, cardiomyopathy, and liver failure (). Two disease-associated assembly factors, COX10 and COX15, are needed for the synthesis of the heme a component of CIV, and their defects caused loss of CIV holoenzyme (Citation38,Citation39). Another example of an essential CIV assembly factor is the 30 kDa Surf1 protein (Citation40). The function of this protein is not completely understood, but it may be involved in the insertion of heme a into COX1 (Citation41).
A new mechanism of CIV deficiency was found in a patient with slowly progressive LS, who had a mutation in a gene encoding a protein of previously unknown function. This protein, named TACO1, was found to be needed for the activation of translation of COX1 (Citation42). TACO1 was suggested to be a member of a group of translational activators needed for the activation of mitochondrial mRNAs, which in contrast to cytoplasmic mRNAs lack 5′ untranslated regions. Furthermore, CIV deficiency and mitochondrial encephalomyopathy was found in patients with mutations in the gene FASTKD2, which encoded a mitochondrial protein potentially involved in apoptosis (Citation43). However, the exact mechanism of CIV deficiency had not been fully elucidated in the setting of FASTKD2 mutation.
Complex V deficiency
Mitochondrial complex V (CV) couples proton current to the phosphorylation of ADP. Known also as ATP synthase, the complex has a membrane part F0 and a peripheral part F1. Two F0 subunits, ATP6 and ATP8, are mtDNA-encoded, whereas all of the F1 subunits are nuclear-encoded. Mutations in the ATP6 gene of mtDNA are well known to cause disease. The T8993G and T8993C mutations in this gene cause disease depending on mutant load: a low load is asymptomatic, an intermediate load causes NARP (neurogenic weakness, ataxia, retinitis pigmentosa) (Citation44), and a high load causes maternally inherited LS, potentially through impaired CV assembly (reviewed in (Citation45)).
Like the other OXPHOS complexes, CV has assembly factors encoded by nuclear genes. Atp12 and the related Atp11 have been characterized as essential factors required for the formation of the F1 component of CV in both yeast and humans (Citation46). Mutations in the ATP12 gene were found in a child with early-onset encephalopathy and lactic acidosis, and the amount of assembled CV was strongly decreased (Citation47). Furthermore, a number of other patients with apparent defects in CV assembly, but no mutations in the known disease genes, have been described. A mutation in a nDNA-encoded structural gene has also been identified recently () (Citation48). The nuclear gene defects leading to CV deficiency generally lead to different phenotypes than NARP or LS (reviewed in (Citation45)).
Disorders of mtDNA maintenance
Defects in the nuclear-encoded proteins responsible for mtDNA maintenance lead to decreased mtDNA copy number, mtDNA point mutations, multiple mtDNA deletions, or combinations thereof. The disorders of mtDNA maintenance form a diverse group that ranges from severe infant-onset multi-system disease, where mtDNA copy number is severely depleted in one or more tissues (mtDNA depletion syndrome, MDS), to mild adult-onset autosomal-dominant (ad) or autosomal-recessive (ar) progressive external ophthalmoplegia (PEO). The genetic causes are heterogeneous (reviewed in (Citation49)). In PEO, mtDNA deletions gradually accumulate in patients’ tissues, but mtDNA copy number usually remains normal. The disease affects particularly external eye muscles, but more generalized myopathy with exercise intolerance is often observed, as well as additional symptoms that include cardiomyopathy, mild ataxia, Parkinsonism and depression (Citation4,Citation49,Citation50). Furthermore, we recently found that a very high mtDNA copy number was associated with a number of detrimental effects on mtDNA maintenance and expression in transgenic mice (Citation51). These results suggested that mitochondrial disease could also be caused by an abnormally high mtDNA copy number. However, this possibility remains unexplored in humans.
Most mtDNA maintenance diseases are caused by mutations in genes encoding proteins with well defined roles in mtDNA replication or mitochondrial deoxynucleoside triphosphate (dNTP) pool maintenance (). Two proteins that are intimately involved in mtDNA replication have particularly strong associations with human disease: the mtDNA polymerase γ (Pol γ), and the replicative helicase Twinkle (reviewed in (Citation52)). Mitochondrial dNTP pools are regulated by a complex system of interlinked pathways localized both in the cytosol and mitochondria (reviewed in (Citation53)). Interestingly, defects in one mtDNA maintenance protein can cause several different types of syndromes. Thus, the clinical picture is not always a good guide to finding the responsible gene.
Table III. The genes listed in this table encode proteins that are essential for mtDNA maintenance. Mutations in these genes cause mtDNA disease through different mechanisms, as indicated.
The mtDNA replisome in disease
A large number of disease-causing mutations have been found in the POLG1 gene, which encodes the catalytic subunit of Pol γ (Pol γ A) (reviewed in (Citation54)). The spectrum of Pol γ-related clinical manifestations is likewise large. The three main Pol γ-related phenotypes are: infant- or childhood-onset hepatocerebral MDS (known as Alpers syndrome), ataxia-neuropathy disorders such as MIRAS (mitochondrial recessive ataxia syndrome) that has a median age of onset of 28 years (range 5–41), and PEO with multiple mtDNA deletions, which can be either dominant or recessive and which rarely presents before 20 years of age (Citation49,Citation55,Citation56). In addition, Pol γ defects have been associated with a number of other phenotypes including Parkinsonism and premature menopause (Citation50).
The positions of POLG1 mutations show correlation with the clinical outcome. Pol γ A has a C-terminal polymerase domain and an N-terminal proof-reading exonuclease domain, separated by a spacer region involved in DNA binding and interaction with the accessory subunit Pol γ B. The latter is encoded by the gene POLG2, and it confers increased DNA binding, processivity, and polymerization rate to the holoenzyme (reviewed in (Citation52)). In general, polymerase domain mutations tend to cause dominant disease, as the DNA binding is not impaired and the mutant can compete with the wild-type protein. Recessive mutations cluster in the spacer and exonuclease regions, often causing reductions in DNA binding ability and accessory subunit interaction (Citation54).
The Pol γ crystal structure was particularly pertinent to the understanding of the spacer regions mutations, since this region does not share homology with the related DNA polymerase of the T7 phage in the manner the polymerase domain does (Citation57). One of the most common Pol γ amino acid changes, A467T, affects a residue that is located in the thumb domain of Pol γ A. This domain was found to be involved in both template binding and the interaction with Pol γ B. Thus, the position of the mutated residue in the enzyme's tertiary structure explained the results from earlier biochemical experiments that the mutation impairs both polymerase activity and processivity (Citation57). Mutation in the POLG2 gene has been associated with PEO in two patients so far (Citation58,Citation59). POLG2 defects may be a rare cause of PEO, but more patients need to be found before the position of POLG2 as a disease gene is firmly established.
Twinkle is the mitochondrial replicative helicase. It was found based on its similarity to the T7 phage helicase primase gp4, and was found to be defective in several families with adPEO and multiple mtDNA deletions in skeletal muscle (Citation60). Recessive Twinkle mutations cause either hepatocerebral MDS that is reminiscent of Alpers syndrome (Citation61,Citation62), or a syndrome of intermediate severity termed infantile-onset spinocerebellar ataxia (IOSCA) (Citation63). IOSCA is part of the Finnish disease heritage. It manifests between ages 1 and 2 years and is slowly progressive with ataxia, neuropathy, ophthalmoplegia, and seizures. Brains from IOSCA patients showed mtDNA depletion but no deletions or point mutations (Citation63). Mitochondrial DNA depletion in IOSCA may be more pronounced in particular neuronal populations, for instance cerebellar Purkinje cells, which could account for the restricted clinical findings and longer life-span compared to other forms of MDS (Citation63).
The mechanisms by which Pol γ and Twinkle mutations cause mtDNA deletions and instability have been studied in various systems, including cultured cells, in-vitro biochemical assays, and a transgenic mouse model (reviewed in (Citation52)). The Twinkle holoenzyme is a homohexamer, and most of the dominant mutations impair subunit interaction (Citation52). PEO mutations are dominant because the presence of just one defective subunit can impair the assembly and loading of the entire holoenzyme. Transgenic expression of Twinkle with a linker region duplication of amino acids 353–364, which was originally described in a Finnish adPEO family (Citation60), caused progressive mtDNA deletion accumulation in skeletal muscle and brain, similar to the patients (Citation64). These so-called Deletor mice provided an ideal tool for studying the in-vivo pathogenic mechanisms of the dominant Twinkle defect. Expression of PEO-related mutants of both Twinkle and Pol γ in human cultured cells induced significant stalling of the mtDNA replisome (Citation65,Citation66). Replication stalling was also observed in the tissues of Deletor mice (Citation65). Stalling could lead to double-strand DNA breaks, which in turn are known to induce deletion formation through double-strand break repair pathways (Citation67). Importantly, replication stalling in Deletor mice was evident already at 6 weeks of age, whereas deletions were detectable starting at ∼12 months age (Citation64,Citation65). This suggested that the underlying replication defect in PEO is present throughout life and that deletions accumulate from initially very small levels, until they eventually reach the threshold that causes disease. Deletion accumulation is enhanced by clonal expansion of deleted molecules within individual muscle cells (Citation68). Clonal expansion may be enhanced by the deleted molecules having a replicative advantage due to their shorter length compared to wild-type mtDNA (Citation69).
Mitochondrial dNTP pool maintenance and disease
The four dNTPs, deoxyadenosine-, deoxyguanosine-, deoxycytidine-, and thymidine triphosphate (dATP, dGTP, dCTP, and dTTP, respectively), are the building blocks of DNA. A sufficiently large dNTP pool is required for replication and repair of both nDNA and mtDNA. The relative proportions of the four dNTPs also need to be balanced in order to avoid mutagenesis (reviewed in (Citation70)). Nuclear DNA replicates only in S-phase of the cell cycle, during which dNTP synthesis is heavily up-regulated. Mitochondrial DNA replicates independently of the cell cycle in replicative cells and continues to be replicated in post-mitotic cells, for instance in neurons and skeletal muscle cells. Therefore, dNTP synthesis is required also in non-proliferative cells. There are two main pathways to supply dNTPs: in the de-novo pathway ribonucleotides, derived from the purine and pyrimidine biosynthetic pathways, are converted into the corresponding deoxyribonucleotides by the cytoplasmic enzyme ribonucleotide reductase (RNR); in the salvage pathway, deoxynucleosides undergo sequential phosphorylations into deoxynucleoside mono- and diphosphates, which are finally phosphorylated into dNTPs (reviewed in (Citation53)).
The de-novo pathway for deoxynucleotide synthesis is located in the cytoplasm, but mitochondria contain their own salvage pathway. Two mitochondrial kinases, thymidine kinase 2 (TK2) and deoxyguanosine kinase (dGK), are able to phosphorylate all four deoxynucleosides and thus form the basis of the mitochondrial deoxynucleoside salvage pathway. Mutations in the genes encoding TK2 or dGK in humans were found to cause recessively inherited, muscle-specific or hepatocerebral MDS, respectively (Citation71,Citation72). This proved the importance of deoxynucleoside salvage for mtDNA maintenance. For some time it was thought that post-mitotic cells almost completely lack RNR, and that mtDNA replication in non-cycling cells relies solely on dNTPs derived from the salvage pathway. However, this view changed when it was found that RNR is in fact also essential for mtDNA maintenance in post-mitotic tissues. Inactivating mutations in the RRM2B gene, encoding the p53-inducible small subunit of RNR (p53R2), caused multi-system, recessively inherited MDS (Citation73). Furthermore, we recently found that a heterozygous mutation of RRM2B, that leads to truncation of the p53R2 protein, causes adPEO with multiple mtDNA deletions (Citation74). Therefore, both salvage and de-novo production of dNTPs is necessary for mtDNA replication.
The patients with homozygous or compound heterozygous defects in the anabolic enzymes of deoxynucleotide synthesis, TK2, dGK, or p53R2, are usually normal at birth, but the MDS manifests abruptly in specific tissues during the first months or years of life. Reasons for the time of onset and tissue selectivity of these diseases are unknown. A recent study on a mouse model of TK2 deficiency suggested that disease onset coincides with down-regulation of the cytoplasmic salvage enzyme TK1 (Citation75). This TK1 down-regulation essentially unmasked a latent TK2 deficiency. The same study also suggested that increased transcription of mtDNA could partially compensate for mtDNA depletion in skeletal muscle (Citation75). Therefore, it is conceivable that tissue-specificities of MDS may be determined by different capacities to compensate for the various genetic defects.
The importance of proper dNTP pool balance is illustrated by the disease MNGIE (mitochondrial neurogastrointestinal encephalomyopathy). This disease is caused by deficiency of the catabolic enzyme thymidine phosphorylase (TP) that degrades thymidine (Citation76). TP deficiency leads to thymidine excess and probably to elevations of the mitochondrial dTTP pool in relation to the other dNTPs. This dNTP imbalance causes mtDNA deletions and point mutations in addition to mtDNA depletion (reviewed in (Citation77)). Furthermore, we recently studied a mouse model over-expressing more than one RNR subunit. The RNR over-expression caused dNTP pool imbalance and led to mtDNA depletion in skeletal muscle, but it did not cause mtDNA deletions or point mutations (Citation78). Therefore, it is clear that mtDNA replication is sensitive to the dNTP pool balance. However, the exact mechanisms by which dNTP pool imbalances cause mtDNA instability remain incompletely understood. It is also unknown why certain changes to the enzymatic pathways regulating dNTP pools lead to mtDNA depletion only, while other changes cause point mutations or deletions.
Disorders of mitochondrial protein synthesis
Mitochondrial translation is more reminiscent of prokaryotic translation than eukaryotic cytoplasmic translation. However, several significant differences exist, and the mitochondrial translation machinery is incompletely characterized to date (reviewed in (Citation79)). Mitochondria use a genetic code that is slightly different from the universal genetic code of other systems. Furthermore, the mitochondrial mRNAs also lack 5′-end untranslated regions and ‘cap’ structures, and the same tRNA-Met is used in both translation initiation and elongation. In addition to the 22 tRNAs and 2 rRNAs that are encoded by mtDNA, mitochondrial translation requires a wealth of nDNA-encoded proteins. Translation initiation requires the initiation factors mtIF2 and mtIF3, whereas elongation of the polypeptide is dependent on the elongation factors mtEFTu, mtEFTs, mtEFG1, and mtEFG2 (reviewed in (Citation80)). The release factors mtRF1 and mtRF1a are needed to terminate translation at stop codons. The mitochondrial ribosomes have particularly high protein content, with up to 81 mitochondrial ribosomal proteins (MRPs). Finally, aminoacylation of mitochondrial tRNAs is performed by mitochondrial aminoacyl-tRNA synthetases, of which 19 have been described (reviewed in (Citation81)).
Defects in nuclear-encoded mitochondrial translation factors
Disease-causing mutations have been identified in several of the nDNA-encoded mitochondrial translation factors (). In general, the associated phenotypes are severe, affecting the synthesis of all five OXPHOS complexes, and causing early death. However, the organ manifestations are diverse, even among patients who carry the same mutation. Understanding the molecular basis of the tissue specificity is challenging, given that all factors are involved in the same molecular machinery.
Table IV. Nuclear-encoded mitochondrial translation factors involved in disease.
Mutations in the PUS1 gene were found in patients with the disease MLASA (mitochondrial myopathy, lactic acidosis, and sideroblastic anemia), which affects bone-marrow, skeletal muscle, and the central nervous system but shows clinical variability (Citation82). PUS1 encodes pseudouridine synthase 1 (Pus1), which is not directly involved in translation but is required for the post-translational modification of tRNAs both in the cytoplasm and in mitochondria (Citation79). It was suggested that modification of the expression level of ribosomal proteins could compensate for the phenotype and account for the variability in phenotypes between tissues and between individuals (Citation83). In addition, Pus1 could have several non-tRNA substrates (Citation83).
Defects in another enzyme involved in tRNA modification, tRNA 5-methylaminomethyl-thiouridylate (Trmu), are also associated with mitochondrial disease. Trmu modifies wobble base positions of three mitochondrial tRNAs: tRNA-Lys, tRNA-Gln, and tRNA-Glu. Several mutations in the TRMU gene caused acute liver failure and were found to render the tRNAs hypomodified, leading to defective mitochondrial protein synthesis (Citation84). One TRMU mutation was not associated with liver failure but caused aggravation of the deafness phenotype associated with mitochondrial 12S rRNA mutations (Citation85).
Mutations in the mitoribosomal proteins MRPS16 and MRPS22 impair assembly and stability of the small ribosomal subunit (Citation86,Citation87). Clinically, the MRPS16 defect led to agenesis of the corpus callosum, dysmorphism, and fatal neonatal lactic acidosis (Citation86). The MRPS22 defect led to antenatal skin edema, hypotonia, cardiomyopathy, and tubulopathy (Citation87).
Disease-causing mutations have been identified in genes encoding translational elongation factors but not in those encoding initiation factors. The syndromes associated with elongation factor mutations exhibit remarkable tissue selectivity. All of these mutations are associated with defective translation of most mitochondrial transcripts, but bicistronic transcripts (e.g. the one encoding the ATP6 and ATP8 genes) can be translated at normal or elevated levels. The mtEFG1 deficiency caused a severe hepato(encephalo)pathy, but the heart was spared despite a high normal expression level of the affected protein in this tissue (Citation88–90). Mutation of mtEFTu in turn was associated with a rapidly progressive encephalopathy (Citation89). However, a single homozygous mutation in mtEFTs caused hypertrophic cardiomyopathy in one patient and encephalomyopathy in another (Citation91). The relatively benign phenotype in some tissues is apparently related to more favorable relative expression levels of the translation factors, as well as adaptive changes in these ratios. For instance, the heart of patients with mtEFG1 mutation had an up-regulation of the mtEFTu:mtEFTs ratio, whereas the reverse was true for the liver (Citation90). In addition, there may be other genetic modifiers that are responsible for tailoring mitochondrial translation to tissue-specific needs. The mtEFTs mutant could be compensated for by over-expression of mtEFTu, probably because the mutation interferes with the formation of the complex between these two proteins and renders both proteins unstable. Bringing in additional mtEFTu thus helps stabilize the complex (Citation91). It is possible that this type of compensatory mechanism accounted for the difference between the two mtEFTs patients.
Mutations in two of the aminoacyl-tRNA synthetases affected the brain. RARS2 (encoding mitochondrial arginyl-tRNA synthase) mutations caused pontocerebellar hypoplasia (Citation92), whereas the DARS2 (encoding mitochondrial aspartyl-tRNA synthetase) mutations led to LSBL (leukoencephalopathy with brain-stem and spinal cord involvement and lactate elevation) (Citation93). The mutations were in introns and affected splice sites. Higher expression of splicing factors in extracerebral tissues was suggested as a reason for brain specificity of the phenotypes in RARS2 and DARS2 mutations (Citation92). The RARS2 mutation caused a decrease in the total abundance of mitochondrial tRNA-Arg, probably because the unloaded tRNA is unstable (Citation92). However, the DARS2 mutation did not lead to a translation deficit or OXPHOS dysfunction in patient fibroblasts, even though the aminoacylation activity was strongly reduced (Citation93). The mechanism by which mutation in this gene causes disease is therefore unclear. It is possible that only a subset of neurons is affected, meaning that studies on whole-brain lysates or cultured fibroblasts are not informative (Citation93).
A mutation in YARS2, which encodes the mitochondrial tyrosyl-tRNA, synthetase was found to cause MLASA with no apparent effects on brain structure or function (Citation94). Since the phenotype was highly similar to that caused by PUS1 mutation, a common mechanism was invoked. The dysfunctional Pus1 may cause disease through hypomodification of tRNA-Tyr, leading to decreased amino acid charging reminiscent of the situation in YARS2 deficiency (Citation94). However, it remains unclear why the brain is not affected. Three further mitochondrial aminoacyl-tRNA synthetases, encoded by SARS2, HARS2, and AARS2, have been associated with disease very recently (Citation95–97). The involved organs appear to be strikingly diverse and specific for each gene ().
Mitochondrial tRNA and rRNA mutations and single mtDNA deletions
Most human mtDNA point mutations occur in tRNA genes (www.mitomap.org). The clinical manifestations of tRNA mutations are manifold. Two of the best studied mitochondrial tRNA mutations are the A3243G mutation in tRNA-Leu(UUR) and the A8344G mutation in tRNA-Lys. The former has a population prevalence of as high as 5.7 per 100,000 and causes a wide range of phenotypes of which MELAS is prominent, and the latter is associated with MERRF (myoclonic epilepsy with ragged-red fibers) (reviewed in (Citation98)). Mutations in other tRNA genes can lead to distinct phenotypes, e.g. deafness due to mutations in tRNA-Ser(UCN) or cardiomyopathy due to mutations in tRNA-Ile (reviewed in (Citation99,Citation100)). A number of explanations have been put forth to explain the clinical heterogeneity of tRNA mutations, including variations in heteroplasmy or mtDNA segregation, different OXPHOS complexes being differently affected depending on their amino acid composition, tissue-specific toxicities of prematurely terminated translation products, or other tissue-specific modifiers that affect mitochondrial protein synthesis or function (Citation100).
Mutations in the mitochondrial rRNA genes are associated with non-syndromic sensorineural deafness or aminoglycoside-induced deafness. The most common mutation is A1555G in the 12S rRNA gene (Citation101). The mutation alters the structure of the rRNA such that it more closely resembles the bacterial 16S rRNA. This facilitates interaction with aminoglycosides, leading to translation defect. Not all people who carry this mutation develop deafness. In addition to aminoglycoside exposure, the penetrance depends on mtDNA haplotype and nuclear modifier genes (Citation79). This is a good example of how a mitochondrial disease is modified by genetic and environmental factors.
The mtDNA deletion syndromes are caused by large-scale (usually several thousand base pairs) deletions of mtDNA, which are usually sporadic (Citation102). In these disorders, the patient tissues typically harbor a single partially deleted species together with the wild-type mtDNA. These diseases are denoted single mtDNA deletion diseases to distinguish them from the multiple deletion diseases, in which deletions of different sizes are present and which are caused by defects in mtDNA maintenance. Deletions usually involve several genes including at least one tRNA, meaning that mitochondrial protein synthesis as a whole will be affected. Clinically, the mtDNA deletion syndromes form a spectrum that ranges from relatively benign adult-onset disorders such as PEO to severe infantile-onset disorders such as Pearson's syndrome ().
Disorders of mitochondrial dynamics
Mitochondrial fusion and fission, collectively mitochondrial dynamics, are essential for mitochondrial distribution and function. Mitochondria form an interconnected network that is constantly reorganized (reviewed in (Citation5)). Defective fusion or fission underlies a number of neurological diseases in humans. Mitochondrial fusion requires three proteins: Mitofusins 1 and 2 (Mfn1 and Mfn2) are needed for outer membrane fusion, and Opa1 has been suggested to be involved in inner membrane fusion (reviewed in (Citation103)). Mutations in the MFN2 gene cause the autosomal-dominant Charcot–Marie–Tooth disease type 2A, which is a peripheral neuropathy caused by degeneration of axons in long sensory and motor nerves of the distal extremities (Citation104). The selective manifestation of Mfn2 mutations in neurons has been attributed to a low relative level of Mfn1 expression. Mfn1 is able to complement Mfn2 functionally (Citation105), and tissues with high endogenous Mfn1 expression may thus be resistant to Mfn2 defects. In addition, peripheral neurons may be particularly susceptible to aberrant mitochondrial dynamics due to the extreme length of their axons (reviewed in (Citation103)).
OPA1 mutations cause autosomal-dominant optic atrophy (Citation106,Citation107). In most cases, this disease specifically involves degeneration of retinal ganglion cells. However, more diverse OPA1-related phenotypes have been described recently, and they include disorders of mtDNA maintenance () (Citation103).
Disorders where OXPHOS is indirectly affected
In most of the disorders described so far, mitochondrial dysfunction arises as a direct consequence of a defect in genes involved in OXPHOS or in the synthesis of OXPHOS subunits. However, mitochondrial dysfunction can arise also through indirect mechanisms.
Ubiquinone or coenzyme Q (CoQ10) shuttles electrons from CI and CII to CIII. It consists of a benzoquinone ring and a tail of typically 10 isoprenyl subunits that anchors it to membranes (reviewed in (Citation108)). Reduced ubiquinone (QH2) also functions as an antioxidant. Disorders of CoQ10 biosynthesis form a group of rare autosomal-recessive disorders. Clinical manifestations are reminiscent of other mitochondrial disorders and are divided into five groups: encephalomyopathy, severe infantile multi-systemic disease, cerebellar ataxia, isolated myopathy, and nephritic syndrome () (Citation108). Correct diagnosis of these patients is essential, as ubiquinone Q10 supplementation may improve the condition of some patients dramatically.
Table V. Disease-associated genes invloved in the biosynthesis of coenzyme Q (CoQ10). Derived from reference (108).
The import of nuclear-encoded proteins into mitochondria requires specialized multi-subunit machineries in the IMM, IMS, and OMM. Defects in the protein translocation machinery have been found to cause disease by impairing the importation of proteins needed for OXPHOS. The consequence of mutation in the deafness/dystonia protein 1/translocase of mitochondrial inner membrane 8a (DDP1/TIMM8a) is Mohr–Tranebjaerg syndrome/deafness-dystonia syndrome, which is an X-linked disorder characterized by progressive sensorineural deafness, cortical blindness, dystonia, dysphagia, and paranoia (Citation109,Citation110). Certain cysteine-rich proteins are imported into the IMS with the aid of a disulfide relay system, which catalyzes the oxidative folding of these proteins. The disulfide relay system donates electrons to oxygen via cytochrome c and CIV, thus connecting it to the RC. Accordingly, mutation in the gene GFER, which encodes one of two key components of the disulfide relay system, was found to cause dysfunction of multiple RC complexes and a progressive mitochondrial myopathy (Citation111).
Another cause of OXPHOS defect was found in the dysfunction of the protein paraplegin in patients with hereditary spastic paraplegia (Citation112). Paraplegin is a subunit of a ubiquitous mitochondrial protease that has a quality control role, suggesting that the patients have problems with mitochondrial protein turn-over (Citation113). Recently, mutation in the X-chromosomal gene AIFM1, encoding the apoptosis-inducing factor (AIF), was found to be the cause of mitochondrial encephalomyopathy in two male infants (Citation114). AIF is an IMS-targeted protein whose function is incompletely understood. The protein can be released into the cytoplasm and nucleus where it induces fragmentation of nuclear DNA, leading to a form of programmed cell death that is independent of caspase. Patient muscle displayed increased sensitivity to this form of programmed cell death, in addition to mtDNA depletion and OXPHOS failure. The latter may have been due to destabilization of the IMM (Citation114).
Finally, severe lack of CIV activity was found in patients with ethylmalonic encephalopathy and mutation in the gene ETHE1. The gene encodes a mitochondrial dioxygenase, and the mutation caused accumulation of sulfide, which is a potent inhibitor of CIV (Citation115). Together, these studies showed that mitochondrial dysfunction can result from a multitude of mechanisms other than direct impairment of OXPHOS.
Consequences of OXPHOS defects
Our understanding of the genetic etiology of mitochondrial diseases is expanding rapidly. However, relatively little is known of the molecular consequences of impaired OXPHOS that account for the development of symptoms in specific tissues. Given the multitude of functions that mitochondria take part in and the universal need of ATP by all cells, the pathogenic mechanisms are likely to be complex. Potential consequences of OXPHOS impairment include metabolite build-up, deficiency of ATP, aberrant calcium handling, and ROS production (reviewed in (Citation116)).
Mitochondria produce ROS as a side product of normal respiration. The production of ROS can be considerably increased in malfunctioning mitochondria, and the effect of ROS is believed to be toxic, since they can irreversibly modify many cellular macromolecules. There are various ROS detoxifying enzymes to mitigate these effects. Moreover, a limited production of ROS also may play an important signaling role, by acting as a retrograde signal that influences gene expression in the nucleus in response to alterations of mitochondrial membrane potential or redox state (reviewed in (Citation117)).
One consequence of OXPHOS dysfunction is that NADH produced by the citric acid cycle cannot be fed forward, leading to elevation of the NADH:NAD + ratio. The level of pyruvate, produced through glycolysis, is increased due to an inhibition of the citric acid cycle. Thus, the equilibrium of the NADH-dependent lactate dehydrogenase is shifted towards the production of lactate from pyruvate. Lactate is released to the blood-stream, which causes a systemic drop in pH. Indeed, lactic acidosis is among the most important symptoms of mitochondrial disease (reviewed in (Citation3)).
ATP deficiency is one of the most obvious consequences of OXPHOS deficiency. Decreased rates of ATP production have been found in mitochondria from CI-deficient patient muscle, but ATP deficiency may not be the most crucial metabolic derangement responsible for cellular dysfunction in these disorders (Citation116). ATP deficiency and aberrant calcium signaling have been proposed as interlinked pathways leading to cell dysfunction in mitochondrial disease: first, because mitochondrial ATP production is needed to fuel calcium pumps in the endoplasmic reticulum, and, second, because mitochondria themselves can buffer calcium and have their function regulated by the influx of calcium (reviewed in (Citation118)). This model could account for the frequent involvement of muscle and nerve tissues in mitochondrial diseases, since these cells rely heavily on ATP and on fluctuating levels of activity mediated by Ca2+ pulses. However, as different mitochondrial disorders are highly tissue-specific, ATP deficiency cannot be the sole underlying factor in mitochondrial diseases.
Finally, OXPHOS dysfunction in individual tissues is likely to have metabolic consequences on the systemic level. For instance, we recently reported that skeletal muscle in mice reacts to OXPHOS dysfunction by developing a starvation-like state that includes up-regulating the expression of the fasting-related hormone FGF21, which is likely to induce the mobilization of energy stores in liver and fat for use in the starved skeletal muscle (Citation119). The starvation-like state could also entail increased turn-over of mitochondria by the process known as mitochondrial autophagy, or mitophagy. Selective removal of dysfunctional mitochondria by mitophagy is likely to be a central mechanism by which cells maintain a healthy mitochondrial population (Citation120,Citation121). Intriguingly, defective mitophagy has been implicated in the development of Parkinson's disease (Citation120). Hence, modification of the physiological responses to OXPHOS dysfunction could provide novel therapeutic tools to combat mitochondrial and neurodegenerative diseases. For instance, we found that high-fat feeding of mice considerably improved the ultrastructural changes of mitochondria in skeletal muscle of late-onset mitochondrial myopathy mice (Citation122). These results suggested the following: 1) Mitochondrial biogenesis is boosted upon lipid feeding. The mitochondria are the major lipid-oxidizing organelle, whereas glucose can be used for ATP production both by mitochondria and by cytoplasmic glycolysis. Use of lipids requires active mitochondria. 2) If glucose is available and glycolytic ATP production is possible, even mild dysfunction in mitochondria may cause their active down-regulation, leading to cells relying on glycolysis. 3) By lipid-feeding mitochondria are forced to be active, and poorly working mitochondria may actually be more beneficial for cells than down-regulated mitochondria, as they produce more ATP than mere glycolysis. Therefore, correct composition of nutrition may be crucial for mitochondrial patients.
Conclusion
The first description of genetic mutations in mitochondrial disease came in 1988 (Citation21,Citation102), and since then the field has expanded exponentially. The molecular pathways required for the synthesis and assembly of functional OXPHOS machinery are remarkably complex, requiring over a thousand genes in two separate genomes. New techniques for gene searching, such as whole-exome sequencing, are likely to expand rapidly the list of genes implicated in mitochondrial diseases, including those with strictly tissue-specific manifestation. However, the real challenge will be to explain the molecular mechanisms responsible for diversity of phenotypes, both on the level of the individual cell and the entire organism.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders—past, present and future. Biochim Biophys Acta. 2004;1659:115–20.
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, . Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9.
- Debray FG, Lambert M, Mitchell GA. Disorders of mitochondrial function. Curr Opin Pediatr. 2008;20:471–82.
- McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol. 2002;1:343–51.
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, . Sequence and organization of the human mitochondrial genome. Nature. 1981;290: 457–65.
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, . A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–23.
- Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, Holt IJ. Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet. 2010;6:pii:e1001066.
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, . A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–54.
- Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2008;40:1484–8.
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, . A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–62.
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, . Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:e10.
- Greaves LC, Turnbull DM. Mitochondrial DNA mutations and ageing. Biochim Biophys Acta. 2009;1790:1015–20.
- Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. 2009;1793:200–11.
- Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724–7.
- Berrisford JM, Sazanov LA. Structural basis for the mechanism of respiratory complex I. J Biol Chem. 2009;284:29773–83.
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–5.
- Hunte C, Zickermann V, Brandt U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science. 2010;329:448–51.
- Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–6.
- Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH, . Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132(Pt 4):833–42.
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, . Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242: 1427–30.
- Antonicka H, Ogilvie I, Taivassalo T, Anitori RP, Haller RG, Vissing J, . Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J Biol Chem. 2003;278:43081–8.
- Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, Nijtmans L. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet. 2004;13:2461–72.
- Vogel RO, Dieteren CE, van den Heuvel LP, Willems PH, Smeitink JA, Koopman WJ, . Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J Biol Chem. 2007;282:7582–90.
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, . The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419: 367–74.
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, . Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11: 144–9.
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, . Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54.
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, . Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51.
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–70.
- Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202.
- Benit P, Lebon S, Rustin P. Respiratory-chain diseases related to complex III deficiency. Biochim Biophys Acta. 2009;1793:181–5.
- Crofts AR. The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol. 2004;66:689–733.
- Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, . Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell. 2004;13:805–15.
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–9.
- Visapaa I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, . GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002;71:863–76.
- Hinson JT, Fantin VR, Schonberger J, Breivik N, Siem G, McDonough B, . Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med. 2007;356:809–19.
- Diaz F. Cytochrome c oxidase deficiency: patients and animal models. Biochim Biophys Acta. 2010;1802:100–10.
- Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, . Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12:2693–702.
- Antonicka H, Mattman A, Carlson CG, Glerum DM, Hoffbuhr KC, Leary SC, . Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101–14.
- Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, . SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–43.
- Smith D, Gray J, Mitchell L, Antholine WE, Hosler JP. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J Biol Chem. 2005;280:17652–6.
- Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, . Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41:833–7.
- Ghezzi D, Saada A, D'Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, . FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet. 2008;83:415–23.
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–33.
- Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006;1757:1400–5.
- Wang ZG, White PS, Ackerman SH. Atp11p and Atp12p are assembly factors for the F(1)-ATPase in human mitochondria. J Biol Chem. 2001;276:30773–8.
- De Meirleir L, Seneca S, Lissens W, De Clercq I, Eyskens F, Gerlo E, . Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J Med Genet. 2004;41:120–4.
- Mayr JA, Havlickova V, Zimmermann F, Magler I, Kaplanova V, Jesina P, . Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet. 2010;19:3430–9.
- Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med. 2008;59:131–46.
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, . Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364: 875–82.
- Ylikallio E, Tyynismaa H, Tsutsui H, Ide T, Suomalainen A. High mitochondrial DNA copy number has detrimental effects in mice. Hum Mol Genet. 2010;19:2695–705.
- Wanrooij S, Falkenberg M. The human mitochondrial replication fork in health and disease. Biochim Biophys Acta. 2010;1797:1378–88.
- Rampazzo C, Miazzi C, Franzolin E, Pontarin G, Ferraro P, Frangini M, . Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat Res. 2010 28;703:2–10.
- Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787:312–9.
- Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, . Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–41.
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–2.
- Lee YS, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–24.
- Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, . Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–34.
- Walter MC, Czermin B, Muller-Ziermann S, Bulst S, Stewart JD, Hudson G, . Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J Neurol. 2010;257:1517–23.
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, . Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–31.
- Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130(Pt 11):3032–40.
- Sarzi E, Goffart S, Serre V, Chretien D, Slama A, Munnich A, . Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann Neurol. 2007;62:579–87.
- Hakonen AH, Goffart S, Marjavaara S, Paetau A, Cooper H, Mattila K, . Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet. 2008;17:3822–35.
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, . Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–92.
- Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–40.
- Wanrooij S, Luoma P, van Goethem G, van Broeckhoven C, Suomalainen A, Spelbrink JN. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32: 3053–64.
- Srivastava S, Moraes CT. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum Mol Genet. 2005;14: 893–902.
- Moslemi AR, Melberg A, Holme E, Oldfors A. Clonal expansion of mitochondrial DNA with multiple deletions in autosomal dominant progressive external ophthalmoplegia. Ann Neurol. 1996;40:707–13.
- Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18:1028–36.
- Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–74.
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, . The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–41.
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–4.
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, . Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–80.
- Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, Suomalainen A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet. 2009;85:290–5.
- Dorado B, Area E, Akman HO, Hirano M. Onset and organ specificity of Tk2 deficiency depends on Tk1 down-regulation and transcriptional compensation. Hum Mol Genet. 2010;20:155–64.
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–92.
- Hirano M, Nishigaki Y, Marti R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10:8–17.
- Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, Ide T, . Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Res. 2010;38:8208–18.
- Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol. 2010; 2010:737385.
- Spremulli LL, Coursey A, Navratil T, Hunter SE. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog Nucleic Acid Res Mol Biol. 2004;77: 211–61.
- Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 2007;35: 4686–703.
- Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004;74:1303–8.
- Bykhovskaya Y, Mengesha E, Fischel-Ghodsian N. Pleiotropic effects and compensation mechanisms determine tissue specificity in mitochondrial myopathy and sideroblastic anemia (MLASA). Mol Genet Metab. 2007;91: 148–56.
- Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, Beinat M, . Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85:401–7.
- Guan MX, Yan Q, Li X, Bykhovskaya Y, Gallo-Teran J, Hajek P, . Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291–302.
- Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, . Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56:734–8.
- Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, . Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–6.
- Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, . Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med. 2004;351:2080–6.
- Valente L, Tiranti V, Marsano RM, Malfatti E, Fernandez-Vizarra E, Donnini C, . Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80:44–58.
- Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15:1835–46.
- Smeitink JA, Elpeleg O, Antonicka H, Diepstra H, Saada A, Smits P, . Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–77.
- Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, . Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81: 857–62.
- Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, . Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534–9.
- Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, McKenzie M, Compton A, . Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am J Hum Genet. 2010;87:52–9.
- Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, Feinstein S, Zeligson S, . Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200.
- Gotz A, Tyynismaa H, Euro L, Ellonen P, Hyotylainen T, Ojala T, . Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–42.
- Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, . Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108:6543–8.
- DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–32.
- Scaglia F, Wong LJ. Human mitochondrial transfer RNAs: role of pathogenic mutation in disease. Muscle Nerve. 2008; 37:150–71.
- Jacobs HT. Disorders of mitochondrial protein synthesis. Hum Mol Genet. 2003;12 Spec No 2:R293–301.
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, . Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–94.
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–9.
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–9.
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, . Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–51.
- Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176: 405–14.
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, . OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–5.
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, . Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–10.
- Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16:183–8.
- Jin H, May M, Tranebjaerg L, Kendall E, Fontan G, Jackson J, . A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness. Nat Genet. 1996;14:177–80.
- Roesch K, Curran SP, Tranebjaerg L, Koehler CM. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum Mol Genet. 2002;11:477–86.
- Di Fonzo A, Ronchi D, Lodi T, Fassone E, Tigano M, Lamperti C, . The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency. Am J Hum Genet. 2009;84:594–604.
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, . Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83.
- Rugarli EI, Langer T. Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia. Trends Mol Med. 2006;12:262–9.
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P, . Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–49.
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, . Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–5.
- Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13.
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95.
- Willems PH, Valsecchi F, Distelmaier F, Verkaart S, Visch HJ, Smeitink JA, . Mitochondrial Ca2 + homeostasis in human NADH:ubiquinone oxidoreductase deficiency. Cell Calcium. 2008;44:123–33.
- Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkila S, Wenz T, Ruhanen H, . Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet. 2010;19:3948–58.
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803.
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, . Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46.
- Ahola-Erkkilä S, Carroll CJ, Peltola-Mjosund K, Tulkki V, Mattila I, Seppanen-Laakso T, . Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–84.
- Zhu X, Peng X, Guan MX, Yan Q. Pathogenic mutations of nuclear genes associated with mitochondrial disorders. Acta Biochim Biophys Sin (Shanghai). 2009;41:179–87.
- Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, . Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–4.
- Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, . SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654–6.
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, . SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–42.
- Barel O, Shorer Z, Flusser H, Ofir R, Narkis G, Finer G, . Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82:1211–6.
- Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, . Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43:259–63.
- Shteyer E, Saada A, Shaag A, Al-Hijawi FA, Kidess R, Revel-Vilk S, . Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet. 2009;84:412–7.
- Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJ, van den Brand MA, Hendriksfranssen MG, . A mutation in c2orf64 causes impaired cytochrome C oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488–93.
- Jonckheere AI, Hogeveen M, Nijtmans LG, van den Brand MA, Janssen AJ, Diepstra JH, . A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet. 2008;45:129–33.
- Cizkova A, Stranecky V, Mayr JA, Tesarova M, Havlickova V, Paul J, . TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet. 2008;40:1288–90.
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, . Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–5.
- Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, . Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–6.
- Ostergaard E, Christensen E, Kristensen E, Mogensen B, Duno M, Shoubridge EA, . Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–7.
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, . OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131(Pt 2):338–51.
- Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D'Adamo P, Calvo S, . MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38: 570–5.
- Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Pedersen AM, . Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87:115–22.