Abstract
Aspirin is an irreversible inhibitor of platelet prostaglandin synthase activity, and is the most widely prescribed drug for the secondary prevention of cardiovascular disease. In recent years, clinical and laboratory evidence has shown significant individual variability in the response to aspirin and its link to clinical outcome. The term ‘aspirin resistance’ has been introduced to describe situations when clinical or ex-vivo effects of aspirin are less than expected. The accumulating evidence of increased risk of major adverse clinical events (MACE) associated with ‘aspirin resistance’ in the settings of acute coronary syndrome (ACS), stroke, and peripheral arterial disease has stimulated the search for ways of overcoming aspirin resistance. Existence of the link between high on-treatment platelet reactivity and atherothrombotic events suggests the common mechanisms for atherosclerosis progression and thrombotic complications with the platelets, being a key cellular interface between coagulation and inflammation.
This review article provides a contemporary view on ‘aspirin resistance’ and discusses its definition, clinical importance, and possible mechanisms in light of recent data on the role of platelets in atherothrombosis.
Key messages
Existence of the link between high on-treatment platelet reactivity and atherothrombotic events suggests the common mechanisms for atherosclerosis progression and thrombotic complications with the platelets, being a key cellular interface between coagulation and inflammation.
Introduction
Platelets have a vital homeostatic function in the prevention of excessive blood loss following injury by the facilitation of haemostasis. However, variation in platelet response to physiological or pathological stimuli may predispose to an increased risk of pathological bleeding or thrombosis.
Current therapeutic strategies for the prevention and treatment of arterial thrombosis are based on coagulation glycoproteins and on the known enzyme and receptor systems of platelets that are involved in the activation of these cells. Three broad groups of antiplatelet agents have been proven to be effective in a clinical setting, as follows: 1) cyclo-oxygenase-1 (COX-1) inhibitors (such as aspirin); 2) adenosine 5-diphosphate (ADP) receptor antagonists, irreversible (ticlopidine, clopidogrel, prasugrel) and reversible (ticagrelor); and 3) glycoprotein IIb/IIIa antagonists (abciximab, eptifibatide, tirofiban). Although aspirin, thienopyridines (ticlopidine, clopidogrel, prasugrel), and two-cyclopentyl-triazolo-pyrimidines (ticagrelor) inhibit a single pathway of platelet activation, their pronounced antithrombotic effect is explained by the fact that both thromboxane A2 (TxA2) pathway and ADP pathway are necessary for the amplification of platelet activation which is essential for the full platelet aggregation.
However, there is growing evidence that not all individuals respond equally to antiplatelet agents, a feature which has led to the introduction of the terms of aspirin and clopidogrel ‘resistance’ (Citation1). Given that ‘resistance’ to antiplatelet drugs can be associated with increased risk of recurrent vascular events, the ‘problem’ of ‘variable platelet response to antiplatelet therapy’ attracts the attention of many investigators.
The aim of this review article is to provide a contemporary overview on concepts and controversies related to a ‘variable platelet response to antiplatelet therapy’.
Search strategy
We performed a comprehensive literature search by using electronic bibliographic databases (i.e. MEDLINE, EMBASE, DARE, Cochrane Database), scanning reference lists from included articles and hand-searching abstracts from national and international cardiovascular meetings. For the search, we had used the search terms ‘aspirin’, ‘acetylsalicylic acid’, ‘resistance’, ‘platelet’, ‘reactivity’, ‘atherothrombosis’, ‘atherosclerosis’, and ‘inflammation’.
Where necessary, study authors were contacted to obtain further data. We have essentially performed a semi-systematic review of the published literature, rather than conduct a formal Cochrane-style systematic review and critical appraisal.
Terminology
The term ‘aspirin resistance’ has been used to describe different manifestations of insufficient inhibitory effect of aspirin on platelets, as reflected by either the inability of aspirin to protect individuals from thrombotic complications (that is, clinically defined ‘aspirin resistance’) or persistently high platelet reactivity in laboratory tests, such as bleeding time, TxA2 production, light aggregometry, etc. (that is, laboratory-defined ‘aspirin resistance’) (Citation1). A pharmacological definition of ‘aspirin resistance’ focuses on the inability of aspirin to inhibit COX-1-dependent TxA2 production and, consequently, the TxA2-dependent pathway of platelet activation (Citation2). Resistance to clopidogrel (or the other thienopyridines) is the failure to inhibit the platelet P2Y12 receptor and, consequently, the P2Y12-dependent pathway of platelet activation.
Some authors distinguish between pharmacodynamic ‘aspirin resistance’ (because of changes in the target enzyme for aspirin, COX-1, or because of transient inaccessibility of the enzyme due to blockade of the active site by non-steroidal anti-inflammatory agents) and pharmacokinetic ‘aspirin resistance’ (due to limited availability of the active drug at the level of its target). The in-vitro addition of aspirin to the blood sample would not significantly change the aggregation or thromboxane B2 (TxB2) levels in the first case and should significantly reduce the aggregation and TxB2 concentration in the second scenario (Citation3–5).
Laboratory-defined ‘aspirin resistance’ or aspirin non- responsiveness is present when in-vitro platelet reactivity is not properly blocked despite the use of aspirin (Citation5–7). Despite attempts to dichotomize patients as either ‘responders’ or ‘non- responders’ using arbitrary cut-off points in platelet function tests, on-treatment platelet reactivity as a measure of effectiveness of antiplatelet therapy is more likely to have a normal, bell-shaped distribution, as many other biological processes (Citation8–11).
‘High on-treatment residual platelet reactivity’ (Citation2), found in vitro in patients on antiplatelet therapy, does not necessarily mean ‘resistance’ to the antiplatelet drug from a pharmacological point of view. The recommendation of the Working Group on antiplatelet drugs resistance (Citation5) is to reserve the term ‘laboratory aspirin resistance’ to situations corresponding to the pharmacodynamic resistance, when an acetyl-salicylic acid (ASA)-specific laboratory test was used (platelet aggregation induced by arachidonic acid (AA) or the measurement of serum level of TxB2) and in-vitro addition of ASA performed to exclude pharmacokinetic resistance. The results obtained with less specific methods used for monitoring the effects of aspirin reflect the reactivity of platelets as a whole and can only be partly attributed to the TxA2 pathway of platelet activation. In this situation, the term ‘high on-treatment residual platelet reactivity’ would be more appropriate to describe insufficient inhibition of platelet function by aspirin.
Clinical ‘aspirin resistance’ or ‘treatment failure’ is the occurrence of thrombotic events in patients treated with antiplatelet agents (Citation2,Citation5,Citation6). However, as thrombogenesis is a complex multifactorial process, even complete inhibition of a single pathway may not prevent recurrence of thrombosis in all cases. This limits the practical value of the clinical definition of ‘aspirin resistance’.
Assessment of platelet response to aspirin
The prevalence of ‘aspirin resistance’ reported in different studies varies significantly and grossly depends on the laboratory techniques used for its diagnosis. Unfortunately pharmacokinetic studies on aspirin are of limited use, as aspirin is rapidly absorbed from the stomach and acetylates platelet COX-1 in the pre-systemic circulation. Furthermore, low doses of aspirin, administered slowly, may reduce TxB2 formation, even being hardly detected in the systemic circulation (Citation12).
Aspirin efficacy is perhaps better determined by assays directly dependent upon COX-1 function, such as TxB2 generation in serum and AA-induced platelet aggregation. Serum TxB2 (a stable metabolite of TxA2) reflects the total capacity of platelets to synthesize TxA2 ex vivo in response to physical and chemical stimuli and can be estimated by measurement of TxB2 in blood clotted at 37°C for 30–60 min.
The capacity of platelets to generate TxA2 is approximately 1,000-fold greater than endogenous plasma level. Normal concentrations of circulating TxB2 are very low, 1–2 pg/mL, and thus plasma samples must be purified and concentrated prior to analysis. Consequently, plasma assays are generally less sensitive and less specific for the estimation of the effect of aspirin. Also, stable levels of thromboxane production in vivo are maintained by different mechanisms, and reduction of thromboxane levels is usually only seen when > 95% inhibition of thromboxane generation is achieved in ex-vivo tests (Citation13). However, even the adequate inhibition of aggregation may require 99% inhibition of TxA2 production by platelets (Citation14), and thus even a minimal residual capacity to generate TxA2 may be sufficient to sustain TxA2- dependent platelet activation (Citation13). Low concentrations of TxA2 (or other bioactive eicosanoids acting through TxA2 receptors) from aspirin-treated stimulated platelets and from monocytes/macrophages may still activate the tyrosine-kinase-based signalling pathway. This pathway cannot trigger full platelet activation on its own but can synergize with the Gz-linked adrenaline receptor and thus activate platelets (Citation15).
Indeed, in patients with stable coronary artery disease (CAD) prominent activity of platelet alpha-2 adrenoceptors was shown to be a considerable factor in high residual platelet reactivity (Citation16). For example, Williams et al. (Citation17) demonstrated that patients with acute coronary syndrome (ACS) have increased platelet sensitivity to adrenergic stimuli despite the effective suppression of platelet release response by aspirin. This may lead to false classification of some patients as ‘aspirin-resistant’ depending upon the particular assay used.
In healthy individuals and patients with CAD treated with aspirin, the rate of residual significant TxB2 production is extremely low (Citation18–20). Only about 2% or less of patients with CAD had evidence of incomplete inhibition of TxB2 production, which was either because of under-dosing or non-compliance (Citation18,Citation19,Citation21).
Levels of the urinary thromboxane metabolite—11-dehydro TxB2—reflects time-integrated in-vivo TxA2 biosynthesis (Citation22,Citation23). It is less specific for TxA2, generated by platelet COX-1, as about 30% of the urinary metabolite is derived from extra-platelet sources or thromboxane generated by COX-2. The latter may also be present in platelets and megakaryocytes. Additionally, in conditions associated with inflammation or increased platelet turn-over, the contribution of extra-platelet sources of TxA2 may even be higher (Citation24–27).
The other most often used tests of platelet function, dependent on COX-1 activity, include agonist-induced platelet aggregation measured by light transmission (turbidimetric) aggregometry (LTA) in platelet-rich plasma (PRP), electrical impedance (whole blood) platelet aggregometry, semi-automated (platelet function analyser (PFA-100-Siemens Healthcare Diagnostics, Deerfield, IL, USA)) and automated (Ultegra rapid platelet function assay (RPFA) - Accumetrics, San Diego, CA, VerifyNow - Accumetrics, San Diego, CA) platelet aggregometry, and impedance aggregometry (Multiple platelet function analyzer (Multiplate® analyzer - Verum Diagnostica GmbH, Munich,Germany)) (). Several agonists of varying concentration have been employed to stimulate platelet aggregation. However, the degree of involvement of COX-1- dependent pathway of platelet activation is not the same for different agonists. AA is the substrate for platelet COX-1, and aggregation response to AA-stimulation directly reflects platelet COX-1 activity. Activation-dependent changes in surface expression of P-selectin, CD63, activated GP IIb/IIIa receptor, and leucocyte-platelet aggregates in response to AA by flow cytometry may also be used to determine platelet inhibition by aspirin (Citation28–32).
Table I. Platelet function tests commonly used for measurement of antiplatelet effect of aspirin.
Comparison of different laboratory methods in assessment of ‘aspirin resistance’ usually shows moderate reproducibility and very weak mutual correlations between them (Citation33–36). This reflects their sensitivity to different pathways of platelet activation, both TxA2-dependent and independent. Accordingly, the reported prevalence of ‘aspirin resistance’ varies widely (e.g. from < 1% to 61%) (Citation37). A systematic review of 42 studies of aspirin use for secondary prevention showed a mean prevalence of ‘aspirin resistance’ of 24% (95% CI 20%–28%) (Citation38). However, studies measuring platelet aggregation using AA-stimulated LTA reported a pooled unadjusted prevalence of ‘aspirin resistance’ of only 6% (95% CI 1%–12%). In studies using point-of-care platelet function-analysing devices, the unadjusted prevalence was significantly higher, being 26% (95% CI 21%–31%), which, to a great extent, can be explained by lower specificity of the point-of-care tests used for COX-1.
In a study that employed several testing approaches simultaneously, the prevalence of ‘aspirin resistance’ was relatively low with the use of more specific tests—for example, AA-induced LTA (4%) and VerifyNow Aspirin (6.7%)—and much higher with non-specific tests, ADP-induced platelet aggregation (51.7%) and PFA-100 (59.5%) (Citation33).
Of note, interindividual variability of base-line platelet reactivity could affect the degree of platelet inhibition by aspirin (Citation39). This is particularly important when tests that are less specific to COX-1 pathway are used to assess platelet response to aspirin (Citation40). Factorial analysis has revealed the presence of a common factor (other than platelet COX-1) which explains almost half of the variation in platelet aggregation induced by collagen, ADP, and collagen-related peptide (Citation21). Furthermore, poor compliance with the medication is a potential reason for ‘aspirin resistance’ and should be considered in patients with high on-treatment platelet responsiveness. The prevalence of ‘aspirin resistance’ in compliant subjects has been reported to be generally low (2% or less) (Citation19).
Clinical implications of aspirin resistance
There is growing evidence that major adverse clinical events (MACE) in the settings of ACS, stroke/transient ischaemic attacks, and peripheral arterial disease can be predicted by the results of some tests evaluating on-treatment platelet reactivity.
Recently, several large meta-analyses have explored the relationship between laboratory data on platelet reactivity and risk of MACE (Citation41). In a meta-analysis of 20 studies including 2,930 patients, 28% of participants were classified as ‘aspirin-resistant’ by laboratory methods. Overall, 39% of ‘aspirin-resistant’ patients compared with 16% of patients responsive to aspirin developed cardiovascular events (odds ratio 3.85; 95% CI 3.08–4.80). The odds ratio for increased mortality in aspirin-resistant patients was even higher, at 5.99 (95% CI 2.28–15.72).
The laboratory method used in various studies is also important—for example, 16% were classified as resistant to aspirin with LTA with relatively small heterogeneity of the results but with the highest odds ratio (3.85; 95% CI 2.5–5.88). In contrast, with the PFA-100, 33% of subjects were identified as aspirin-resistant, with a higher heterogeneity of the results and a lower predictive value for thrombosis (odds ratio for cardiovascular outcomes 2.94; 95% CI 1.88–4.55) (Citation42). In another meta-analysis of 12 studies comprising 1,813 patients, the mean prevalence of aspirin resistance was 27%, although there was significant statistical heterogeneity amongst the studies (Citation43). The pooled odds ratio of cardiovascular outcomes was 3.8 (95% CI 2.3–6.1) independently of the aspirin dose used (≤ 100 mg, 101–299 mg, and ≥ 300 mg).
Two other meta-analyses focused on studies where PFA-100 with the collagen/epinephrine cartridge was used for definition of non-responders. In the analysis by Crescente et al. (Citation44) of 64 patient populations with 6,450 patients (2,283 subjects without history of vascular events and 4,167 patients affected by vascular events), the prevalence of aspirin non-responders was significantly higher in populations with vascular events (0.28 (95% CI 0.26–0.30) versus 0.23 (95% CI 0.21–0.25); P = 0.0002), and among them it was significantly higher in the acute phase of the disease. The factors associated with increased rate of poor responsiveness to aspirin were advanced age and type 2 diabetes. Pooled analysis found a relative risk of MACE amongst patients with ‘aspirin resistance’ of 1.63 (95% CI 1.16–2.28). Comparable results were obtained in another meta-analysis of studies based on the PFA-100 with an odds ratio of 2.1 (95% CI 1.4–3.4) for the increased recurrence of ischaemic events in aspirin non-responders (Citation45). Taken together, these data indicate that the PFA-100 may be less useful for the prediction of MACE relevant to ‘aspirin resistance’.
In a large prospective study, Frelinger et al. (Citation46) demonstrated that serum TxB2 levels (a marker of COX-1-dependent platelet inhibition) and platelet aggregation with PFA-100 and collagen/ADP cartridge (i.e. aggregation non-specific to COX-1 pathway) were significantly correlated with subsequent MACE. These observations indicate that poor clinical outcomes in the aspirin-treated may be partly related to incomplete COX-1 inhibition, but also to COX-1-independent platelet hyper-reactivity.
In the Heart Outcomes Prevention Evaluation (HOPE) study (Citation47), the 5-year follow-up of 976 patients with high risk of cardiovascular events revealed an increasing risk of MACE with each increasing quartile of 11-dehydro-TxB2 levels, with patients in the upper quartile having a 1.8-fold higher risk of all MACE, as well as a 2-fold higher risk of myocardial infarction and a 3.5-fold higher risk of cardiovascular death compared to those in the lower quartile. This association was independent of conventional cardiovascular risk factors. Similarly, being in the upper quartile of LTA levels was an independent risk factor for developing cardiovascular events within 12 months (hazard ratio 7.76; 95% CI 1.72–35.3) (Citation22).
In the pre-specified substudy of the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) (Citation48), base-line urinary 11-dehydro-TxB2 concentrations in the highest quartile were associated with an increased risk for MI, stroke, or cardiovascular death (adjusted hazard ratio 1.66; 95% CI 1.06–2.61). Increasing age, female gender, history of peripheral artery disease, current smoking, and oral hypoglycaemic or angiotensin-converting enzyme inhibitor therapy were also independently associated with higher urinary concentrations of 11-dehydro-TxB2, whereas aspirin dose ≥ 150 mg, history of treatment with non-steroidal anti-inflammatory drugs, history of hypercholesterolaemia, and statin treatment were associated with lower concentrations. However, urinary 11-dehydro-TxB2 is not a specific marker of platelet TxA2 formation as it may also be produced by monocytes/macrophages in inflamed atherosclerotic plaques (Citation49).
In a study of 1,069 patients taking clopidogrel and undergoing elective PCI, residual platelet reactivity has been detected using LTA, VerifyNow, and Plateletworks (Helena Laboratories, Beaumont, TX, USA) (Citation50). Although the predictive accuracy of these tests was modest, high on-treatment platelet reactivity confirmed by any of these methods was associated with increased incidence of primary composite point of all-cause death, non-fatal acute MI, stent thrombosis, and ischaemic stroke. Many of the above studies had certain limitations, sometimes being underpowered and inadequately adjusted for potential confounders (age, gender, haemoglobin and platelet levels, hyperlipidaemia, etc.) or even compliance (Citation41,Citation51). Nevertheless the conclusions are generally uniform, that is, high platelet reactiveness on the antiplatelet therapy is linked to an increased risk of atherothrombosis (Citation41).
However, there is still some debate on the relative importance of response to aspirin itself and high background platelet reactivity which cannot be sufficiently inhibited by aspirin. In fact, whereas the true ‘aspirin resistance’ appears to be a relatively rare phenomenon, residual on-treatment platelet reactivity can have greater importance. Indeed, in patients with ACS, high on-treatment platelet reactivity according to LTA with three agonists (AA, ADP, collagen) had the highest predictive value for cardiovascular death and non-fatal MI within 12 months (OR 4.7; 95% CI 2.9–7.7), whilst isolated platelet hyper-reactivity to only one agonist had no predictive value (Citation52). In a study of 326 stable CAD patients, there was an association of ‘aspirin resistance’ with increased risk for all-cause death, MI, or stroke with both LTA with AA (i.e. COX-1-dependent pathway) and 10 µmol/L ADP (i.e. largely independent of TxA2 production) (Citation2,Citation53). Also, Mueller et al. have demonstrated an 87% higher risk of arterial reocclusion after peripheral angioplasty in patients with ‘aspirin resistance’ as defined by response to ADP which parallels with appropriate aggregation in response to AA, an aspirin-specific stimulator (Citation54). This observation indirectly confirms that background platelet reactivity may be more important rather than just the response to aspirin alone. Of note, in the cases of so-called ‘dual resistance’ to both aspirin and ADP receptor inhibitors, poor compliance with the medications should be considered (Citation40,Citation55–58).
‘Aspirin resistance’, residual platelet reactivity, or higher activity of inflammatory-coagulation response in severe vascular disease
Various genetic, biological, clinical factors affecting pharmacokinetics and pharmacodynamics of aspirin and modulating platelet sensitivity to the drug are likely to play a role in ‘aspirin resistance’ (). Alongside well known factors contributing to poor response to aspirin treatment, such as factors reducing bioavailability of aspirin or alternative pathways of platelet activation, the variable effectiveness of aspirin in terms of clinical outcome and laboratory tests may be related to additional antithrombotic effects of aspirin such as inhibition of thrombin generation and attenuation of factor XIII activation (Citation62–64). These effects of aspirin are linked with common genetic polymorphisms such as the Leu33Pro β3-integrin or Val34Leu factor XIII mutations (Citation62–64). In addition, elevated levels of cholesterol (more than 240 mg/dL) appear to diminish the influence of aspirin on thrombin formation at the site of microvascular injury (Citation62,Citation64). Also, hypercholesterolaemia and smoking increase platelet response to other agonists, TxA2 production from AA via lipid peroxidation, thus further contributing to ‘aspirin resistance’ (Citation64).
Figure 1. Potential mechanisms of aspirin resistance.
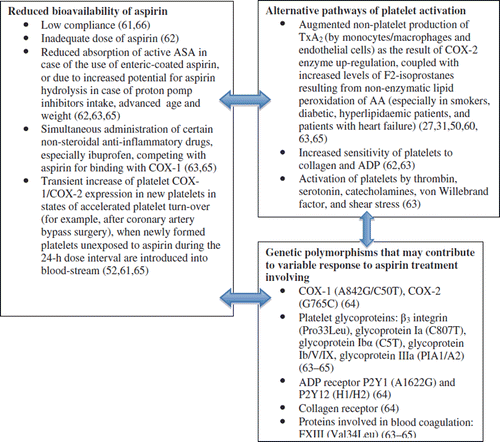
There is also growing evidence that platelets play an intimate role in the inflammatory changes that lead to atherosclerosis, being a key cellular interface between coagulation and inflammation. Platelets are activated by inflammatory mediators and propagate inflammation by the further release of inflammatory substances, such as platelet factor 4 (PF4), IL-1β, RANTES (CCL5), and the surface expression of adhesion molecules such as GP Ib/IX/V, P-selectin, CD40L, and TNF superfamily 14 (TNFSF14) (Citation66,Citation67). Platelet receptors and released molecules also play an important role in acceleration of atherosclerosis by mediating leucocyte recruitment and adhesion to the vascular wall. Soluble CD40L (sCD40L) binds to platelet integrin glycoprotein IIb/IIIa, amplifying platelet activation in an autocrine fashion (Citation66).
Platelet interactions with the monocytes—in the circulation or at the vessel wall itself—result in monocyte activation, which subsequently become more adhesive, more migratory, more procoagulant (increased tissue factor expression), more proinflammatory, and prone to macrophage differentiation.
Additionally, both monocytes and platelets contribute to an inflammatory phenotype of endothelium with enhanced expression of adhesion molecules and the release of IL-8, monocyte chemoattractant (MCP-1), matrix metalloproteases, and reactive oxygen species (Citation66–69). This further promotes adhesion and activation of platelets and monocytes. At the same time, a dysfunctional endothelium, an early feature of atherosclerosis, produces less NO and prostacyclin, and releasing inflammatory molecules itself contributes to platelet activation and adhesion to the vascular wall.
Oxidized low-density lipoproteins (LDL) and high-density lipoproteins (HDL) have been shown to bind to platelet CD36, thus, in a dose-dependent way, provoking the release of significant amounts of CD40L as well as phosphatidylserine on the platelet surface (Citation70–72). Stimulation both with thrombin and with aggregated IgG complexes induces similar release of sCD40L and RANTES from platelets, indicating common activation pathways of the coagulation system and the innate inflammatory response (Citation73).
Whole blood assays of platelet function, examining platelet response in conditions of cell interactions, usually show a higher rate of increased on-treatment platelet reactivity than aggregation in PRP per se. For example, Chen et al. demonstrated that ‘aspirin resistance’ as measured by VerifyNow Aspirin was associated with impaired coronary flow reserve in patients who underwent elective PCI (Citation74). In animal experiments, interactions between white blood cells (polymorphonuclear cells and monocytes) and platelets were suggested to play an important role in the pathogenesis of the ‘no-reflow’ phenomenon seen in interventional coronary procedures (Citation75). In 1,600 healthy individuals with a family history of CAD, whole blood aggregation (but not aggregation in PRP depleted of leucocytes) increased with increasing quartile of leucocyte count both at base-line and after low-dose aspirin treatment, although the absolute magnitude of platelet reactivity was suppressed by aspirin therapy (Citation76).
Tantry et al. (Citation77) demonstrated incremental changes in platelet function, as measured by GP IIb/IIIa expression and increased release of RANTES, IL-8, and MCP-1, as CAD progresses from the asymptomatic state to stable angina and, finally, to unstable angina. The demonstration of significant correlations between inflammation, platelet function, and hypercoagulability markers has been interpreted by some as support for the hypothesis that reactive and activated platelets influence inflammation and other cells involved in low-grade inflammation of the vascular wall, thus creating a vicious cycle leading to a prothrombotic state, atherosclerosis progression, and/or plaque destabilization (Citation78–80).
There is growing evidence that high platelet reactivity and atherosclerosis severity are closely related. Indeed, a high degree of platelet reactivity has been demonstrated in patients with more extensive coronary atherosclerosis and more severe peripheral arterial disease (Citation81,Citation82). Platelet activation reflects the stability of the CAD, with the greatest degree of platelet activation occurring in patients with onset of MI < 48 h (Citation83). Frossard et al. (Citation84) reported that the PFA-100 closure time (CT) at presentation may be an independent predictor of the severity of a STEMI (perhaps reflecting the ‘stability’ of a STEMI presentation), as measured by markers of myonecrosis. Also, Campo et al. (Citation85) demonstrated that in patients undergoing PCI, base-line platelet reactivity (as measured by PFA-100 CT) influences the angiographic success of the procedure, the degree of ST-segment resolution, the extent of myonecrosis, and the short- and mid-term clinical outcomes.
Whilst ACS is independently associated with increased on-treatment platelet reactivity (Citation86), this may be due to a higher pretreatment platelet activity in coronary ischaemic events (Citation11). A significant association between interferon-inducible protein (IP-10), interferon-gamma (IFN-gamma), IL-4, and residual platelet reactivity can be demonstrated in patients with ACS (Citation87). Elevated levels of CRP, leucocyte count, and fibrinogen are also significantly associated with high platelet reactivity in stable CAD patients on chronic clopidogrel treatment (Citation88). The rate of platelet aggregation is also significantly related to monocyte count, high-sensitivity CRP (hs-CRP), mean platelet volume, vWF levels, and activity (Citation89,Citation90).
These findings indicate the strong impact of the systemic inflammatory properties on the pattern of platelet activity independently of the administration of aspirin or other antiplatelet agents. Indeed, there is a clear relationship between elevated inflammatory biomarkers and the risk of future atherothrombotic events (Citation91–96), and dual antiplatelet therapy with aspirin and clopidogrel seems to bring more benefit to those with increased levels of inflammatory biomarkers (Citation97,Citation98).
Recent data show that obese individuals and patients with metabolic syndrome have greater native platelet reactivity and even retain greater reactivity after administration of aspirin (Citation99). This parallels signs of chronic inflammation, such as increased leucocyte count and levels of CRP and sCD40L in these patients (Citation100). Patients with diabetes have increased oxidative stress, inflammation, and endothelial dysfunction, and these may be related to higher pretreatment platelet activity, higher number of platelet-monocyte aggregates, enhanced biosynthesis of TxA2 by monocytes, higher rate of non-responsiveness to antiplatelet therapy, and a higher rate of atherothrombotic complications in these patients (Citation101–105).
It has been suggested that higher rates of non-responsiveness to antiplatelet therapy among diabetic patients may contribute to the lack of efficacy of aspirin for primary prevention of cardiovascular events in this population, and in contrast to the diabetes-free patients (Citation106–108). Current prevention strategy in diabetic patients includes wide use of statins and inhibitors of the angiotensin system. The diverse effects of these agents include some anti-inflammatory properties and the ability to improve endothelial function. This may indirectly reduce platelet reactivity and blur the effect of aspirin on clinical outcomes in primary prevention amongst diabetics.
Shall we treat ‘aspirin resistance’?
The demonstration of the link between suboptimal platelet responsiveness to aspirin and increased risk of cardiovascular events prompted the concept of antiplatelet therapy monitoring. However, what should be the target of such monitoring? Shall we identify patients with true resistance to antiplatelet drugs, or shall we identify patients with high on-treatment platelet reactivity who may be at higher risk of ischaemic events?
We can only identify true non-responders using laboratory tests that are specific for the pharmacological target of an antiplatelet drug (Citation30). The possibility of in-vitro testing of platelet resistance to aspirin has been demonstrated, but its clinical utility is still being established (Citation4). For identifying patients with high residual platelet reactivity we need a well standardized test which is inexpensive, easy to perform, and reproducible, that may be used in different laboratories and results may be compared. This test must prove its utility in large prospective clinical studies.
To date, there are no robust clinical data obtained from large prospective trials with sufficient numbers of patients proving that the routine determination and/or monitoring of platelet function and consequent clinical decisions concerning antiplatelet therapy would lead to clinically relevant advantages and changes in patient management that are cost-effective. Consequently, no specific recommendations have been published for platelet function testing in routine clinical practice (Citation5,Citation109). However, class IIB recommendations from the American College of Cardiology/American Heart Association Task Force on Practice Guidelines have stated that platelet aggregation studies are warranted in patients undergoing PCI who are at risk of subacute stent thrombosis and in whom possible stent thrombosis might lead to devastating complications, with the option to increase the maintenance dose of clopidogrel from 75 to 150 mg/day in patients with < 50% blockade of platelet aggregation despite antiplatelet therapy (Citation110).
The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS) states that monitoring of antiplatelet response by platelet function assays is currently used for clinical research, but not in daily clinical practice (Citation111). At the same time, the use of a higher maintenance dose (150 mg) of clopidogrel is mentioned in patients with high thrombotic risk (e.g. in diabetics, patients after recurrent MI, after early and late stent thrombosis, for complex lesions, or in life-threatening situations should occlusion occur), undergoing elective PCI (Citation111). Similarly, there are no recommendations for treatment of incomplete response to antiplatelet therapy ‘discovered’ using one of the platelet function tests (Citation5).
On-going clinical trials aim to evaluate the effect of modifying therapy on clinical outcome for patients deemed non-responsive to antiplatelet therapy according to platelet function tests (). In the ASCET study, 1,001 patients with documented coronary heart disease were randomized to either continued treatment with aspirin 160 mg/d or change to clopidogrel 75 mg/d. Clinical end-points (the composite of unstable angina, myocardial infarction, stroke, or death) were recorded for at least 2 years and related to the initial aspirin response, assessed by the PFA-100 method (Citation113,Citation118). In all, 26% of patients had high on-treatment platelet reactivity at randomization, but this did not significantly influence occurrence of the primary end-point (13.1% in non-responders versus 10.5% in responders; P = 0.41). Among aspirin non-responders who switched to clopidogrel, there was a 40% reduction in the combined end-point compared with those who stayed on aspirin, but this finding was not statistically significant, and there was no difference in the number of primary end-point events between the randomized groups (56/503 on aspirin versus 50/498 on clopidogrel; P = 0.57) (Citation119). Recently published data of the BOCLA-Plan trial indicate that low responsiveness to aspirin or clopidogrel, defined with a platelet function test, can be overcome by escalating of the aspirin maintenance dose in case of low response to aspirin—or by an increase of the maintenance dose of clopidogrel, or switching to ticlopidine, or including prasugrel in the treatment plan, if low response to clopidogrel is detected (Citation120). However, this study did not aim to investigate if the strategy to improve biochemical response provides improved MACE rates without causing an increased risk of bleeding. Future randomized clinical trials are needed to prove the clinical benefits of an individually tailored antiplatelet therapy.
Table II. Studies evaluating the effect of modifying therapy based on platelet responsiveness to aspirin (113–122).
Thus, the optimal treatment, if any, of aspirin resistance is unknown. Therefore, it is not currently recommended to test for aspirin resistance in patients or to change therapy based on these tests, in routine clinical practice (Citation5). Patients that have experienced a recurrent vascular event require detailed evaluation of the causes of the initial and recurrent events. Whenever the suspicion of aspirin resistance is raised by laboratory tests, compliance and potentially modifiable causes of aspirin failure should be considered and optimized (e.g. concurrent intake of non-steroidal anti-inflammatory drugs, reduced absorption of aspirin, or inadequate dose of aspirin), and other risk factors potentially influencing platelet function (e.g. smoking, hypercholesterolaemia, inadequate glucose control, obesity) should be managed appropriately.
The experts of the Working Group on antiplatelet drug resistance admit the possibility of individual antiplatelet dosing in individual cases (e.g. patients with multiple cardiovascular risk factors and recurrent thrombotic events despite proven compliance to standard antiplatelet drugs doses). However, the Working Group states that those measures should be undertaken only as a research activity in academic centres with experience in platelet function testing, and currently there is not sufficient evidence of their efficacy (Citation5).
New, more potent P2Y12 receptor antagonists are currently available, and on-going clinical trials aim to justify an individualized approach to antiplatelet therapy, based on on-treatment platelet reactivity. Cholesterol-independent effects of statins, which include improvement of endothelial function, decrease of oxidative stress, and inflammation, are thought to inhibit indirectly platelet activity (Citation48). Recent studies showed that statins significantly reduce platelet TxA2 formation in patients taking low-dose aspirin (Citation122), prompt overcoming of aspirin resistance in 65% of the patients after a therapy of 3 months (Citation123), and may have a direct effect on platelet eicosanoid synthesis (Citation124). Combined with aspirin, omega-3 polyunsaturated fatty acids facilitate aspirin-induced platelet inhibition (Citation125), improving response to aspirin comparably with the aspirin dose increase (Citation126). The addition of omega-3 polyunsaturated fatty acids to dual antiplatelet therapy significantly potentiates platelet response to clopidogrel after PCI (Citation127).
Some existing evidence links inflammation and thrombosis, as well as anti-inflammatory properties of antiplatelet drugs (Citation128), which supports the hypothesis that agents with both anti- inflammatory and antiplatelet effects may reduce vascular inflammation and platelet reactivity and limit acute and long-term thrombotic events.
Conclusion
Currently, extensive literature exists about aspirin resistance, its mechanisms, detection, and its treatment. Existence of a link between high on-treatment platelet reactivity and atherothrombotic events suggests common mechanisms for atherosclerosis progression and thrombotic complications, with the platelets being a key cellular interface between coagulation and inflammation. A change in the approach to prevention of atherothrombotic events may ultimately include all the components of atherothrombogenesis, including platelets, monocytes, and endothelial cells.
Nonetheless, tailoring antiplatelet therapy in accordance with the presence of aspirin resistance is one possible solution. However, the evidence in favour of this strategy is insufficient.
Acknowledgements
The work was supported by the European Society of Cardiology Atherothrombosis Fellowship.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Patrono C. Aspirin resistance: definition, mechanisms and clinical read-outs. J Thromb Haemost. 2003;1:1710–3.
- Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24:1980–7.
- Weber AA, Przytulski B, Schanz A, Hohlfeld T, Schrör K. Towards a definition of aspirin resistance: a typological approach. Platelets. 2002;13:37–40.
- Pulcinelli FM, Riondino S. More on aspirin resistance: position paper of the Working Group on Aspirin Resistance. Proposal for a Laboratory Test Guiding Algorithm. J Thromb Haemost. 2006;4:485–7.
- Kuliczkowski W, Witkowski A, Polonski L, Watala C, Filipiak K, Budaj A, . Interindividual variability in the response to oral antiplatelet drugs: a position paper of the Working Group on antiplatelet drugs resistance appointed by the Section of Cardiovascular Interventions of the Polish Cardiac Society, endorsed by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J. 2009;30:426–35.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2:15–28.
- Ben-Dor I, Kleiman NS, Lev E. Assessment, mechanisms, and clinical implication of variability in platelet response to aspirin and clopidogrel therapy. Am J Cardiol. 2009;104:227–33.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AI, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol. 2005;45:246–51.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramírez C, Sabaté M, Jimenez-Quevedo P, . Influence of aspirin resistance on platelet function profiles in patients on long-term aspirin and clopidogrel after percutaneous coronary intervention. Am J Cardiol. 2006;97:38–43.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramírez C, Cavallari U, Trabetti E, . Variability in platelet aggregation following sustained aspirin and clopidogrel treatment in patients with coronary heart disease and influence of the 807 C/T polymorphism of the glycoprotein Ia gene. Am J Cardiol. 2005;96:1095–9.
- Gurbel PA, Bliden KP, Hiatt BL, O’Connor CM. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation. 2003;107:2908–13.
- Pedersen AK, FitzGerald GA. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–11.
- Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–6.
- Maree AO, Curtin RJ, Dooley M, Conroy RM, Crean P, Cox D, . Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J Am Coll Cardiol. 2005;46:1258–63.
- Minuz P, Fumagalli L, Gaino S, Tommasoli RM, Degan M, Cavallini C, . Rapid stimulation of tyrosine phosphorylation signals downstream of G-protein-coupled receptors for thromboxane A2 in human platelets. Biochem J. 2006;400:127–34.
- Béres BJ, Tóth-Zsámboki E, Vargová K, László A, Masszi T, Kerecsen G, . Analysis of platelet alpha2-adrenergic receptor activity in stable coronary artery disease patients on dual antiplatelet therapy. Thromb Haemost. 2008;100:829–38.
- Williams MS, Kickler TS, Vaidya D, Ng'alla LS, Bush DE. Evaluation of platelet function in aspirin treated patients with CAD. J Thromb Thrombolysis. 2006;21:241–7.
- Fontana P, Nolli S, Reber G, de Moerloose P. Biological effects of aspirin and clopidogrel in a randomized cross-over study in 96 healthy volunteers. J Thromb Haemost. 2006;4:813–9.
- Frelinger AL 3rd, Furman MI, Linden MD, Li Y, Fox ML, Barnard MR, . Residual arachidonic acid-induced platelet activation via an adenosine diphosphate-dependent but cyclooxygenase-1- and cyclooxygenase-2-independent pathway: a 700-patient study of aspirin resistance. Circulation. 2006;113:2888–96.
- Hedegaard SS, Hvas AM, Grove EL, Refsgaard J, Rocca B, Daví G, . Optical platelet aggregation versus thromboxane metabolites in healthy individuals and patients with stable coronary artery disease after low-dose aspirin administration. Thromb Res. 2009;124:96–100.
- Ohmori T, Yatomi Y, Nonaka T, Kobayashi Y, Madoiwa S, Mimuro J, . Aspirin resistance detected with aggregometry cannot be explained by cyclooxygenase activity: involvement of other signaling pathway(s) in cardiovascular events of aspirin-treated patients. J Thromb Haemost. 2006;4:1271–8.
- Patrono C, Ciabattoni G, Pugliese F, Pierucci A, Blair IA, FitzGerald GA. Estimated rate of thromboxane secretion into the circulation of normal humans. J Clin Invest. 1986;77:590–4.
- Chiabrando C, Rivoltella L, Martelli L, Valzacchi S, Fanelli R. Urinary excretion of thromboxane and prostacyclin metabolites during chronic low-dose aspirin: evidence for an extrarenal origin of urinary thromboxane B2 and 6-keto-prostaglandin F1 alpha in healthy subjects. Biochim Biophys Acta. 1992;1133:247–54.
- Catella F, FitzGerald GA. Paired analysis of urinary thromboxane B2 metabolites in humans. Thromb Res. 1987;47:647–56.
- Maclouf J, Folco G, Patrono C. Eicosanoids and iso-eicosanoids: constitutive, inducible and transcellular biosynthesis in vascular disease. Thromb Haemost. 1998;79:691–705.
- Karim S, Habib A, Lévy-Toledano S, Maclouf J. Cyclooxygenase-1 and -2 of endothelial cells utilize exogenous or endogenous arachidonic acid for transcellular production of thromboxane. J Biol Chem. 1996;271:12042–8.
- Weber AA, Zimmermann KC, Meyer-Kirchrath J, Schrör K. Cyclooxygenase-2 in human platelets as a possible factor in aspirin resistance. Lancet. 1999;353:900.
- Michelson AD. Platelet function testing in cardiovascular diseases. Circulation. 2004;110:e489–93.
- Harrison P, Frelinger AL 3rd, Furman MI, Michelson AD. Measuring antiplatelet drug effects in the laboratory. Thromb Res. 2007;120:323–36.
- Cattaneo M. Resistance to antiplatelet drugs: molecular mechanisms and laboratory detection. J Thromb Haemost. 2007;5 Suppl 1:230–7.
- Coleman JL, Wang JC, Simon DI. Determination of individual response to aspirin therapy using the Accumetrics Ultegra RPFA-ASA System. Point Care. 2004;3:77–82.
- Schmitz G, Rothe G, Ruf A, Barlage S, Tschöpe D, Clemetson KJ, . European Working Group on Clinical Cell Analysis: Consensus protocol for the flow cytometric characterisation of platelet function. Thromb Haemost. 1998;79:885–96.
- Lordkipanidzé M, Pharand C, Schampaert E, Turgeon J, Palisaitis DA, Diodati JG. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J. 2007;28:1702–8.
- Grove EL, Hvas AM, Johnsen HL, Hedegaard SS, Pedersen SB, Mortensen J, . A comparison of platelet function tests and thromboxane metabolites to evaluate aspirin response in healthy individuals and patients with coronary artery disease. Thromb Haemost. 2010;103:1245–53.
- Muir AR, McMullin MF, Patterson C, McKeown PP. Assessment of aspirin resistance varies on a temporal basis in patients with ischaemic heart disease. Heart. 2009;95:1225–9.
- Chakroun T, Addad F, Abderazek F, Ben-Farhat M, Hamdi S, Gamra H, . Screening for aspirin resistance in stable coronary artery patients by three different tests. Thromb Res. 2007;121:413–8.
- Campbell CL, Steinhubl SR. Variability in response to aspirin: do we understand the clinical relevance? J Thromb Haemost. 2005;3:665–9.
- Hovens MM, Snoep JD, Eikenboom JC, van der Bom JG, Mertens BJ, Huisman MV. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175–81.
- Guthikonda S, Mangalpally K, Vaduganathan M, Patel R, Delao T, Bergeron AL, . Increased platelet sensitivity among individuals with aspirin resistance—platelet aggregation to submaximal concentration of arachidonic acid predicts response to antiplatelet therapy. Thromb Haemost. 2008;100:83–9.
- Dichiara J, Bliden KP, Tantry US, Chaganti SK, Kreutz RP, Gesheff TB, . Platelet function measured by VerifyNow identifies generalized high platelet reactivity in aspirin treated patients. Platelets. 2007;18:414–23.
- Zimmermann N, Hohlfeld T. Clinical implications of aspirin resistance. Thromb Haemost. 2008;100:379–90.
- Krasopoulos G, Brister SJ, Beattie WS, Buchanan MR. Aspirin ‘resistance’ and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336:195–8.
- Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med. 2007;167:1593–9.
- Crescente M, Di Castelnuovo A, Iacoviello L, Vermylen J, Cerletti C, de Gaetano G. Response variability to aspirin as assessed by the platelet function analyzer(PFA)-100. A systematic review. Thromb Haemost. 2008;99:14–26.
- Reny JL, De Moerloose P, Dauzat M, Fontana P. Use of the PFA-100 closure time to predict cardiovascular events in aspirin-treated cardiovascular patients: a systematic review and meta-analysis. J Thromb Haemost. 2008;6:444–50.
- Frelinger AL 3rd, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, . Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–96.
- Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–5.
- Eikelboom JW, Hankey GJ, Thom J, Bhatt DL, Steg PG, Montalescot G, . Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation. 2008;118:1705–12.
- Halushka MK, Halushka PV. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2? Circulation. 2002;105:1620–2.
- Breet NJ, van Werkum JW, Bouman HJ, Kelder JC, Ruven HJ, Bal ET, . Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. 2010;303:754–62.
- Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006;367 606–17.
- Marcucci R, Gori AM, Paniccia R, Giusti B, Valente S, Giglioli C, . High on-treatment platelet reactivity by more than one agonist predicts 12-month follow-up cardiovascular death and non-fatal myocardial infarction in acute coronary syndrome patients receiving coronary stenting. Thromb Haemost. 2010;104:279–86.
- Gum PA, Kottke-Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–5.
- Mueller MR, Salat A, Stangl P, Murabito M, Pulaki S, Boehm D, . Variable platelet response to low-dose ASA and the risk of limb deterioration in patients submitted to peripheral arterial angioplasty. Thromb Haemost. 1997;78:1003–7.
- Lev EI, Patel RT, Maresh KJ, Guthikonda S, Granada J, DeLao T, . Aspirin and clopidogrel drug response in patients undergoing percutaneous coronary intervention: the role of dual drug resistance. J Am Coll Cardiol. 2006;47:27–33.
- Gori AM, Marcucci R, Migliorini A, Valenti R, Moschi G, Paniccia R, . Incidence and clinical impact of dual nonresponsiveness to aspirin and clopidogrel in patients with drug-eluting stents. J Am Coll Cardiol. 2008;52:734–9.
- Cuisset T, Frere C, Quilici J, Morange PE, Camoin L, Bali L, . Relationship between aspirin and clopidogrel responses in acute coronary syndrome and clinical predictors of non response. Thromb Res. 2009;123:597–603.
- Fontana P, Berdagué P, Castelli C, Nolli S, Barazer I, Fabbro-Peray P, . Clinical predictors of dual aspirin and clopidogrel poor responsiveness in stable cardiovascular patients from the ADRIE study. J Thromb Haemost. 2010;8:2614–23.
- Gasparyan AY, Watson T, Lip GY. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008;51:1829–43.
- Hanjis C, Frishman WH, Lerner RG. Aspirin resistance: mechanisms and clinical implications. Cardiol Rev. 2006;14:18–25.
- Sanderson S, Emery J, Baglin T, Kinmonth AL. Narrative review: aspirin resistance and its clinical implications. Ann Intern Med. 2005;142:370–80.
- Undas A, Brummel-Ziedins KE, Mann KG. Antithrombotic properties of aspirin and resistance to aspirin: beyond strictly antiplatelet actions. Blood. 2007;109:2285–92.
- Goodman T, Ferro A, Sharma P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review. Br J Clin Pharmacol. 2008;66:222–32.
- Szczeklik A, Musiał J, Undas A, Sanak M, Nizankowski R. Aspirin resistance. Pharmacol Rep. 2005;57 Suppl:33–41.
- Schwartz KA, Schwartz DE, Ghosheh K, Reeves MJ, Barber K, DeFranco A. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol. 2005;95:973–5.
- Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood. 2009;114:2367–74.
- Van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204.
- Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–57.
- Shantsila E, Lip GY. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb Haemost. 2009;102:916–24.
- Assinger A, Koller F, Schmid W, Zellner M, Babeluk R, Koller E, . Specific binding of hypochlorite-oxidized HDL to platelet CD36 triggers proinflammatory and procoagulant effects. Atherosclerosis. 2010;212:153–60.
- Assinger A, Koller F, Schmid W, Zellner M, Koller E, Volf I. Hypochlorite-oxidized LDL induces intraplatelet ROS formation and surface exposure of CD40L-A prominent role of CD36. Atherosclerosis. 2010;213:129–34.
- Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, . Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–95.
- Antczak AJ, Singh N, Gay SR, Worth RG. IgG-complex stimulated platelets: a source of sCD40L and RANTES in initiation of inflammatory cascade. Cell Immunol. 2010;263:129–33.
- Chen WH, Lee PY, Ng W, Kwok JY, Cheng X, Lee SW, . Relation of aspirin resistance to coronary flow reserve in patients undergoing elective percutaneous coronary intervention. Am J Cardiol. 2005;96:760–3.
- Charron T, Jaffe R, Segev A, Bang KW, Qiang B, Sparkes JD, . Effects of distal embolization on the timing of platelet and inflammatory cell activation in interventional coronary no-reflow. Thromb Res. 2010;126:50–5.
- Faraday N, Yanek LR, Vaidya D, Kral B, Qayyum R, Herrera-Galeano JE, . Leukocyte count is associated with increased platelet reactivity and diminished response to aspirin in healthy individuals with a family history of coronary artery disease. Thromb Res. 2009;124:311–7.
- Tantry US, Bliden KP, Suarez TA, Kreutz RP, Dichiara J, Gurbel PA. Hypercoagulability, platelet function, inflammation and coronary artery disease acuity: results of the Thrombotic RIsk Progression (TRIP) study. Platelets. 2010;21:360–7.
- Jurk K, Ritter MA, Schriek C, Van Aken H, Droste DW, Ringelstein EB, . Activated monocytes capture platelets for heterotypic association in patients with severe carotid artery stenosis. Thromb Haemost. 2010;103:1193–202.
- Zalai CV, Kolodziejczyk MD, Pilarski L, Christov A, Nation PN, Lundstrom-Hobman M, . Increased circulating monocyte activation in patients with unstable coronary syndromes. J Am Coll Cardiol. 2001;38:1340–7.
- Liuzzo G, Angiolillo DJ, Buffon A, Rizzello V, Colizzi C, Ginnetti F, . Enhanced response of blood monocytes to in vitro lipopolysaccharide-challenge in patients with recurrent unstable angina. Circulation. 2001;103:2236–41.
- Mangiacapra F, De Bruyne B, Muller O, Trana C, Ntalianis A, Bartunek J, . High residual platelet reactivity after clopidogrel: extent of coronary atherosclerosis and periprocedural myocardial infarction in patients with stable angina undergoing percutaneous coronary intervention. JACC Cardiovasc Interv. 2010;3:35–40.
- Smith T, Dhunnoo G, Mohan I, Charlton-Menys V. A pilot study showing an association between platelet hyperactivity and the severity of peripheral arterial disease. Platelets. 2007;18:245–8.
- Linden MD, Furman MI, Frelinger AL 3rd, Fox ML, Barnard MR, Li Y, . Indices of platelet activation and the stability of coronary artery disease. J Thromb Haemost. 2007;5:761–5.
- Frossard M, Fuchs I, Leitner JM, Hsieh K, Vlcek M, Losert H, . Platelet function predicts myocardial damage in patients with acute myocardial infarction. Circulation. 2004;110:1392–7.
- Campo G, Valgimigli M, Gemmati D, Percoco G, Tognazzo S, Cicchitelli G, . Value of platelet reactivity in predicting response to treatment and clinical outcome in patients undergoing primary coronary intervention: insights into the STRATEGY Study. J Am Coll Cardiol. 2006;48:2178–85.
- Geisler T, Kapp M, Göhring-Frischholz K, Daub K, Dösch C, Bigalke B, . Residual platelet activity is increased in clopidogrel- and ASA-treated patients with coronary stenting for acute coronary syndromes compared with stable coronary artery disease. Heart. 2008;94:743–7.
- Gori AM, Cesari F, Marcucci R, Giusti B, Paniccia R, Antonucci E, . The balance between pro- and anti-inflammatory cytokines is associated with platelet aggregability in acute coronary syndrome patients. Atherosclerosis. 2009;202:255–62.
- Bernlochner I, Steinhubl S, Braun S, Morath T, Jaitner J, Stegherr J, . Association between inflammatory biomarkers and platelet aggregation in patients under chronic clopidogrel treatment. Thromb Haemost. 2010;104:1193–200.
- Elsenberg EH, van Werkum JW, van de Wal RM, Zomer AC, Bouman HJ, Verheugt FW, . The influence of clinical characteristics, laboratory and inflammatory markers on ‘high on-treatment platelet reactivity’ as measured with different platelet function tests. Thromb Haemost. 2009;102:719–27.
- Chakroun T, Gerotziafas G, Robert F, Lecrubier C, Samama MM, Hatmi M, . In vitro aspirin resistance detected by PFA-100 closure time: pivotal role of plasma von Willebrand factor. Br J Haematol. 2004;124:80–5.
- Elesber AA, Conover CA, Denktas AE, Lennon RJ, Holmes DR Jr, Overgaard MT, . Prognostic value of circulating pregnancy- associated plasma protein levels in patients with chronic stable angina. Eur Heart J. 2006;27:1678–84.
- Ferrante G, Niccoli G, Biasucci LM, Liuzzo G, Burzotta F, Galiuto L, . Association between C-reactive protein and angiographic restenosis after bare metal stents: an updated and comprehensive meta-analysis of 2747 patients. Cardiovasc Revasc Med. 2008;9:156–65.
- Huang PH, Lu TM, Wu TC, Lin FY, Chen YH, Chen JW, . Usefulness of combined high-sensitive C-reactive protein and N-terminal-probrain natriuretic peptide for predicting cardiovascular events in patients with suspected coronary artery disease. Coron Artery Dis. 2008;19:187–93.
- Ortolani P, Marzocchi A, Marrozzini C, Palmerini T, Saia F, Taglieri N, . Predictive value of high sensitivity C-reactive protein in patients with ST-elevation myocardial infarction treated with percutaneous coronary intervention. Eur Heart J. 2008;29:1241–9.
- Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, . C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–97.
- Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, . Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355:2631–9.
- Dosh K, Berger PB, Marso S, van Lente F, Brennan DM, Charnigo R, . Relationship between baseline inflammatory markers, antiplatelet therapy, and adverse cardiac events after percutaneous coronary intervention: an analysis from the clopidogrel for the reduction of events during observation trial. Circ Cardiovasc Interv. 2009;2:503–12.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–9.
- Bordeaux BC, Qayyum R, Yanek LR, Vaidya D, Becker LC, Faraday N, . Effect of obesity on platelet reactivity and response to low-dose aspirin. Prev Cardiol. 2010;13:56–62.
- Unek IT, Bayraktar F, Solmaz D, Ellidokuz H, Yuksel F, Sisman AR, . Enhanced levels of soluble CD40 ligand and C-reactive protein in a total of 312 patients with metabolic syndrome. Metabolism. 2010;59:305–13.
- Schneider DJ. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care. 2009;32:525–7.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost. 2008;100:76–82.
- Kaplar M, Kappelmayer J, Veszpremi A, Szabo K, Udvardy M. The possible association of in vivo leukocyte-platelet heterophilic aggregate formation and the development of diabetic angiopathy. Platelets. 2001;12:419–22.
- Danenberg HD, Kantak N, Grad E, Swaminathan RV, Lotan C, Edelman ER. C-reactive protein promotes monocyte-platelet aggregation: an additional link to the inflammatory-thrombotic intricacy. Eur J Haematol. 2007;78:246–52.
- Konieczkowski M, Skrinska VA. Increased synthesis of thromboxane A(2) and expression of procoagulant activity by monocytes in response to arachidonic acid in diabetes mellitus. Prostaglandins Leukot Essent Fatty Acids. 2001;65:133–8.
- Bartolucci AA, Howard G. Meta-analysis of data from the six primary prevention trials of cardiovascular events using aspirin. Am J Cardiol. 2006;98:746–50.
- Ogawa H, Nakayama M, Morimoto T, Uemura S, Kanauchi M, Doi N, .; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300:2134–41.
- Belch J, MacCuish A, Campbell I, Cobbe S, Taylor R, Prescott R, .; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ. 2008;337:a1840.
- Michelson AD, Cattaneo M, Eikelboom JW, Gurbel P, Kottke-Marchant K, Kunicki TJ, . Platelet Physiology Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis; Working Group on Aspirin Resistance. Aspirin resistance: position paper of the Working Group on Aspirin Resistance. J Thromb Haemost. 2005;3:1309–11.
- Smith SC Jr, Feldman TE, Hirshfeld JW Jr, Jacobs AK, Kern MJ, King SB 3rd, . ACC/AHA/SCAI 2005 Guideline Update for Percutaneous Coronary Intervention-Summary Article: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/SCAI Writing Committee to Update the 2001 Guidelines for Percutaneous Coronary Intervention). J Am Coll Cardiol. 2006;47:216–35.
- ; European Association for Percutaneous Cardiovascular InterventionsWijns W, Kolh P, Danchin N, Di Mario C, Falk V, Folliguet T, . Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2010;31:2501–55.
- Optimizing Aspirin and Clopidogrel Therapy (BOchum CLopidogrel and Aspirin Plan) (BOCLAplan). ClinicalTrials.gov Identifier NCT 01212302. Available at: clinicaltrials.gov (accessed December 2010).
- Aspirin Non-Responsiveness and Clopidogrel Endpoint Trial (ASCET). ClinicalTrials.gov Identifier NCT 00222261. Available at: clinicaltrials.gov (accessed December 2010).
- Aspirin Resistance and Percutaneous Coronary Intervention (PCI) (RESIST). ClinicalTrials.gov Identifier NCT 01103440. Available at: clinicaltrials.gov (accessed December 2010).
- Dual Antiplatelet Therapy in Patients With Aspirin Resistance Following Coronary Artery Bypass Grafting. clinicalTrials.gov Identifier NCT 01159639. Available at: clinicaltrials.gov (accessed December 2010).
- Double Randomization of a Monitoring Adjusted Antiplatelet Treatment Versus a Common Antiplatelet Treatment for DES Implantation, and Interruption Versus Continuation of Double Antiplatelet Therapy (ARCTIC). clinicalTrials.gov Identifier NCT 00827411. Available at: clinicaltrials.gov (accessed December 2010).
- ThrombElastoGraphic Coagulation Status and Antiplatelet Therapy After Coronary Artery Bypass Graft Surgery (TEG-CABG). clinicalTrials.gov Identifier NCT 01046942. Available at: clinicaltrials.gov (accessed December 2010).
- Pettersen AA, Seljeflot I, Abdelnoor M, Arnesen H. Unstable angina, stroke, myocardial infarction and death in aspirin non-responders. A prospective, randomized trial. The ASCET (Aspirin non-responsiveness and Clopidogrel Endpoint Trial) design. Scand Cardiovasc J. 2004;38:353–6.
- ASCET: Single antiplatelet therapies compared in stable CAD. Available at: http://www.theheart.org/article/1157257.do accessed 12 April 2011.
- Neubauer H, Kaiser AF, Endres HG, Kruger JC, Engelhardt A, Lask S, . Tailored antiplatelet therapy can overcome clopidogrel and aspirin resistance —The BOchum CLopidogrel and Aspirin Plan (BOCLA-Plan) to improve antiplatelet therapy. BMC Med. 2011;9:3.
- The ITALIC Study: Is There A Life for Drug-eluting Stents (DES) After Discontinuation of Clopidogrel. clinicalTrials.gov Identifier NCT00780156. Available at: clinicaltrials.gov (accessed May 2011).
- Undas A, Siudak Z, Topór-Mądry R, Leśniak M, Tracz W. Simvastatin administration reduces thromboxane production in subjects taking aspirin: Links between aspirin resistance and thrombin generation. Int J Cardiol. 2010 Oct 28 (Epub ahead of print).
- Tirnaksiz E, Pamukcu B, Oflaz H, Nisanci Y. Effect of high dose statin therapy on platelet function; statins reduce aspirin-resistant platelet aggregation in patients with coronary heart disease. J Thromb Thrombolysis. 2009;27:24–8.
- Santos MT, Fuset MP, Ruano M, Moscardó A, Valles J. Effect of atorvastatin on platelet thromboxane A(2) synthesis in aspirin-treated patients with acute myocardial infarction. Am J Cardiol. 2009;104:1618–23.
- Larson MK, Ashmore JH, Harris KA, Vogelaar JL, Pottala JV, Sprehe M, . Effects of omega-3 acid ethyl esters and aspirin, alone and in combination, on platelet function in healthy subjects. Thromb Haemost. 2008;100:634–41.
- Lev EI, Solodky A, Harel N, Mager A, Brosh D, Assali A, . Treatment of aspirin-resistant patients with omega-3 fatty acids versus aspirin dose escalation. J Am Coll Cardiol. 2010;55:114–21.
- Gajos G, Rostoff P, Undas A, Piwowarska W. Effects of polyunsaturated omega-3 fatty acids on responsiveness to dual antiplatelet therapy in patients undergoing percutaneous coronary intervention: the OMEGA-PCI (OMEGA-3 fatty acids after pci to modify responsiveness to dual antiplatelet therapy) study. J Am Coll Cardiol. 2010;55:1671–8.
- Muhlestein JB. Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb Haemost. 2010;103:71–82.