Abstract
Mitochondrial disorders are a heterogeneous group of disorders resulting from primary dysfunction of the respiratory chain. Muscle tissue is highly metabolically active, and therefore myopathy is a common element of the clinical presentation of these disorders, although this may be overshadowed by central neurological features. This review is aimed at a general medical and neurologist readership and provides a clinical approach to the recognition, investigation, and treatment of mitochondrial myopathies. Emphasis is placed on practical management considerations while including some recent updates in the field.
| Abbreviations | ||
| ANS | = | ataxia-neuropathy syndromes |
| CoQ10 | = | coenzyme Q10, also known as ubiquinone |
| COX | = | cytochrome c oxidase |
| EM | = | electron microscopy |
| KSS | = | Kearns–Sayre syndrome |
| MELAS | = | myopathy, encephalopathy, lactic acidosis and stroke-like episodes |
| MEMSA | = | myoclonic epilepsy myopathy sensory ataxia |
| MERRF | = | myoclonus, epilepsy, and ragged red fibres |
| MIRAS | = | mitochondrial recessive ataxia syndrome |
| MM | = | mitochondrial myopathy |
| MNGIE | = | myopathy, neurogastrointestinal encephalopathy |
| mtDNA | = | mitochondrial DNA |
| NADH | = | nicotinamide adenine dinucleotide dehydrogenase |
| nDNA | = | nuclear (or chromosomal) DNA |
| PEO | = | progressive external ophthalmoplegia |
| POLG | = | polymerase gamma |
| RCE | = | respiratory chain enzyme |
| RRF | = | ragged red fibres |
| SANDO | = | sensory ataxic neuropathy, dysarthria, ophthalmoplegia |
| SCAE | = | spinocerebellar ataxia and epilepsy |
| SDH | = | succinate dehydrogenase |
| TP | = | thymidine phosphorylase |
| tRNA | = | transfer RNA |
Key messages
Mitochondrial myopathies frequently present with multi-system dysfunction and have a broad variety of phenotypes and genetic aetiologies.
Although no disease-modifying therapy exists, it is important to address disease complications which are often treatable and have an important impact on patient care.
Further study is required to assess the efficacy of various treatments, but a trial of coenzyme Q10 is reasonable as it may be useful for the rare patients with coenzyme Q10 biosynthetic defects.
Introduction
Mitochondrial myopathies (MM) comprise a large heterogeneous group of disorders resulting from primary dysfunction of the mitochondrial respiratory chain and causing muscle disease. These disorders are characterized by dysfunction in multiple organ systems, extensive variability in clinical presentation, and generally poor genotype–phenotype correlation.
Several characteristics of mitochondria produce features which differentiate mitochondrial disorders from other genetic diseases and are relevant for clinical practice. Mitochondria are intracellular organelles which contain their own genetic material (mtDNA), in the form of a 16.5-kb genome. However, most of the mitochondrial proteins are encoded by the nuclear genome (nDNA). Therefore, mitochondrial myopathies can result from abnormalities of mtDNA or nDNA. Abnormalities of mtDNA include point mutations, single large-scale deletions, mtDNA depletion, and multiple deletions. Point mutations of mtDNA affect the protein-coding regions of the genome and tRNA genes which alter intramitochondrial protein synthesis. Single large-scale deletions are typically due to sporadic events and are usually not inherited, although inherited deletions have been described (Citation1). Depletion (loss of mtDNA) and multiple deletions are typically secondary effects of faulty mtDNA maintenance, from mutation of nDNA-encoded mitochondrial proteins.
Each cell contains several mitochondria, and each mitochondrion contains numerous genomes. In this setting, the phenomenon of genetic heteroplasmy arises, where a proportion of genomes contain a mutation, and a proportion are wild-type (normal). The degree of heteroplasmy affects the likelihood and severity of the disease phenotype. The major factor affecting the inherited level of heteroplasmy of mutations occurs during oogenesis and is referred to as the ‘bottle-neck’ effect, in which the organism's entire repertoire of mitochondria is replicated from a small pool of genomes.However, the transmission of mtDNA mutations is complex and incompletely understood; women with mtDNA mutations pass their mutations forward at a level of heteroplasmy which is unpredictable, and apparently random (Citation2).
Although the focus of this article is on primary mitochondrial disorders which feature myopathy, it is important to mention that in many of these syndromes the myopathy component is overshadowed by other aspects of the clinical presentation. Disorders which will not be considered in this review include primary mitochondrial disorders which do not have myopathy as a clinical feature (for example, Leber optic neuropathy), and diseases due to secondary mitochondrial dysfunction, or which otherwise involve the mitochondria in their pathogenesis (several inherited and acquired muscle diseases, and numerous common neurodegenerative and other genetic diseases (Citation3)). The reader should also be cautioned that syndromes resembling primary mitochondrial disorders may be produced from secondary mitochondrial dysfunction and acquired mitochondrial toxicity. Furthermore, acquired mitochondrial toxicity and medication effects may also affect the muscle biopsy or other test results to mimic those seen in a primary mitochondrial disorder, as may the effects of healthy ageing (Citation4).
Clinical features
The prevalence of mitochondrial disorders as a whole is approximately 1 in 10,000 (Citation5), although the carrier frequency of mtDNA mutations is about 1 in 200 (Citation6). Onset can occur at any age, although typically the more severe phenotypes present earlier in life, and milder phenotypes present later in life. As a prototype example of this, the so-called deletion syndromes (caused by sporadic, large-scale deletions of mtDNA) exist on a disease spectrum in which the most severe syndrome presents in infancy (Pearson syndrome), a more moderate syndrome in early childhood or adolescence (Kearns–Sayre syndrome (KSS)), and a milder syndrome in childhood up to late adulthood (progressive external ophthalmoplegia (PEO)). MM are usually progressive conditions which produce significant disability and, in some instances, premature death, often due to non- muscle involvement such as cardiac conduction defects or seizures (Citation7).
Because mitochondria are the main source of energy production in mammalian cells, clinical features typically involve tissues with the highest energy requirements. Furthermore, the presence of mtDNA in all human tissues means that dysfunction occurs in multiple organ systems. The most commonly affected organ systems are the nervous system (central, peripheral, autonomic, as well as optic nerve and retina), muscles (and in particular extra-ocular muscle), cardiac, and endocrine systems. The clinical presentation is highly variable with regard to onset age, symptoms, signs, severity, and prognosis. The clinician should consider a diagnosis of MM when myopathy is accompanied by clinical features of multi-organ dysfunction, which are summarized in . It is common for MM to present with constellations of symptoms, which allow them to be categorized into one of several syndromes. Ocular myopathy, which is manifested by ptosis and ophthalmoparesis, is an important feature in various MM syndromes, which are summarized in . In this sense, PEO is both a syndrome on its own but also a component of other syndromes when certain combinations of other features are present. The ocular muscle weakness develops gradually, and presentation may be delayed by years or decades until the signs are noticed by family members. Isolated PEO, which is the mildest syndrome, may still carry significant visual and other disability (Citation8) and often presents with multiple features of mitochondrial disease (Citation9). PEO deserves special mention because of its extensive genetic heterogeneity, which has implications for the risk to other family members of inheriting the disease. PEO may be sporadic (due to single deletions), maternally inherited (due to mtDNA mutation), autosomal dominant, or recessive (due to nDNA mutations). These are often clinically indistinguishable, emphasizing the importance of obtaining a molecular diagnosis. The phenotypic variability should also be emphasized, since this condition may present at any age, the ophthalmoparesis may be anywhere along a spectrum of subtle to complete, and associated features of the condition may appear in any combination.
Figure 1. Clinical features of mitochondrial myopathies, by organ system.
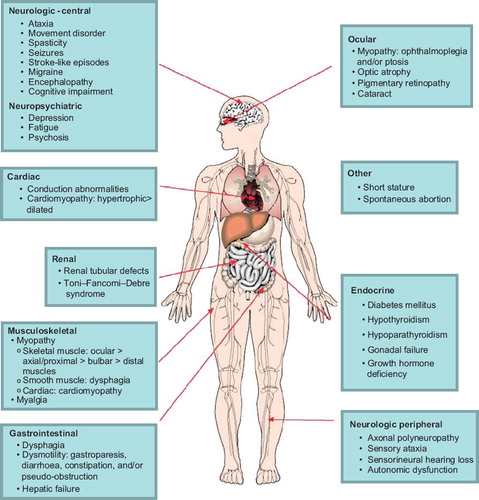
Table I. Mitochondrial myopathy syndromes presenting with ocular myopathy.
For the ataxia-neuropathy syndromes (ANS), this includes an overlapping group of disorders, about half of which have PEO as part of their clinical presentation. These syndromes are variably referred to as spinocerebellar ataxia with epilepsy (SCAE), myoclonic epilepsy myopathy sensory ataxia (MEMSA), sensory ataxia, neuropathy, dysarthria, ophthalmoplegia (SANDO), or mitochondrial recessive ataxia syndrome (MIRAS). These syndromes have in common the presence of an axonal sensory neuropathy, affecting proprioceptive function in combination with variable degrees of cerebellar ataxia, making them part of a disease spectrum which is caused by nDNA mutations affecting mtDNA maintenance.
Other syndromes present with multi-organ dysfunction, without ocular myopathy, and these are summarized in . In myoclonus, epilepsy, and ragged red fibres (MERRF), myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), and some of ANS, central nervous system dysfunction predominates on a background of dysfunction in other organ systems. For these syndromes, genotype–phenotype correlations are somewhat better, where the majority of MELAS and MERRF patients have common tRNA mutations (respectively, m.3243A > G and m.8344A > G, and ANS are most often caused by mutations in the gene encoding the mtDNA polymerase gamma, POLG).
Table II. Mitochondrial myopathy typically presenting without PEO.
Isolated MM typically presents with axial and proximal weakness, variable age of onset or severity, and variable co-occurrence of other features of mitochondrial dysfunction. As in PEO, genotype– phenotype correlation is poor, and as it stands this condition is already difficult to distinguish from other types of acquired or genetic myopathy, due to its fairly non-specific presentation.
The infant-onset mitochondrial myopathies have a severe clinical presentation, although it is important to be aware of a subset of patients with infantile cytochrome c oxidase (COX)-deficiency myopathy with reversible disease, whose molecular defect has recently been described (Citation10,Citation11).
Another rare but important subgroup of patients with MM are due to defects in coenzyme Q10 (CoQ10) biosynthesis. These disorders are important to recognize because of their partial responsiveness to CoQ10 supplementation. The infantile-onset form of CoQ10 deficiency is a multi-systemic disorder with encephalopathy and nephropathy. Typically this is steroid-resistant and may progress to renal failure (Citation12,Citation13). In adults, CoQ10 deficiency manifests as adult-onset myopathy or ataxia with variable myopathy, peripheral neuropathy, and/or seizures (Citation14–16).
Numerous patients with MM do not fit into the described syndromes. Many features of MM are uncommon, or recently described (such as distal myopathy from certain POLG mutations (Citation17)). These ill-defined syndromes may have a novel or unique molecular basis, or they may be due to mutations previously described to cause specific syndromes. For example, the m.3243A > G mutation has been implicated in a broad variety of atypical clinical presentations (Citation18).
Diagnostic testing
For patients with suspected MM, diagnostic tests fall into two broad categories. The first category of testing confirms the presence of dysfunction in various organ systems (summarized in ) and does not as such confirm a diagnosis of MM. These tests are nonetheless important to define the extent of the phenotype, to exclude other disorders, and to increase or decrease the clinical suspicion of a MM diagnosis. The tests selected are guided by the pattern of organ involvement in each individual patient. Some of these tests deserve special mention, because of their potential to alter patient management. Cardiac investigations are of particular importance because cardiac conduction defects can be fatal if not identified and are treatable with cardiac pacemakers. Endocrine investigations may identify diabetes mellitus, hypothyroidism, or growth hormone deficiency, all of which are treatable. Patients with hearing or visual symptoms should be investigated in order to obtain appropriate aids if required. Dysphagia is common in some mitochondrial syndromes and can be managed with dietary modification.
Table III. Confirmatory tests for organ dysfunction in mitochondrial myopathy.
The second category of tests definitively addresses whether the patient is affected by a MM, and these mainly include muscle biopsy and molecular genetic studies. These studies are performed in combination, since the assessment of mtDNA should ideally be done from DNA extracted from muscle (the high replication rate of blood cells selects against pathogenic mtDNA abnormalities, therefore many mtDNA abnormalities are not detectable in blood).
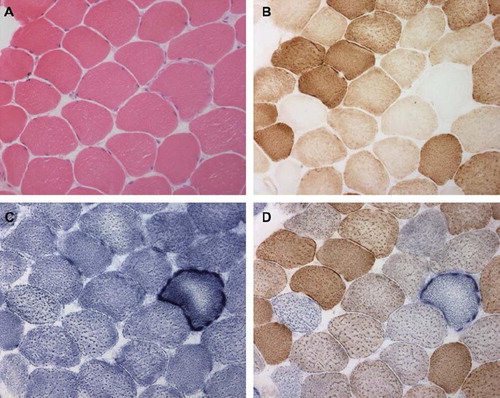
Muscle biopsy is typically performed from a limb muscle, such as quadriceps femoris or deltoid, and examples of characteristic abnormalities are provided in . The testing should include a variety of histochemical functional assays and be performed in a centre with experience in mitochondrial disease diagnosis. The major diagnostic feature is the presence of fibres deficient for COX activity, which represents poor activity of complex IV of the respiratory chain (and is encoded by both mtDNA and nDNA genes). However, a low frequency of COX-deficient fibres is a normal finding in healthy aged individuals. In general, the detection of any COX-deficient fibres in individuals <50 years of age, or a higher frequency of COX-deficient fibres at any age (>5%), is strongly suggestive of a mitochondrial disorder. The identification of COX-deficient fibres is greatly helped by serially staining muscle for COX followed by succinate dehydrogenase (SDH), which stains for complex II (and is encoded entirely by nuclear genes). The demonstration of COX-deficient, SDH-positive muscle fibres is thought to have the best sensitivity and specificity for MM (Citation19). The sub-sarcolemmal accumulation of mitochondria is a classic feature of MM, and can be demonstrated by SDH histochemistry (so-called ‘raggedblue fibres’), or the Gomori trichrome stain (so-called ‘ragged red fibres’ or RRF). Again, a low frequency of RRFs (<5%) can be seen in healthy aged individuals. However, the detection of RRFs in individuals <50 years of age, or >5% RRF at any age, is highly suggestive of MM, although even high levels can be secondary to other pathologies, such as inclusion body myositis.
Figure 2. Abnormalities on skeletal muscle biopsy in mitochondrial myopathy. Serial sections through vastus lateralis in a patient with mitochondrial myopathy showing: (A) haematoxylin and eosin, (B) cytochrome c oxidase histochemistry (COX) (note the COX deficient fibres), (C) succinate dehydrogenase histochemistry (SDH) (note the sub-sarcolemmal accumulation of mitochondria analogous to a ragged red fibre), and (D) sequential COX-SDH histochemistry showing a mosaic COX defect as seen in patients with mtDNA disorders.
Electron microscopy (EM) may also be performed on muscle specimens and demonstrate a variety of abnormalities associated with MM, although these are rarely specific for mitochondrial diseases. These include enlarged pleiomorphic mitochondria and paracrystalline inclusions. At present EM is thought to provide minor criteria for the diagnosis of MM (Citation20). However, EM may provide minor diagnostic criteria for mitochondrial disease in some patients with normal histochemistry (Citation9), therefore EM may contribute additional information in selected cases. Some caveats to diagnosis with muscle biopsy should be discussed.
Muscle histochemistry and/or EM may be normal even in the context of genetically proven mitochondrial syndromes (Citation21), particularly early in the disease course or when the biochemical defect does not involve complex IV (COX). Furthermore, for certain MM syndromes (mainly PEO), unconventional muscle biopsy sites have been studied for their utility in providing a diagnosis of MM. These include levator palpebrae superioris (Citation22) and orbicularis oculi (Citation23,Citation24), which are easily accessible muscles during corrective ocular surgery for ptosis in PEO. For patients requiring ocular surgery, biopsy of ocular muscle may be able to provide a diagnosis and avoid a separate procedure for limb muscle biopsy—however, limited information from healthy controls can limit interpretation from these unconventional sites.
Another test available from muscle tissue is respiratory chain enzyme (RCE) analysis. This testing must be done either on fresh or snap-frozen muscle samples. RCE is technically difficult to perform, even in specialist laboratories (Citation19,Citation25,Citation26), and the results should be interpreted in the context of the other investigations. Demonstrating a RCE defect is a crucial diagnostic step in patients with normal or near-normal muscle histochemistry, particularly children.
The genetic tests should be guided based on the muscle biopsy findings, the MM syndrome which is suspected, and, if present, the inheritance pattern. As a general rule, mosaic appearance of COX-negative fibres suggests a mtDNA mutation (due to the variable degrees of heteroplasmy between muscle cells), whereas uniformly decreased COX activity suggests a nDNA mutation (which would be equally present in all muscle cells). If only a single respiratory chain complex has decreased activity, this suggests a mutation in a structural gene for the relevant complex, which may be in mtDNA or nDNA, or a specific complex assembly factor in nDNA. However, these general principles are not invariably true, because patients with mtDNA depletion may have isolated complex deficiencies early in the disease course. Other characteristic features include the presence of strongly succinate dehydrogenase-positive blood vessels (SSVs) seen in patients with MELAS harbouring m.3243A > G (Citation27). If the patient fits into a particular clinical syndrome, this can be helpful in deciding testing, and common mutations for different phenotypes are listed in and . Cases in which genetic testing may precede muscle biopsy include syndromes and/or inheritance history that implicate nDNA mutations, which may be tested in blood. Characteristic examples include syndromes caused by POLG mutations (autosomal dominant or recessive PEO, the ANS, and hepatocerebral syndromes such as Alpers syndrome) (Citation28). The m.3243A >G mutation is easily detected in urine, may cause a variety of mitochondrial syndromes (MELAS, maternally inherited diabetes and deafness, PEO, isolated MM, and cardiomyopathy), and its mutation load may even provide prognostic information (Citation29). Elevations of plasma and urine thymidine are seen in myopathy, neuropathy, gastrointestinal encephalopathy (MNGIE) syndrome due to mutations in TP.
If a genetic diagnosis is not reached after eliminating common molecular defects, more extensive testing should be carried out. This may involve sequencing the mitochondrial genome and/or known nuclear disease genes. In the case of mtDNA genome sequencing, previously described mutations may be identified in this manner, or novel mutations, although distinguishing mutations from the high level of variability in the mtDNA sequence in the general population is a challenging exercise (Citation30).
Tissues aside from muscle may be biopsied to support a diagnosis of MM. Skin biopsy is a non-invasive procedure which is used to obtain fibroblasts for RCE and DNA for genetic studies. However, the RCE defect or molecular genetic defect may not be present in fibroblasts in all patients, and as a result this method has lower sensitivity than muscle biopsy (Citation31). Liver biopsy is appropriate in selected situations with an important component of hepatic failure, providing there is no coagulopathy. In these situations the biopsy is helpful to exclude other disorders and is a tissue source for histological, EM, RCE, and DNA analysis.
Diagnosis of MM due to CoQ10 biosynthetic defects are made by the demonstration of CoQ10 deficiency in muscle tissue and may be supported by decreased levels in other tissues such as fibroblasts and white blood cells (Citation12,Citation26). Plasma levels have a broad reference range and may be normal in this condition (Citation26). RCE analysis may demonstrate the combination of either complex I + III deficiency or complex II + III deficiency, since these complexes are CoQ10-dependent (Citation16).
Another category of diagnostic tests for MM includes exercise testing. There are numerous described protocols for testing using cycle ergometry or treadmill exercise (Citation32). The diagnostic usefulness of these investigations is controversial, given reports revealing low specificity (Citation33,Citation34) and sensitivity (Citation35). Protocols for measuring venous pO2 during handgrip testing have demonstrated excellent specificity, and for practical purposes these work well as non-invasive screening tests for MM (Citation36,Citation37).
Treatment
There is currently no available disease-modifying therapy for MM. Several agents (mostly nutritional supplements) have been investigated with double-blind, placebo-controlled studies. These include carnitine (Citation38), creatine (Citation39–41), CoQ10 (Citation42,Citation43), cysteine (Citation44), dichloroacetate (Citation42,Citation45–48), dimethylglycine (Citation49), and the combination of creatine, CoQ10, and lipoic acid (Citation50). None has demonstrated efficacy in clinical disease end-points, although numerous non-blinded studies and case reports have suggested efficacy. Examples of treatments with reported benefit in MM that have not yet been evaluated in placebo-controlled trials are summarized in . Further study is required to identify whether any of these agents have therapeutic benefit.
Table IV. Treatments with reported benefit in mitochondrial myopathy which may benefit from further study in blinded placebo-controlled trials.
Although extremely rare, MM caused by CoQ10 deficiency will sometimes respond to CoQ10 supplementation (Citation51,Citation52), therefore a trial on this agent is appropriate for patients who have a possible phenotype of these conditions. Anecdotal and open-labelled case series report improvements with the CoQ10 analogue idebenone in mitochondrial myopathy (Citation53–55). Otherwise, due to the lack of available treatments, numerous experimental treatments are in development, and these were reviewed recently (Citation56). These include the PPAR/PGC-1α activator bezafibrate, which increased mitochondrial biogenesis and delayed the onset of myopathy in transgenic mice with a COX defect (Citation57), and the mitochondrially targeted antioxidant MitoQ which has been used safely in several common human diseases (Citation58).
There has been great interest in exercise programmes and their benefit on both biochemical and clinical end-points in MM. Aerobic (Citation59,Citation60), endurance (Citation61,Citation62), and resistance (Citation63) training programmes have been studied. It is currently not clear whether the benefits of exercise in MM are simply reversing the de-conditioning, which is a common feature of many muscle diseases, or whether the exercise affects the underlying pathology. In any event, evidence is mounting that exercise programmes are safe and beneficial for numerous end-points, including strength, fatigue, and quality of life.
Treatment of MM concentrates on the management of disease complication. A diseasecomplication which is particular to MM is the stroke-like episodes of MELAS. A non-blinded study of 24 MELAS patients compared L-arginine with placebo as acute treatment for stroke-like episodes (Citation64). Symptoms improved 30 minutes and 24 hours after administration. A portion of the study also followed six patients on daily treatment with L-arginine for 18 months. The frequency and severity of stroke-like episodes were significantly decreased. However, these findings have not been replicated, and the non-blinded nature of the study may have biased the results. Recently, a small study suggested a benefit of L-arginine in cardiomyopathy due to MM (Citation65), and so further study of this agent would be of interest. Finally, status epilepticus was successfully treated with intravenous magnesium in two teenage girls with juvenile-onset Alpers syndrome due to POLG mutations (Citation66). One died 2 weeks after treatment from pneumonia. The other remained seizure-free 8 months after treatment.
Other disease complications of MM are due to dysfunction of various organ systems, which are important to recognize and treat because they are potentially preventable causes of death and disability in MM patients (Citation67). provides a summary of these as well as examples of possible treatments. Cardiac dysfunction will be discussed in further detail because it is common, frequently asymptomatic, and potentially fatal. Cardiac dysfunction can take many forms, namely cardiac conduction defects and cardiomyopathy. Abnormalities of cardiac conduction are common, even among asymptomatic patients (Citation68), and although they are a central feature of KSS they may occur in any MM. Cardiomyopathy may be hypertrophic or dilated and is most commonly present in MELAS, MERRF, and KSS (Citation69). Figures are not available for adults, although one study in children demonstrated hypertrophic cardiomyopathy to be present in 17%, often asymptomatic, and associated with higher mortality than MM patients without cardiomyopathy (Citation70). Patients with MM should therefore be screened with 12-lead ECG and transthoracic echocardiogram irrespective of whether they are symptomatic for cardiac disease. Guidelines do not exist as to whether investigations should be repeated in the event that they are normal, although repeating investigations every 1–3 years may be reasonable. Other cardiac investigations which may be considered include Holter monitoring in patients with symptoms suggestive of arrhythmia. Cardiac MRI is a new imaging modality, but from case reports the findings in MM may be characteristic (Citation71,Citation72).
Endocrinopathy is another common finding in MM patients and is treatable. The classic endocrine manifestation is diabetes mellitus, which is particularly associated with KSS and MELAS (Citation73). Hypothyroidism may also be a common endocrinopathy in milder syndromes (Citation9), and the clinician should be aware that any type of endocrine abnormality is possible, particularly growth hormone deficiency, which is treatable.
Respiratory dysfunction can have serious ramifications (Citation74), although it has received limited study in MM. Recent clinical series have queried the presence of respiratory symptoms but found them to be similar to controls in a large series of MELAS patients (Citation73), and present in only 1 patient in a series of 40 PEO patients (Citation9). The only major study on this subject investigated respiratory parameters during sleep in eight patients with PEO (Citation75). Although all had no respiratory symptoms, during the sleep studies four of the patients had central apnoeic episodes and/or poor responsivity to CO2. This was postulated to be due to the chronic adaptation to respiratory and laryngeal muscle weakness, or due to a central mechanism. While it is unclear what effect these asymptomatic respiratory disturbances had on these patients, clinicians should have a low threshold to investigate respiratory- and sleep-related symptoms in MM patients. Another series reporting more severe respiratory disturbances suggested a central mechanism that could be episodic and exacerbated by metabolic stressors, such as infection or anaesthesia (Citation76).
Other management considerations in MM include the avoidance of agents which may worsen the patient's condition. While the list of medications with theoretical toxicity is massive (Citation77), a few agents of clinical importance will be discussed here. Statin medications are thought to cause toxic effects on skeletal muscle through a disturbance of mitochondrial function (Citation78), although the precise mechanisms remain unclear. Statins appear to cause muscle symptoms in 10% of patients who receive the drugs (Citation79), have been reported to unmask symptoms of metabolic myopathies in patients who were previously asymptomatic (Citation80), and occasionally these agents are associated with syndromes resembling PEO (Citation81,Citation82). They should therefore be used cautiously in MM, with careful monitoring of symptoms and the serum creatine kinase. Antiretroviral agents are known to cause reversible and dose-dependent mitochondrial toxicity (Citation83). If necessary for the treatment of HIV, it appears that certain agents have less mitochondrial toxicity (Citation84) and should be used preferentially. Small series have documented the development of PEO-like syndromes in patients on antiretrovirals (Citation85–87), although whether this is due to an unmasking effect or whether the disease is caused by cumulative mitochondrial toxicity is unknown. This is especially the case for nucleotide reverse transcriptase inhibitors. Valproic acid is known to interfere with mitochondrial function and in clinical practice can aggravate symptoms in patients with MM (Citation88), and valproate-induced hepatotoxicity may be more common in MM patients (Citation89,Citation90). Genetic variation in POLG is strongly associated with increased risk of hepatotoxicity due to valproic acid (Citation91).
In summary, the identification of patients with possible MM depends upon the investigation of multiple organ dysfunction in the clinical history, examination, and clinical tests. Although there is no disease-modifying therapy for MM, there are numerous points of clinical relevance that can reduce morbidity and improve quality of life for patients with these disorders.
Acknowledgements
G.P. is the recipient of funding from the Clinician Investigator Program from the University of British Columbia, and from a Bisby Fellowship from the Canadian Institutes of Health Research.
P.F.C. is an Honorary Consultant Neurologist at Newcastle upon Tyne Foundation Hospitals NHS Trust. He is a Wellcome Trust Senior Fellow in Clinical Science and a UK NIHR Senior Investigator who also receives funding from the Medical Research Council (UK), the Association Française contre les Myopathies, and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust.
Declaration of interest: No competing interests are reported.
References
- Chinnery PF, DiMauro S, Shanske S, , SchonEAZeviani M, Mariotti C, . Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–6.
- Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl HM, Turnbull DM, . The inheritance of mitochondrial DNA heteroplasmy: random drift, selection or both? Trends Genet. 2000;16:500–5.
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008; 60:748–66.
- Sarnat HB, Marin-Garcia J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci. 2005;32:152–66.
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, . Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9.
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–60.
- Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82.
- Yu Wai Man CY, Smith T, Chinnery PF, Turnbull DM, Griffiths PG. Assessment of visual function in chronic progressive external ophthalmoplegia. Eye. 2006;20:564–8.
- Pfeffer G, Sirrs S, Wade NK, Mezei MM. Multisystem disorder in late-onset chronic progressive external ophthalmoplegia. Can J Neurol Sci. 2011;38:119–23.
- Mimaki M, Hatakeyama H, Komaki H, Yokoyama M, Arai H, Kirino Y, . Reversible infantile respiratory chain deficiency: a clinical and molecular study. Ann Neurol. 2010;68: 845–54.
- Horvath R, Kemp JP, Tuppen HA, Hudson G, Oldfors A, Marie SK, . Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain. 2009; 132(Pt 11):3165–74.
- Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, . Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–5.
- Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM, . Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65:606–8.
- Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, . The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007; 130(Pt 8):2037–44.
- Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol. 2005; 62:317–20.
- Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A. 1989;86:2379–82.
- Giordano C, Pichiorri F, Blakely EL, Perli E, Orlandi M, Gallo P, . Isolated distal myopathy of the upper limbs associated with mitochondrial DNA depletion and polymerase gamma mutations. Arch Neurol. 2010;67:1144–6.
- Moraes CT, Ciacci F, Silvestri G, Shanske S, Sciacco M, Hirano M, . Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul Disord. 1993;3:43–50.
- Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord. 2004;14:237–45.
- Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–11.
- Schaefer AM, Blakely EL, Griffiths PG, Turnbull DM, Taylor RW. Ophthalmoplegia due to mitochondrial DNA disease: the need for genetic diagnosis. Muscle Nerve. 2005; 321:104–7.
- Greaves LC, Yu-Wai-Man P, Blakely EL, Krishnan KJ, Beadle NE, Kerin J, . Mitochondrial DNA defects and selective extraocular muscle involvement in CPEO. Invest Ophthalmol Vis Sci. 2010;51:3340–6.
- Eshaghian J, Anderson RL, Weingeist TA, Hart MN, Cancilla PA. Orbicularis oculi muscle in chronic progressive external ophthalmoplegia. Arch Ophthalmol. 1980;98:1070–3.
- Almousa R, Charlton A, Rajesh ST, Sundar G, Amrith S. Optimizing muscle biopsy for the diagnosis of mitochondrial myopathy. Ophthal Plast Reconstr Surg. 2009;25:366–70.
- Wibrand F, Jeppesen TD, Frederiksen AL, Olsen DB, Duno M, Schwartz M, . Limited diagnostic value of enzyme analysis in patients with mitochondrial tRNA mutations. Muscle Nerve. 2010;41:607–13.
- Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, . Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. 2009;9:331–9.
- Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29: 601–5.
- Milone M, Massie R. Polymerase gamma 1 mutations: clinical correlations. Neurologist. 2010;16:84–91.
- Whittaker RG, Blackwood JK, Alston CL, Blakely EL, Elson JL, McFarland R, . Urine heteroplasmy is the best predictor of clinical outcome in the m.3243A >G mtDNA mutation. Neurology. 2009;72:568–9.
- DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106:18–26.
- van den Heuvel LP, Smeitink JA, Rodenburg RJ. Biochemical examination of fibroblasts in the diagnosis and research of oxidative phosphorylation (OXPHOS) defects. Mitochondrion. 2004;4:395–401.
- Tarnopolsky M. Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion. 2004;4:529–42.
- Dandurand RJ, Matthews PM, Arnold DL, Eidelman DH. Mitochondrial disease. Pulmonary function, exercise performance, and blood lactate levels. Chest. 1995;108:182–9.
- Hammaren E, Rafsten L, Kreuter M, Lindberg C. Modified exercise test in screening for mitochondrial myopathies—adjustment of workload in relation to muscle strength. Eur Neurol. 2004;51:38–41.
- Jeppesen TD, Olsen D, Vissing J. Cycle ergometry is not a sensitive diagnostic test for mitochondrial myopathy. J Neurol. 2003;250:293–9.
- Jensen TD, Kazemi-Esfarjani P, Skomorowska E, Vissing J. A forearm exercise screening test for mitochondrial myopathy. Neurology. 2002;58:1533–8.
- Taivassalo T, Abbott A, Wyrick P, Haller RG. Venous oxygen levels during aerobic forearm exercise: An index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol. 2002;51:38–44.
- Gimenes AC, Napolis LM, Silva NL, Siquiera GO, Bulle AS, Neder JA, . The effect of L-carnitine supplementation on respiratory muscle strength and exercise tolerance in patients with mitochondrial myopathies. [Abstract]. Eur Respir J 2007;51(Suppl):21S[E297].
- Kornblum C, Schroder R, Muller K, Vorgerd M, Eggers J, Bogdanow M, . Creatine has no beneficial effect on skeletal muscle energy metabolism in patients with single mitochondrial DNA deletions: a placebo-controlled, double-blind 31P-MRS crossover study. Eur J Neurol. 2005;12:300–9.
- Klopstock T, Querner V, Schmidt F, Gekeler F, Walter M, Hartard M, . A placebo-controlled crossover trial of creatine in mitochondrial diseases. Neurology. 2000;55: 1748–51.
- Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502–9.
- Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, . Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–31.
- Bresolin N, Doriguzzi C, Ponzetto C, Angelini C, Moroni I, Castelli E, . Ubidecarenone in the treatment of mitochondrial myopathies: a multi-center double-blind trial. J Neurol Sci. 1990;100:70–8.
- Mancuso M, Orsucci D, Logerfo A, Rocchi A, Petrozzi L, Nesti C, . Oxidative stress biomarkers in mitochondrial myopathies, basally and after cysteine donor supplementation. J Neurol. 2010;257:774–81.
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, . Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–30.
- De Stefano N, Matthews PM, Ford B, Genge A, Karpati G, Arnold DL. Short-term dichloroacetate treatment improves indices of cerebral metabolism in patients with mitochondrial disorders. Neurology. 1995;45:1193–8.
- Vissing J, Gansted U, Quistorff B. Exercise intolerance in mitochondrial myopathy is not related to lactic acidosis. Ann Neurol. 2001;49:672–6.
- Duncan GE, Perkins LA, Theriaque DW, Neiberger RE, Stacpoole PW. Dichloroacetate therapy attenuates the blood lactate response to submaximal exercise in patients with defects in mitochondrial energy metabolism. J Clin Endocrinol Metab. 2004;89:1733–8.
- Liet JM, Pelletier V, Robinson BH, Laryea MD, Wendel U, Morneau S, . The effect of short-term dimethylglycine treatment on oxygen consumption in cytochrome oxidase deficiency: a double-blind randomized crossover clinical trial. J Pediatr. 2003;142:62–6.
- Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35:235–42.
- Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, . Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–55.
- Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358:2849–50.
- Ikejiri Y, Mori E, Ishii K, Nishimoto K, Yasuda M, Sasaki M. Idebenone improves cerebral mitochondrial oxidative metabolism in a patient with MELAS. Neurology. 1996;47:583–5.
- Seki A, Nishino I, Goto Y, Maegaki Y, Koeda T. Mitochondrial encephalomyopathy with 15915 mutation: clinical report. Pediatr Neurol. 1997;17:161–4.
- Haginoya K, Miyabayashi S, Kikuchi M, Kojima A, Yamamoto K, Omura K, . Efficacy of idebenone for respiratory failure in a patient with Leigh syndrome: a long-term follow-up study. J Neurol Sci. 2009;278:112–4.
- Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies: developments in treatment. Curr Opin Neurol. 2010;23:459–65.
- Wenz T, Diaz F, Hernandez D, Moraes CT. Endurance exercise is protective for mice with mitochondrial myopathy. J Appl Physiol. 2009;106:1712–9.
- Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103.
- Jeppesen TD, Schwartz M, Olsen DB, Wibrand F, Krag T, Duno M, . Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. 2006;129(Pt 12):3402–12.
- Taivassalo T, De Stefano N, Argov Z, Matthews PM, Chen J, Genge A, . Effects of aerobic training in patients with mitochondrial myopathies. Neurology. 1998;50:1055–60.
- Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, . Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129(Pt 12):3391–401.
- Cejudo P, Bautista J, Montemayor T, Villagomez R, Jimenez L, Ortega F, . Exercise training in mitochondrial myopathy: a randomized controlled trial. Muscle Nerve. 2005;32: 342–50.
- Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, . Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. 2008; 131(Pt 11):2832–40.
- Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, . L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–2.
- Arakawa K, Kudo T, Ikawa M, Morikawa N, Kawai Y, Sahashi K, . Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to L-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circ J. 2010;74:2702–11.
- Visser NA, Braun KP, Leijten FS, van Nieuwenhuizen O, Wokke JH, van den Bergh WM. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J Neurol. 2011;258:218–22.
- Klopstock T, Jaksch M, Gasser T. Age and cause of death in mitochondrial diseases. Neurology. 1999;53:855–7.
- Baik R, Chae JH, Lee YM, Kang HC, Lee JS, Kim HD. Electrocardiography as an early cardiac screening test in children with mitochondrial disease. Korean J Pediatr. 2010;53:644–7.
- Marin-Garcia J, Goldenthal MJ. Mitochondrial cardiomyopathy: molecular and biochemical analysis. Pediatr Cardiol. 1997;18:251–60.
- Holmgren D, Wahlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J. 2003;24:280–8.
- Jose T, Gdynia HJ, Mahrholdt H, Vohringer M, Klingel K, Kandolf R, . CMR gives clue to “ragged red fibers” in the heart in a patient with mitochondrial myopathy. Int J Cardiol. 2011;149:e24–7.
- Partington SL, Givertz MM, Gupta S, Kwong RY. Cardiac magnetic resonance AIDS in the diagnosis of mitochondrial cardiomyopathy. Circulation. 2011;123:e227–9.
- Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Battista V, . Protean phenotypic features of the A3243G mitochondrial DNA mutation. Arch Neurol. 2009;66:85–91.
- Cros D, Palliyath S, DiMauro S, Ramirez C, Shamsnia M, Wizer B. Respiratory failure revealing mitochondrial myopathy in adults. Chest. 1992;101:824–8.
- Manni R, Piccolo G, Banfi P, Cerveri I, Bruschi C, Zoia C, . Respiratory patterns during sleep in mitochondrial myopathies with ophthalmoplegia. Eur Neurol. 1991;31:12–17.
- Barohn RJ, Clanton T, Sahenk Z, Mendell JR. Recurrent respiratory insufficiency and depressed ventilatory drive complicating mitochondrial myopathies. Neurology. 1990; 40:103–6.
- Finsterer J, Segall L. Drugs interfering with mitochondrial disorders. Drug Chem Toxicol. 2010;33:138–51.
- Sirvent P, Bordenave S, Vermaelen M, Roels B, Vassort G, Mercier J, . Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun. 2005;338:1426–34.
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14.
- Baker SK, Vladutiu GD, Peltier WL, Isackson PJ, Tarnopolsky MA. Metabolic myopathies discovered during investigations of statin myopathy. Can J Neurol Sci. 2008;35:94–7.
- Elsais A, Lund C, Kerty E. Ptosis, diplopia and statins: an association? Eur J Neurol. 2008;15:e90–1.
- Fraunfelder FW, Richards AB. Diplopia, blepharoptosis, and ophthalmoplegia and 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor use. Ophthalmology. 2008;115:2282–5.
- Venhoff N, Setzer B, Melkaoui K, Walker UA. Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antivir Ther. 2007;12:1075–85.
- Ananworanich J, Nuesch R, Cote HC, Kerr SJ, Hill A, Jupimai T, . Changes in metabolic toxicity after switching from stavudine/didanosine to tenofovir/lamivudine—a Staccato trial substudy. J Antimicrob Chemother. 2008;61:1340–3.
- Dinges WL, Witherspoon SR, Itani KM, Garg A, Peterson DM. Blepharoptosis and external ophthalmoplegia associated with long-term antiretroviral therapy. Clin Infect Dis. 2008;47:845–52.
- Zannou DM, Azon-Kouanou A, Bashi BJ, Gougounon A, Zinsou R, Ade G, . Mitochondrial toxicity: a case of palpebral ptosis in a woman infected by HIV and treated with HAART including zidovudine. Bull Soc Pathol Exot. 2009;102:97–8.
- Pfeffer G, Cote HC, Montaner JS, Li CC, Jitratkosol M, Mezei MM. Ophthalmoplegia and ptosis: mitochondrial toxicity in patients receiving HIV therapy. Neurology. 2009;73:71–2.
- Lin CM, Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab Brain Dis. 2007;22:105–9.
- Krahenbuhl S, Brandner S, Kleinle S, Liechti S, Straumann D. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver. 2000;20:346–8.
- McFarland R, Hudson G, Taylor RW, Green SH, Hodges S, McKiernan PJ, . Reversible valproate hepatotoxicity due to mutations in mitochondrial DNA polymerase gamma (POLG1). Arch Dis Child. 2008;93:151–3.
- Stewart JD, Horvath R, Baruffini E, Ferrero I, Bulst S, Watkins PB, . Polymerase gamma gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010;52:1791–6.
- Eleff S, Kennaway NG, Buist NR, Darley-Usmar VM, Capaldi RA, Bank WJ, . 31PNMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc Natl Acad Sci U S A. 1984;81:3529–33.
- Mowat D, Kirby DM, Kamath KR, Kan A, Thorburn DR, Christodoulou J. Respiratory chain complex III [correction of complex] in deficiency with pruritus: a novel vitamin responsive clinical feature. J Pediatr. 1999;134:352–4.
- Panetta J, Smith LJ, Boneh A. Effect of high-dose vitamins, coenzyme Q and high-fat diet in paediatric patients with mitochondrial diseases. J Inherit Metab Dis. 2004;27:487–98.
- Remes AM, Liimatta EV, Winqvist S, Tolonen U, Ranua JA, Reinikainen K, . Ubiquinone and nicotinamide treatment of patients with the 3243A– > G mtDNA mutation. Neurology. 2002;59:1275–7.
- Majamaa K, Rusanen H, Remes A, Hassinen IE. Metabolic interventions against complex I deficiency in MELAS syndrome. Mol Cell Biochem. 1997;174:291–6.
- Penn AM, Lee JW, Thuillier P, Wagner M, Maclure KM, Menard MR, . MELAS syndrome with mitochondrial tRNA(Leu)(UUR) mutation: correlation of clinical state, nerve conduction, and muscle 31P magnetic resonance spectroscopy during treatment with nicotinamide and riboflavin. Neurology. 1992;42:2147–52.
- Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace DC. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: a slip-replication model and metabolic therapy. Proc Natl Acad Sci U S A. 1989;86: 7952–6.
- Oguro H, Iijima K, Takahashi K, Nagai A, Bokura H, Yamaguchi S, . Successful treatment with succinate in a patient with MELAS. Intern Med. 2004;43:427–31.