Abstract
Cardiac remodelling is defined as changes in the size, shape, and function of the heart, which are most commonly caused by hypertension-induced left ventricular hypertrophy and myocardial infarction. Both neurohumoral and inflammatory factors have critical roles in the regulation of cardiac remodelling. A characteristic feature of cardiac remodelling is modification of the extracellular matrix (ECM), often manifested by fibrosis, a process that has vital consequences for the structure and function of the myocardium. In addition to established modulators of the ECM, the matricellular protein thrombospondin-4 (TSP-4) as well as the tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 has been recently shown to modulate cardiac ECM. TSP-4 null mice develop pronounced cardiac hypertrophy and fibrosis with defects in collagen maturation in response to pressure overload. TWEAK and Fn14 belong to the tumour necrosis factor superfamily of proinflammatory cytokines. Recently it was shown that elevated levels of circulating TWEAK via Fn14 critically affect the cardiac ECM, characterized by increasing fibrosis and cardiomyocyte hypertrophy in mice.
Here we review the literature concerning the role of matricellular proteins and inflammation in cardiac ECM remodelling, with a special focus on TSP-4, TWEAK, and its receptor Fn14.
| Abbreviations | ||
| ACTA2 | = | α-smooth muscle actin 2 |
| EC | = | endothelial cell |
| ECM | = | extracellular matrix |
| HF | = | heart failure |
| IEG | = | immediate early gene |
| KO | = | knock-out |
| LV | = | left ventricular |
| MI | = | myocardial infarction |
| MMP | = | matrix metalloproteinase |
| SMC | = | smooth muscle cell |
| SPARC | = | secreted protein acidic and rich in cysteine |
| STEMI | = | ST elevation myocardial infarction |
| TAC | = | transverse aortic constriction |
| TGF | = | transforming growth factor |
| TIMP | = | tissue inhibitor of matrix metalloproteinase |
| TNF | = | tumour necrosis factor |
| TSP | = | thrombospondin |
| TWEAK | = | tumour necrosis factor-like weak inducer of apoptosis |
Key messages
The extracellular matrix is both the target of modifications as well as an active regulator of the cardiac remodelling process, and thus contributes significantly to the development of heart failure.
Inflammation and proinflammatory cytokines have a critical role in regulating cardiac structure and function during cardiac remodelling.
Matricellular proteins modulate cellular function and behaviour by regulating cell–cell and cell–matrix interactions in cardiac remodelling.
Cardiac remodelling-related inflammatory modulators and matricellular proteins are potential targets for therapeutic interventions.
Introduction
Cardiac remodelling is an adaptive process in which the heart changes its size, shape, and function (Citation1). The aim of remodelling is to maintain adequate cardiac output despite an altered cardiac load and is most commonly due to continuous cardiac overload. Conditions that generate cardiac overload include chronic hypertension, volume overload due to mitral valve regurgitation, or the loss of viable myocardium after myocardial infarction (MI) (). Cardiac remodelling also occurs under physiological conditions, such as during pregnancy or in response to exercise, but characteristically only the pathological remodelling due to intrinsic or acquired heart diseases can lead to heart failure (HF) if left untreated (Citation1).
Figure 1. Cardiac remodelling plays a crucial role during development of heart failure. Cardiac remodelling occurs most commonly as a result of either prolonged hypertension or myocardial infarction and also in physiological conditions, such as during pregnancy.
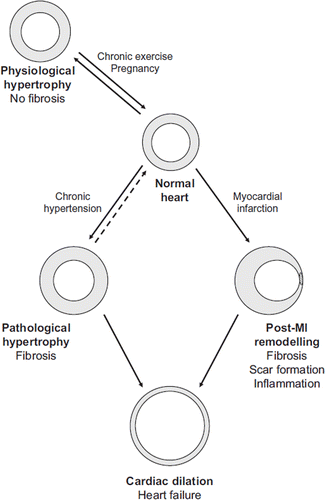
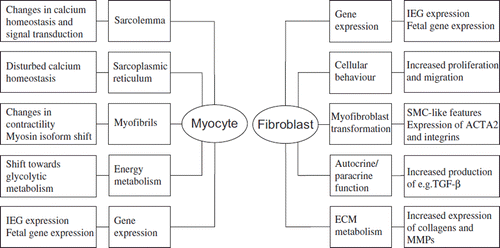
Common features in the process of pathological cardiac remodelling regardless of the aetiology are molecular, cellular, and interstitial changes of the myocardium such as myocyte hypertrophy, disorganization of the sarcomere, cell loss, and deposition of extracellular matrix (ECM) (). Cellular and molecular changes of the myocyte include e.g. changes in gene expression patterns such as activation of immediate early genes (IEGs) (c-jun, c-fos) followed by the reactivation of a fetal gene programme (atrial natriuretic peptide (ANP), β-myosin heavy chain), a shift from fatty acid oxidation to using carbohydrates as energy substrates, and alterations in calcium handling. There is also extensive myofibrillar remodelling that leads to altered contractile properties of the myocyte and subsequently to altered contractile function of the whole myocardium () (Citation1,Citation2). The response of cardiac fibroblasts to stress or injury includes alterations in proliferation, migration, ECM metabolism, and release of autocrine/paracrine mediators (Citation3). A key event in fibroblast activation is their transformation into myofibroblasts, which have smooth muscle cell (SMC)-like features and exhibit increased migration, proliferation, and secretion () (Citation4). Myofibroblasts have a contractile apparatus and express proteins distinct from those of undifferentiated fibroblasts, e.g. α-smooth muscle actin (ACTA2), α5β1 and αvβ3 integrins, tensin, and focal adhesion kinase (Citation5). Myofibroblasts are not normally found in the healthy heart muscle (Citation6) but can be detected in hypertrophic and fibrotic hearts and in the scar tissue following MI (Citation4,Citation7). In response to specific hormones, growth factors, and cytokines, the myofibroblasts become highly proliferative. They also secrete ECM proteins, predominantly type I and III collagens, and matrix metalloproteinases (MMPs) as well as bioactive molecules and therefore are a major contributor to the cardiac remodelling process. These fibroblast-stimulating factors are released from inflammatory cells, cardiac myocytes, and fibroblasts themselves and include angiotensin II (Ang II), transforming growth factor (TGF)-β1, and endothelin-1 (ET-1). Mechanical stretch can also directly or indirectly modulate fibroblast phenotype. Fibroblasts are capable of secreting a wide array of bioactive molecules themselves, such as TGF-β1, interleukin (IL)-1β, Ang II, as well as ANP and B-type natriuretic peptide (BNP). The initiating stimulus determines which factors are secreted, whereas the response depends on the receptors expressed in the target cells. The ECM produced by myofibroblasts also differs from that of undifferentiated fibroblasts; modification of the balance between MMPs and their inhibitors causes fibrosis (Citation4,Citation7). In addition to cardiac cells, the ECM plays an important role in cardiac remodelling, as it is both the target of modifications and an active regulator of the process. ECM modifications have severe consequences on the structure and function of the myocardium and contribute significantly to the development of HF (Citation8,Citation9).
Figure 2. Schematic representation of cellular and molecular changes in the heart during hypertensive hypertrophy and after myocardial infarction. In the hypertensive heart, the hypertrophy of cardiomyocytes and the transition of fibroblasts to myofibroblasts are accompanied by an increase in the amount of ECM, which leads to fibrosis. Inflammation, cell death, and scarring are integral parts of post-MI remodelling associated with subsequent hypertrophy and fibrosis of the non-infarcted area.
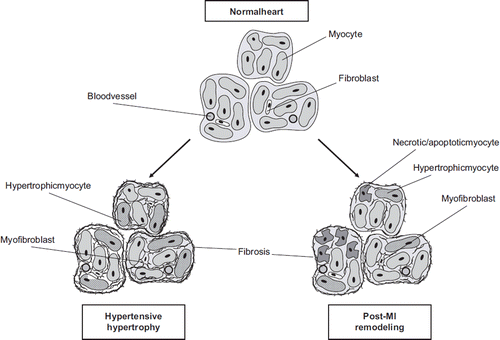
Figure 3. Subcellular targets in myocytes and fibroblasts during cardiac remodelling.
Recently, non-structural ECM proteins such as matricellular proteins have gained increasing attention in the cardiac remodelling process. They modulate cellular function and behaviour by regulating cell–cell and cell–matrix interactions, and many of these proteins appear to affect ECM composition (Citation10). Inflammation and proinflammatory cytokines also have critical roles in regulating cardiac remodelling and in the development of HF (Citation11). This interaction between inflammation and the ECM is reciprocal, in that while inflammation modulates the structure of the ECM, e.g. by affecting matrix turn-over, the ECM also regulates the inflammatory response, for instance by releasing fragments that can affect cytokine synthesis (Citation12). There are also connections between inflammation and matricellular proteins, some of which have been shown to regulate inflammation (Citation12). The following review of the literature concerning the role of matricellular proteins and inflammation in cardiac ECM remodelling will focus on novel ECM-modulating factors, i.e. the matricellular protein thrombospondin (TSP)-4 and the proinflammatory cytokine tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14.
Cardiac ECM remodelling
In cardiac ECM, structural proteins such as collagen and elastin provide support for the cells and participate in transmission of the mechanical force generated by myocytes (Citation13). Adhesive proteins like fibronectin and laminin as well as proteoglycans modulate cardiac cell growth, proliferation, and migration as well as regulate growth factor activity and collagen network organization (Citation14). In addition, a wide array of bioactive molecules participates in the complex signalling between the cells and the ECM (Citation15).
The balance between ECM synthesis and degradation determines the amount of cardiac ECM, which is produced mainly by (myo)fibroblasts but additionally by myocytes, endothelial cells (ECs), and SMCs (Citation16). The synthesis of cardiac ECM is influenced by a variety of mechanical and biochemical stimuli such as stretch, circulating hormones, as well as autocrine and paracrine factors generated in the myocardium (Citation4,Citation15). Major effectors of ECM synthesis are Ang II and TGF-β that in addition to stimulating its production can also inhibit its degradation (Citation4). The ECM is degraded mainly by MMPs. MMPs are secreted by all the major cardiac cell types as inactive precursors, which can be activated by proteases or membrane-bound MMPs (Citation17). MMP expression is increased during cardiac remodelling in response to cytokine, vasoactive peptide, and growth factor activation, e.g. to tumour necrosis factor (TNF)-α, one of the most important cytokines affecting ECM composition (Citation4,Citation17). MMPs not only regulate the degradation of the ECM but can also stimulate matrix synthesis by releasing matrix-bound growth factors and other profibrotic molecules from the ECM (Citation18). MMPs are inhibited by tissue inhibitors of MMPs (TIMPs), and ultimately it is the balance between MMPs and TIMPs together with ECM synthesis that determines the rate of matrix turn-over (Citation17). The rate of ECM turn-over in the heart is normally low, but in pathological situations both the synthesis and degradation increase (Citation8).
The most common pathological situations that result in cardiac remodelling are hypertensive hypertrophy and MI (). In the former the net effect is an increase in the amount of ECM, which leads to fibrosis, a process characteristic of pathological remodelling since it is absent in physiological hypertrophy (Citation19). Fibrosis is detrimental because the accumulation of excessive ECM impairs diastolic function, diminishes coronary flow reserve, and predisposes to arrhythmias (Citation19). In the reverse situation, when cardiac ECM degradation exceeds its synthesis, such as after MI, left ventricular (LV) dilatation, sphericalization, and increased compliance ensue (Citation20). In addition to thinning and scarring of the infarcted wall, post-MI remodelling causes complex time- and region-dependent changes including also subsequent hypertrophy and fibrosis of the non-infarcted area (Citation21).
In addition to the quantity of the ECM, its quality also affects the physical properties of the myocardium, in particular its compliance. The size and alignment of the collagen fibres as well as altered ratios of diverse collagen types may affect myocardial stiffness, and especially the post-translational modifications of collagen fibrils contribute to the compliance of the myocardium (Citation22,Citation23). An important type of collagen modification is the formation of intermolecular cross-links that make the collagen fibres less soluble and resistant to degradation. Consequently, myocardial collagen cross-linking stiffens the myocardium and is accelerated in situations with increased collagen deposition, such as in LV hypertrophy (Citation23,Citation24). In contrast, poorly cross-linked collagen is more rapidly degraded, which predisposes to LV dilatation (Citation22) as reported in association with dilated myocardium in HF (Citation25,Citation26).
Inflammation and proinflammatory cytokines in cardiac ECM remodelling
As a classical biological response to injury and a mechanism for adaptation and tissue repair (Citation11), inflammation is an integral part of post-MI remodelling (Citation27). A transient myocardial inflammatory reaction with release of inflammatory mediators can also be seen in response to pressure overload in vivo (Citation28,Citation29). A well characterized mechanism for the activation of the inflammatory response is the cell destruction after MI that exposes antigens capable of triggering the immune response, but inflammation can also be provoked by myocardial cytokine release in response to haemodynamic stress (Citation30). Since the initial observation that circulating levels of a classical proinflammatory cytokine TNF-α are increased in HF patients (Citation31), a number of studies have established a critical role for proinflammatory cytokines in the development of HF as well as in regulating cardiac structure and function in other forms of cardiac remodelling. This has led to a theory called the ‘cytokine hypothesis’ which claims that HF progresses because cytokine cascades that are activated following myocardial injury exert deleterious effects on the heart (Citation32).
Cytokines are a group of relatively small molecular weight proteins that are secreted by a broad variety of cells in response to different types of stimuli such as environmental stress. They tend to act over short distances in a juxtacrine, paracrine, or autocrine manner, but in some cases they can also have endocrine effects (Citation32). In the heart they are rapidly up-regulated in response to myocardial injury and act as part of the intrinsic cardiac stress response system. This up-regulation has been shown to be beneficial when short-term and limited but has deleterious effects when sustained (Citation33); e.g. TNF-α modulates fibroblast function by stimulating MMP expression and decreasing collagen synthesis, although long-term stimulation may promote accumulation of collagen and fibrosis (Citation4,Citation33).
Cardiac injury activates immunomodulatory mechanisms initiating an inflammatory reaction; e.g. the complement cascade up-regulates chemokine and cytokine synthesis in the infarcted heart (Citation34). Complement activation may play an important role in mediating neutrophil and monocyte recruitment in the injured myocardium (Citation35). Furthermore, cytokines like TNF-α promote fibroblast proliferation and transformation into myofibroblasts (Citation13). In addition to cardiac fibroblasts, myofibroblasts can also be derived from monocytes, bone-marrow-derived cells, and ECs (Citation4).
Matricellular proteins in cardiac ECM remodelling
Matricellular proteins are non-structural ECM proteins that modulate cellular function and behaviour by regulating cell–cell and cell–matrix interactions. Characteristically matricellular proteins are minimally expressed in normal adult tissue but are up-regulated during development and in tissue injury (Citation10). The matricellular proteins involved in cardiac remodelling include e.g. secreted protein acidic and rich in cysteine (SPARC), tenascin-C, osteopontin, periostin, and TSPs. The importance of these proteins in cardiac remodelling has been demonstrated in knock-out (KO) mice which have a mild cardiovascular phenotype but have increased adverse remodelling and decreased survival in response to MI or pressure overload (Citation36,Citation37). Based on these studies, many of the matricellular proteins seem to have a role in myocardial collagen deposition since e.g. osteopontin, SPARC, and periostin deficiency result in reduced accumulation or maturation of collagen after MI (Citation12).
Thrombospondins
TSPs are a family of five multimeric multidomain matricellular proteins (). They are capable of interacting with a large number of proteins and proteoglycans which enables their various actions and tissue and cell type-specific functions depending on the bioavailability of TSP receptors and binding partners in the given environment (Citation38). Additionally, despite the structural relatedness, their functions have diverged due to their different transcriptional regulation and functional capabilities (Citation38,Citation39). In heart, increased expression of TSPs-1–4 has been shown during cardiac remodelling (Citation37) ().
Figure 4. Schematic structures of TSP proteins, TWEAK, and Fn14. A: TSP-1 and TSP-2; B: TSP-3 and TSP-4; C: TSP-5; D: TWEAK; and E: Fn14 protein. OM = oligomerization domain; VWC = von Willebrand factor C type domain; TSR = thrombospondin repeats; EGF = EGF-like repeats; Cyt = cytoplasmic domain; TM = transmembrane domain; EC = extracellular domain; RB = receptor binding domain; Sig = signal peptide. Arrow indicates a cleavage site. Modified from (Citation88–90).
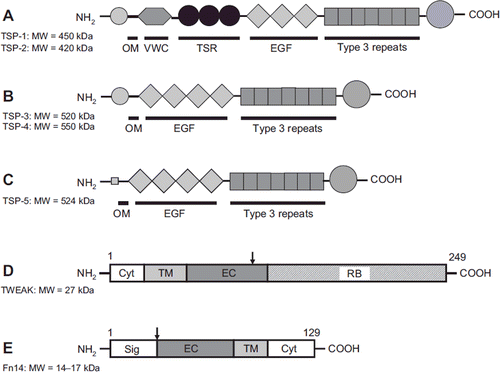
Table I. Expression and localization of TSPs in adult heart in vivo (modified from (Citation37)).
TSP-1 is a major activator of TGF-β, and a suggested function of TSP-1 in cardiac remodelling is limitation of the inflammatory reaction after MI (Citation40). TSP-2, on the other hand, has been shown to regulate myocardial matrix integrity during cardiac remodelling and ageing (Citation41). The absence of TSP-2 results in cardiac rupture after MI (Citation42) or in response to pressure overload (Citation43) probably through increased MMP activity, which also contributes to the age-dependent dilated cardiomyopathy in TSP-2 KO mice (Citation41). In addition, TSP-2 has been shown to inhibit the ageing-associated local myocardial inflammatory response (Citation41). There is hardly any evidence regarding TSP-3 expression in the heart. In a DNA microarray study of Schroen et al., they show that TSP-3 mRNA levels are increased in hearts from hypertensive homozygous renin over-expressing (Ren-2) rats that had progressed to early HF compared to Ren-2 rats that had remained compensated (Citation43). In another study, very low expression levels of TSP-3 in human heart were reported (Citation44). In conclusion, whether TSP-3 has a role with respect to fibrosis or myocardial remodelling remains to be studied.
TSP-4 gene expression has been shown to be increased in DNA microarray studies of ischaemic and failing human myocardium (Citation45,Citation46) and in rats with hypertensive HF (Citation47). According to recent reports, TSP-4 null mice develop pronounced cardiac hypertrophy and fibrosis as well as LV dilatation and depressed systolic function in response to transverse aortic constriction (TAC) (Citation48,Citation49). Defects in collagen maturation were also observed, which suggests that TSP-4 is involved in the post-translational modification of collagen (Citation48). This is supported by the fact that TSP-4 binds to the fibrillar collagens I and III in vitro (Citation50) and has previously been shown to be associated with collagen fibrils in vivo (Citation51). Indeed, it has been suggested that TSP-4 assists in collagen fibril assembly (Citation50,Citation51) possibly in the same way that TSP-5 (i.e. cartilage oligomeric matrix protein (COMP)) has been shown to facilitate collagen–collagen interactions and fibrillogenesis (Citation52).
LV TSP-4 is rapidly up-regulated in rats in response to acute pressure overload well before the development of LV hypertrophy and fibrosis (Citation53), but this induction is attenuated in hypertrophied heart (Citation53). In heart, localization of TSP-4 is restricted to ECs (Citation53,Citation54) and SMCs derived from coronary arteries (Citation54). This distinguishes TSP-4 from e.g. TSP-1, which is expressed in several cell types including myocytes, ECs, SMCs, and fibroblasts (Citation40,Citation55,Citation56). In addition, TSP-4 expression correlated with LV remodelling in hypertensive hypertrophy as well as after MI, when the beta-blocker metoprolol elicited beneficial structural remodelling in parallel with a reduction in TSP-4 gene expression (Citation57). In contrast, induction of TSP-1 gene expression was linked exclusively to the LV remodelling process after MI but not in hypertensive hypertrophy (Citation57), indicating (together with a different expression pattern) that TSPs may have unique roles in the ECM remodelling process. Although a lack of TSP-4 in cardiac remodelling seems to be detrimental, increased TSP-4 expression during hypertensive hypertrophy and ageing might also be harmful due to e.g. increased collagen cross-linking and LV stiffness (Citation47,Citation53).
The data describing TSP-5 expression in the heart are contradictory. Fang et al. reported that TSP-5 is expressed in the adult mouse heart (Citation58), but TSP-5 was not detected in the hearts of newborn mice (Citation59). Moreover, the hearts of COMP-deficient mice showed normal cardiac morphology (Citation60). Altogether, the role of TSP-5, if there is any, is not well defined in the heart, and, to our knowledge, there are no studies describing TSP-5 during cardiac remodelling.
TSP-1 and -2 have been implicated in tissue injury and inflammation such as in wound healing and foreign body reaction (Citation61). Although TSP-1 and TSP-2 have been associated with inhibition of the cardiac inflammatory response (Citation40,Citation41), TSP-4 has been shown to be associated only with vascular inflammation in cardiovascular pathologies (Citation62). In vasculature, TSP-4 influences the recruitment of macrophages by activating endothelial cells and directly interacting with macrophages to increase their adhesion and migration (Citation62).
In conclusion, the recently reported role of TSP-4 in limiting fibrosis and LV dilatation after TAC in mice (Citation48,Citation49) agrees with the positive correlations of TSP-4 with both remodelling after MI and in hypertensive hypertrophy and provides interesting insights for the development of anti-fibrotic therapy targeting TSP-4. Further studies are needed to evaluate whether TSP-4 expression correlates directly with fibrosis in cardiac remodelling. TSP-4 may have a unique role in the ECM remodelling process, which differs from that of other thrombospondins such as TSP-1. Moreover, TSP-4 might be an endothelial-specific marker of pressure overload. Whether TSP-4 has an effect on the myocardial inflammatory reaction also needs to be investigated.
TWEAK and Fn14
Similar to the other TNF family members, TWEAK is initially synthesized as a transmembrane protein and cleaved intracellularly to a soluble form that can trimerize (Citation63). Cells can express both membrane-bound and soluble TWEAK, and recently it was shown that the membrane-bound form can also bind and activate its receptor Fn14 and thus have juxtacrine effects (Citation64). Fn14 was first discovered as a fibroblast growth factor (FGF)-inducible gene (Citation65) then later classified as a member of the TNF receptor family and identified as the receptor for TWEAK (Citation66). Fn14 is the smallest member of the TNF receptor family and lacks the characteristic death domain but has binding sites for TNF receptor-associated factors that activate intracellular signalling pathways (Citation66). The structures of TWEAK and Fn14 are depicted in . As a widely expressed cytokine, TWEAK can also be detected in the heart (Citation67), where it is expressed by various cell types, including myocytes, as is its receptor Fn14 (Citation67,Citation68). Basal Fn14 expression is usually relatively low, but is induced by many substances and tissue injury (Citation66,Citation69,Citation70).
The most established role of TWEAK is the promotion of the inflammatory reaction since it is able to up-regulate, also in myocytes (Citation67), several proinflammatory cytokines, chemokines, cell adhesion molecules, and matrix-degrading enzymes (Citation71). It has been suggested that infiltrating inflammatory cells produce TWEAK in the context of tissue injury, and the selective up-regulation of Fn14 limits its actions to the injured tissue. The TWEAK/Fn14 pathway probably has a physiological role in the facilitation of tissue repair after injury, but its sustained activation may be pathological (Citation71).
The TWEAK-Fn14 axis has previously been implicated in autoimmune diseases, cancer, and ischaemic stroke (Citation70), but recent reports also associate it with cardiac remodelling (Citation67,Citation68,Citation72). Serum levels of TWEAK (sTWEAK) have been measured in human MI and HF patients, and low sTWEAK levels were reported to predict an adverse prognosis in HF patients (Citation73,Citation74). On the other hand, decreased (Citation73) or elevated (Citation68) sTWEAK levels have been measured in HF patients compared with healthy controls. Increased sTWEAK levels have been reported also in patients with acute ST elevation MI (STEMI) in which the sTWEAK levels correlated with an adverse outcome (Citation75). Since acute STEMI is a thromboembolic process including platelet activation, increased sTWEAK levels might be explained by the observation that sTWEAK is released by activated platelets, at least in vitro (Citation76). Altogether, these studies demonstrate that serum levels of TWEAK are modulated in situations requiring cardiac remodelling, although its causes, sources, and consequences remain to be studied.
TWEAK and Fn14 KO mice are viable and lack any significant abnormalities (Citation77,Citation78). However, transgenic mice over-expressing TWEAK in the liver with elevated levels of circulating TWEAK develop dilated cardiomyopathy with subsequent HF characterized by increased fibrosis and eccentric cardiomyocyte hypertrophy. These effects were shown to be mediated via Fn14 (Citation68). Interestingly, increased plasma levels of TWEAK did not result in an increased myocardial inflammatory response in mice (Citation68). In our experiments, TWEAK protein was mainly localized to rat cardiomyocytes and ECs and, notably, not to inflammatory cells (Citation72). This is in contrast to a previous report that TWEAK was mainly expressed by infiltrating inflammatory cells in response to various forms of tissue injury (Citation71). Furthermore, Fn14 immunoreactive cells were mainly fibroblasts in the inflammatory area, and Fn14 immunoreactivity increased as fibrosis progressed (Citation72).
Our experimental studies show that cardiac Fn14 expression was increased both acutely and chronically in rats in response to pressure overload and MI, while TWEAK expression remained relatively constant (Citation72), which distinguishes the expression of these cytokines from TNF-α and its receptors. Similarly, Fn14 expression was also induced in myocytes in vitro by hypertrophic agonists and other chemical stimuli as well as by mechanical stretch, in contrast to stable or decreased expression of TWEAK (Citation72). Interestingly, in the study by Mittal et al. of denervation-induced skeletal muscle atrophy, TWEAK expression did not change while Fn14 was up-regulated (Citation79). This is similar to our results in the heart. Yet, the absence of TWEAK or treatment with TWEAK antibody was able to rescue the muscle loss (Citation79), which supports the hypothesis that a reduction in Fn14 expression at the base-line limits the actions of TWEAK in healthy tissue and the up-regulation of Fn14 renders tissues more sensitive to TWEAK (Citation80). Moreover in renal ischaemia–reperfusion injury, although TWEAK expression was up-regulated only at one time point, Fn14 antibody attenuated the inflammatory reaction and the development of fibrosis (Citation81). The results from these studies demonstrate a profoundly disparate regulation of TWEAK and Fn14 expressions in the heart and emphasize the importance of Fn14 as a mediator of TWEAK/Fn14 signalling and as a potential target of therapeutic interventions.
In the study of Chorianopoulos et al., increased expression of Fn14 was noted in the border zone and the remote non-ischaemic part of infarcted myocardium in mice at day 28 post-MI, when the myocardium was cleared from an inflammatory cell infiltrate (Citation67). Moreover, this remote non-ischaemic myocardium is subjected to a remodelling process with fibrosis (and myocyte hypertrophy), which supports the hypothesis that TWEAK and Fn14 may also modulate ECM factors and affect ECM remodelling beyond inflammation (). On the other hand, studies in murine NIH 3T3 fibroblasts indicated that Fn14 may play a role in cell–matrix interactions (Citation65). In these experiments, Fn14 expression decreased cellular adhesion to the extracellular matrix proteins fibronectin and vitronectin. Whether this also happens in the heart remains to be studied. Furthermore, TWEAK can induce MMP-9 production in skeletal muscle (Citation82), and this increased production of MMP-9 contributed to the loss of skeletal muscle mass in vivo. This suggests that MMP-9 is involved in the degradation of the components of extracellular matrix in skeletal muscle.
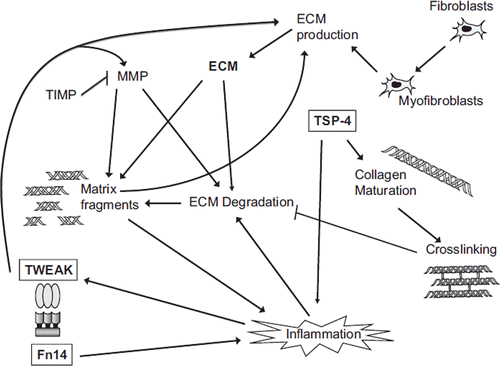
Figure 5. Schematic view of selected factors and pathways involved in the regulation of cardiac ECM remodelling and the potential roles of TSP and TWEAK/Fn14 in the process. The amount of cardiac ECM is determined by the balance between ECM synthesis, deposited mainly by myofibroblasts, and degradation, and regulated by factors like MMPs and TIMPs. The quality of the myocardium is affected by several factors such as intermolecular cross-links between collagen fibres. In addition to established ECM modulating factors, the matricellular protein TSP-4 and the TWEAK/Fn14 pathway seem to have important effects on cardiac ECM structure and homeostasis.
However, further investigation of TWEAK–Fn14 signalling mechanisms is needed, since it is not known whether TWEAK exerts its function on the heart in an autocrine–paracrine fashion like cytokines usually do or in an endocrine manner as proposed by Jain et al. (Citation68). In the former case, it is not known whether the uninducible, basal levels of TWEAK are capable of mediating its effects in the heart or, as is hypothesized, increased Fn14 expression results in ligand-independent Fn14 signalling (Citation70). Neither is it certain if TWEAK is the only ligand of Fn14 (Citation83). TWEAK does not bind to any other TNF receptor superfamily member, including DR3 (Citation70), but it can bind to CD163, a cysteine-rich scavenger receptor that is expressed exclusively on the monocyte/macrophage lineage (Citation84). Although Fn14 is the only recognized receptor mediating the actions of TWEAK, there is evidence that TWEAK can exert its functions without interacting with Fn14 (Citation85,Citation86). Due to the distinct expression of TWEAK and Fn14, it would be interesting to compare the difference between the absence of TWEAK and the absence of Fn14 during cardiac remodelling in order to evaluate the function of this cytokine receptor pair in the heart.
In conclusion, cardiac remodelling influences the TWEAK/Fn14 pathway and thereby justifies it as a potential molecular target in the prevention of pathological remodelling of the heart. Further studies are needed to test whether inhibition of TWEAK signalling is sufficient to block actions of the pathway in the heart or whether direct inhibition of Fn14 downstream signalling is essential to prevent the potentially unfavourable consequences of Fn14 receptor activation.
Conclusions
In addition to the already established ECM-modulating factors, like TSP-1 and TNF-α, recently recognized novel factors TSP-4 and TWEAK/Fn14 provide new insights into the cardiac remodelling process (). A common feature of the matricellular protein TSP-4 and TNF-receptor family member Fn14 is that they seem to have important effects on cardiac ECM structure and homeostasis especially in response to increased load (Citation41,Citation42,Citation68). Another feature these molecules share is that their basal expression is low but they are up-regulated upon cardiac injury. This would make at least Fn14 inhibition an attractive candidate for therapy due to potentially minor harmful effects in extracardiac tissues. TSP-4 augmentation, on the other hand, might potentially be beneficial in the setting of acute heart failure (Citation87).
The modifications of ECM affect cardiac structure and function profoundly, and therefore ECM remodelling is an essential component of cardiac remodelling. Understanding the mechanisms controlling ECM remodelling is important for the development of novel therapeutic interventions or diagnostic and prognostic markers for cardiovascular diseases.
Acknowledgements
This study was financially supported by the Academy of Finland (Center of Excellence funding), Aarne Koskelo Foundation, Aarne and Aili Taponen Foundation, Astra-Zeneca Research Foundation, Biocenter Oulu, Emil Aaltonen Foundation, the Finnish Foundation for Cardiovascular Research, the Finnish Medical Foundation, Ida Montin Foundation, Instrumentarium Research Foundation, Maire Taponen Foundation, Oulu University Scholarship Foundation, Paavo Ilmari Ahvenainen Foundation, the Research and Science Foundation of Farmos, and the Sigrid Jusélius Foundation.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–82.
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008; 358:1370–80.
- Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45: 657–87.
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123: 255–78.
- Eyden B. The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med. 2008;12:22–37.
- Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006; 291:H1015–26.
- Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–75.
- Fedak PW, Verma S, Weisel RD, Li RK. Cardiac remodeling and failure: from molecules to man (Part II). Cardiovasc Pathol. 2005;14:49–60.
- Graham HK, Horn M, Trafford AW. Extracellular matrix profiles in the progression to heart failure. European Young Physiologists Symposium Keynote Lecture—Bratislava 2007. Acta Physiol (Oxf). 2008;194:3–21.
- Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002; 14:608–16.
- Fedak PW, Verma S, Weisel RD, Li RK. Cardiac remodeling and failure: from molecules to man (Part I). Cardiovasc Pathol. 2005;14:1–11.
- Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–11.
- Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3: 1–30.
- Jane-Lise S, Corda S, Chassagne C, Rappaport L. The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart Fail Rev. 2000;5:239–50.
- Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–13.
- Corda S, Samuel JL, Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev. 2000; 5:119–30.
- Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–342.
- Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res. 2000;46:214–24.
- Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227.
- Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, . The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. 2006;30:604–10.
- Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–403.
- Janicki JS, Brower GL. The role of myocardial fibrillar collagen in ventricular remodeling and function. J Card Fail. 2002;8:S319–25.
- Lopez B, Gonzalez A, Hermida N, Valencia F, de Teresa E, Diez J. Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects. Am J Physiol Heart Circ Physiol. 2010;299:H1–9.
- Zieman S, Kass D. Advanced glycation end product cross-linking: pathophysiologic role and therapeutic target in cardiovascular disease. Congest Heart Fail. 2004;10:144–9; quiz 150–1.
- Gunja-Smith Z, Morales AR, Romanelli R, Woessner JF Jr. Remodeling of human myocardial collagen in idiopathic dilated cardiomyopathy. Role of metalloproteinases and pyridinoline cross-links. Am J Pathol. 1996;148:1639–48.
- Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, . Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation. 2001;103: 155–60.
- Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13: 1877–93.
- Baumgarten G, Knuefermann P, Kalra D, Gao F, Taffet GE, Michael L, . Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation. 2002;105:2192–97.
- Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–81.
- Celis R, Torre-Martinez G, Torre-Amione G. Evidence for activation of immune system in heart failure: is there a role for anti-inflammatory therapy? Curr Opin Cardiol. 2008;23: 254–60.
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41.
- Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–49.
- Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol. 2003;65: 81–101.
- Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111.
- Dreyer WJ, Michael LH, Nguyen T, Smith CW, Anderson DC, Entman ML, . Kinetics of C5a release in cardiac lymph of dogs experiencing coronary artery ischemia-reperfusion injury. Circ Res. 1992;71:1518–24.
- Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. 2004;64:24–31.
- Schellings MW, van Almen GC, Sage EH, Heymans S. Thrombospondins in the heart: potential functions in cardiac remodeling. J Cell Commun Signal. 2009;3:201–13.
- Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51.
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–8.
- Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, . Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–42.
- Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D'hooge J, . Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation. 2009;120:1585–97.
- Blankesteijn WM, Creemers E, Lutgens E, Cleutjens JP, Daemen MJ, Smits JF. Dynamics of cardiac wound healing following myocardial infarction: observations in genetically altered mice. Acta Physiol Scand. 2001;1731:75–82.
- Schroen B, Heymans S, Sharma U, Blankesteijn WM, Pokharel S, Cleutjens JP, . Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res. 2004;95: 515–22.
- Adolph KW. Relative abundance of thrombospondin 2 and thrombospondin 3 mRNAs in human tissues. Biochem Biophys Res Commun. 1999;258:792–6.
- Tan FL, Moravec CS, Li J, Apperson-Hansen C, McCarthy PM, Young JB, . The gene expression fingerprint of human heart failure. Proc Natl Acad Sci U S A. 2002;99: 11387–92.
- Gabrielsen A, Lawler PR, Yongzhong W, Steinbruchel D, Blagoja D, Paulsson-Berne G, . Gene expression signals involved in ischemic injury, extracellular matrix composition and fibrosis defined by global mRNA profiling of the human left ventricular myocardium. J Mol Cell Cardiol. 2007;42: 870–83.
- Rysä J, Leskinen H, Ilves M, Ruskoaho H. Distinct upregulation of extracellular matrix genes in transition from hypertrophy to hypertensive heart failure. Hypertension. 2005;45: 927–33.
- Moens AL, Cingolani O, Spinale FS, Kass DA. Exacerbated cardiac remodeling to pressure-overload in mice lacking thrombospondin-4. Hypertension. 2008;52:747–75.
- Frolova E, Sopko N, Penn M, Stenina O, Plow E. Heart hypertrophy in response to pressure overload in TSP4 knockout mice. FASEB J. 2009;23(Meeting Abstracts):642. 5–642.5.
- Narouz-Ott L, Maurer P, Nitsche DP, Smyth N, Paulsson M. Thrombospondin-4 binds specifically to both collagenous and non-collagenous extracellular matrix proteins via its C-terminal domains. J Biol Chem. 2000;275:37110–7.
- Sodersten F, Ekman S, Niehoff A, Zaucke F, Heinegard D, Hultenby K. Ultrastructural immunolocalization of cartilage oligomeric matrix protein, thrombospondin-4, and collagen fibril size in rodent achilles tendon in relation to exercise. Connect Tissue Res. 2007;48:254–62.
- Halasz K, Kassner A, Morgelin M, Heinegard D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282:31166–73.
- Mustonen E, Aro J, Puhakka J, Ilves M, Soini Y, Leskinen H, . Thrombospondin-4 expression is rapidly upregulated by cardiac overload. Biochem Biophys Res Commun. 2008;373:186–91.
- Stenina OI, Desai SY, Krukovets I, Kight K, Janigro D, Topol EJ, . Thrombospondin-4 and its variants: expression and differential effects on endothelial cells. Circulation. 2003;108:1514–9.
- Zhao XM, Hu Y, Miller GG, Mitchell RN, Libby P. Association of thrombospondin-1 and cardiac allograft vasculopathy in human cardiac allografts. Circulation. 2001;103: 525–31.
- Sezaki S, Hirohata S, Iwabu A, Nakamura K, Toeda K, Miyoshi T, . Thrombospondin-1 is induced in rat myocardial infarction and its induction is accelerated by ischemia/reperfusion. Exp Biol Med (Maywood). 2005;230:621–30.
- Mustonen E, Leskinen H, Aro J, Luodonpää M, Vuolteenaho O, Ruskoaho H, . Metoprolol treatment lowers thrombospondin-4 expression in rats with myocardial infarction and left ventricular hypertrophy. Basic Clin Pharmacol Toxicol. 2010;107:709–17.
- Fang C, Carlson CS, Leslie MP, Tulli H, Stolerman E, Perris R, . Molecular cloning, sequencing, and tissue and developmental expression of mouse cartilage oligomeric matrix protein (COMP). J Orthop Res. 2000;18:593–603.
- Kipnes JR, Xu L, Han F, Rallapalli R, Jimenez S, Hall DJ, . Molecular cloning and expression patterns of mouse cartilage oligomeric matrix protein gene. Osteoarthritis Cartilage. 2000;8:236–9.
- Svensson L, Aszódi A, Heinegård D, Hunziker EB, Reinholt FB, Fässler R, . Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol. 2002;22:4366–71.
- Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell–matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol. 2004;36:1115–25.
- Frolova E, Pluskota E, Krukovets I, Burke T, Drumm C, Smith JD, . Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res. 2010;107: 1313–25.
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, . TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272;51:32401–10.
- Brown SA, Ghosh A, Winkles JA. Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-kappaB pathway. J Biol Chem. 2010;285:17432–41.
- Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, . The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274: 33166–76.
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, . A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;155:837–46.
- Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, . FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010;105:301–13.
- Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, . A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–68.
- Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, . The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol. 2000;156:1253–61.
- Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;75:411–25.
- Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine. 2007;40:1–16.
- Mustonen E, Säkkinen H, Tokola H, Isopoussu E, Aro J, Leskinen H, . Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol (Oxf). 2010;199: 11–22.
- Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail. 2009;11:1050–6.
- Richter B, Rychli K, Hohensinner PJ, Berger R, Mortl D, Neuhold S, . Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis. 2010;213:545–8.
- Chorianopoulos E, Jarr K, Steen H, Giannitsis E, Frey N, Katus HA. Soluble TWEAK is markedly upregulated in patients with ST-elevation myocardial infarction and related to an adverse short-term outcome. Atherosclerosis. 2010;211: 322–26.
- Meyer T, Amaya M, Desai H, Robles-Carrillo L, Hatfield M, Francis JL, . Human platelets contain and release TWEAK. Platelets. 2010;21:571–4.
- Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, . TWEAK attenuates the transition from innate to adaptive immunity. Cell. 2005;123:931–44.
- Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, . TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115:2330–40.
- Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, . The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol. 2010;188:833–49.
- Ortiz A, Sanz AB, Munoz Garcia B, Moreno JA, Sanchez Nino MD, Martin-Ventura JL, . Considering TWEAK as a target for therapy in renal and vascular injury. Cytokine Growth Factor Rev. 2009;20:251–8.
- Hotta K, Sho M, Yamato I, Shimada K, Harada H, Akahori T, . Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 2011;79:179–88.
- Li H, Mittal A, Paul PK, Kumar M, Srivastava DS, Tyagi SC, . Tumor necrosis factor-related weak inducer of apoptosis augments matrix metalloproteinase 9 (MMP-9) production in skeletal muscle through the activation of nuclear factor-κB-inducing kinase and p38 mitogen-activated protein kinase: a potential role of MMP-9 in myopathy. J Biol Chem. 2009;284:4439–50.
- Dogra C, Hall SL, Wedhas N, Linkhart TA, Kumar A. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J Biol Chem. 2007;282:15000–10.
- Bover LC, Cardo-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, . A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178:8183–94.
- Polek TC, Talpaz M, Darnay BG, Spivak-Kroizman T. TWEAK mediates signal transduction and differentiation of RAW264.7 cells in the absence of Fn14/TweakR. Evidence for a second TWEAK receptor. J Biol Chem. 2003;278:32317–23.
- De Ketelaere A, Vermeulen L, Vialard J, Van De Weyer I, Van Wauwe J, Haegeman G, . Involvement of GSK-3beta in TWEAK-mediated NF-kappaB activation. FEBS Lett. 2004;566:60–4.
- Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, . Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009;112:119–29.
- Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189–200.
- Carlson CB, Lawler J, Mosher DF. Structures of thrombospondins. Cell Mol Life Sci. 2008;65:672–86.
- Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003; 14:241–9.
- Lawler J, Duquette M, Whittaker CA, Adams JC, McHenry K, DeSimone DW. Identification and characterization of thrombospondin-4, a new member of the thrombospondin gene family. J Cell Biol. 1993:120;1059–67.
- Wang D, Oparil S, Feng JA, Li P, Perry G, Chen LB, . Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension. 2003;42:88–95.
- Zhang XM, Shen F, Xv ZY, Yan ZY, Han S. Expression changes of thrombospondin-1 and neuropeptide Y in myocardium of STZ-induced rats. Int J Cardiol. 2005;105: 192–7.
- van Almen GC, Swinnen M, Carai P, Verhesen W, Cleutjens JP, D'hooge J, . Absence of thrombospondin-2 increases cardiomyocyte damage and matrix disruption in doxorubicin-induced cardiomyopathy. J Mol Cell Cardiol. 2011;51:318–28.