Abstract
There is increasing evidence that frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) represent a continuum of neurodegenerative diseases. FTLD is complicated by ALS in a significant proportion of patients, and neuropsychological studies have demonstrated frontotemporal dysfunction in up to 50% of ALS patients. More recently, advances in neuropathology and molecular genetics have started to disclose the biological basis for the observed clinical concurrence. TDP-43 and FUS have been discovered as key pathological proteins in both FTLD and ALS. The most recent discovery of a pathological hexanucleotide repeat expansion in the gene C9orf72 as a frequent cause of both FTLD and ALS has eventually confirmed the association of these two at first sight distinct neurodegenerative diseases. Mutations in the TARDBP, FUS, and VCP genes had previously been associated with different phenotypes of the FTLD-ALS spectrum, although in these cases one end of the spectrum predominates. Whilst on the one hand providing evidence for overlap, these discoveries have also highlighted that FTLD and ALS are etiologically diverse. In this review, we review the recent advances that support the existence of an FTLD-ALS spectrum, with particular emphasis on the molecular genetic aspect.
Key messages
FTLD and ALS are considered as two extremes of a clinical continuum.
TDP-43 and FUS are key pathological proteins in both FTLD and ALS
Common genetic factors underlie FTLD and ALS, most notably the gene C9orf72.
Introduction
Frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) are two related and devastating neurodegenerative diseases. FTLD is a form of dementia clinically characterized by progressive changes in behavior, personality, and/or language. ALS is a neurodegenerative disorder in which loss of motor neurons leads to progressive weakness of the voluntary muscles. While traditionally they were considered as two separate identities, it is now thought that FTLD and ALS form one clinical continuum, in which pure forms are linked by overlap syndromes. Neuropsychological testing shows normal cognition in the majority of ALS patients. However, in up to 50% of ALS patients some degree of cognitive or behavioral impairment can be documented, which is in 15%–18% sufficient to meet the criteria of FTLD (Citation1,Citation2). Recent advances in neuropathology and molecular genetics have started to disclose the biological basis for this clinical overlap. Neuronal aggregations of dysfunctional proteins are a key neuropathological feature in many forms of neurodegenerative diseases. In both FTLD and ALS the proteins TDP-43 and FUS have been identified as the main components of these inclusions, thereby providing a strong molecular link between the two disorders. Insights into the underlying biological mechanisms have been provided by the identification and functional characterization of mutations in a small fraction of patients with autosomal dominant FTLD and/or ALS. While some genetic causes are specific for either FTLD or ALS, others were found to contribute to both ends of the disease continuum. In this review, we summarize the evidence for the overlap between FTLD and ALS at the different levels, with particular emphasis on the molecular genetic aspect.
Frontotemporal lobar degeneration
FTLD is defined by the focal neurodegeneration in the frontal and anterior temporal lobes of the brain. It is, after Alzheimer's disease (AD), one of the leading causes of dementia, particularly in patients with a disease onset before 65 years (Citation3). FTLD occurs with an estimated prevalence of 10–20 per 100,000 and incidence of 3.5–4.1 per 100,000/y in the 45–64 year age-group (Citation4–7). It can manifest as three clinically recognized subtypes, depending on the early and dominating symptoms: a behavioral variant FTLD (bvFTD) and two language variants, progressive non-fluent aphasia (PNFA) and semantic dementia (SD). The bvFTD is the most frequent diagnosed variant, accounting for approximately two-thirds of FTLD patients, while PNFA and SD represent more rare forms (Citation8). The distinct phenotypes are associated with specific areas of neurodegeneration in the brain, which can be revealed by neuroimaging; bvFTD shows predominant prefrontal neurodegeneration, SD atrophy affects mainly the anterior temporal lobes, and PNFA is associated with left perisylvian atrophy (Citation9,Citation10). Patients with bvFTD present clinically with marked changes in behavior and personality, displaying a combination of disinhibition, apathy, lack of emotional concern, hyperorality, and stereotypic behavior (Citation11,Citation12). Neuropsychological testing reveals deficits on frontal or executive tasks, with relative sparing of episodic memory and visuospatial abilities (Citation13,Citation14). Patients with SD develop progressive comprehension deficits and naming errors with otherwise spared speech production (Citation11,Citation15). In contrast, PNFA is characterized by loss of motor speech fluency and agrammatism, with relatively intact language comprehension (Citation11,Citation15). The clinical subtypes usually start to show overlap with the progression of the disease, likely reflecting the more diffuse spreading of the disease in the frontal and temporal regions (Citation16). Besides the clinical overlap with ALS, symptoms of the atypical Parkinsonian disorders corticobasal syndrome (CBS) and progressive supranuclear palsy (PSP) are not uncommon in FTLD (Citation17). The median disease duration of FTLD is 6–8 years (Citation18,Citation19). Unfortunately, no cure exists for FTLD at the moment (Citation20).
Amyotrophic lateral sclerosis
ALS is a progressive neurodegenerative condition affecting the motor neurons of the cerebral cortex (the upper motor neurons (UMN)), brain-stem, and the anterior horn of the spinal cord (the lower motor neurons (LMN)). ALS has, similar to FTLD, a peak incidence around the age of 60 years. The incidence of ALS is reported to be 1.5–2.7 per 100,000/y in Western countries (Citation21). The prevalence ranges from 2.7 to 7.4 per 100,000 (Citation21). The clinical picture of ALS consists of a characteristic combination of signs of UMN degeneration, which include muscular spasticity and hyperreflexia, together with signs of LMN degeneration, which comprise muscular atrophy and fasciculations. In about two-thirds of ALS patients the disease starts in the muscles of the arms or legs and in one-third of patients in those required for swallowing or speech production (bulbar onset) (Citation22). A respiratory onset ALS is considered rare (Citation23). The motor symptoms progressively spread to include other anatomical regions; a diagnosis of ‘clinical definite ALS’ according to the revised El Escorial criteria requires the presence of UMN and LMN signs in three of four anatomical regions (Citation24). Electromyographic studies in patients suspected for ALS allow documentation of the LMN degeneration and exclusion of other disease processes that may result in muscular weakness. Clinical variants of ALS include progressive muscular atrophy (pure LMN disorder) (Citation25) and primary lateral sclerosis (pure UMN disorder) (Citation26). The majority of ALS patients die within a few years after onset of first symptoms, mostly due to aspiration pneumonia (Citation27). The drug riluzole has been shown to slow down the progression of the disease by a few months (Citation28).
Clinical continuum between FTLD and ALS
Reports of dementia or psychiatric disturbances in association with clinical features of ALS date back to the late nineteenth century (Citation29,Citation30). However, it was only together with the increased recognition of FTLD as a separate form of dementia in the early 1990s that the cognitive and behavioral abnormalities that occur in ALS started to gain more detailed attention. Currently it is estimated that approximately 15% of FTLD patients have motor dysfunction meeting the criteria of ALS (Citation31,Citation32). Similarly, in two large ALS patient series, the prevalence of FTLD was 15% and 18% (Citation1,Citation2). The pattern of cognitive changes in patients with both FTLD and ALS (FTLD-ALS) usually closely resembles that of bvFTD, and the motor neuron disease is similar to that of classic ALS (Citation33,Citation34). FTLD-ALS patients sometimes develop delusions or hallucinations, features that are relatively uncommon in pure FTLD (Citation34–36). A few reports describing a PNFA or SD phenotype in association with ALS have also been published (Citation37–39). The first symptoms in FTLD-ALS patients occur on average in the late 50s, and progress is rapid with death occurring within 2–3 years (Citation18,Citation40). In most of cases, the FTLD symptoms precede those of ALS (Citation41).
Adding further to the evidence of a clinical overlap is the observation in ALS patients without dementia of specific difficulties in performing cognitive tasks sensitive to frontal lobe dysfunction, suggesting a continuum of involvement (, ). Neuropsychological studies in series of ALS patients have most frequently documented abnormalities in the executive functions, which are higher-order mental processes implicated in planning, problem-solving, shifting of attention, and inhibiting behavior and thought to be located in the frontal lobes. Rates of executive function deficits in ALS patients without dementia have ranged from 22% to 36% (Citation1,Citation42). Impaired verbal fluency, related to executive dysfunction, has been a frequent finding in cognitively impaired ALS and has in consequence been proposed as a relatively sensitive screening method (Citation43). The performance on these tests may, however, be confounded by the reduced motor abilities in ALS, and adaptations have been developed to eliminate this effect, for example, through performing a written instead of an oral fluency task in ALS patients with impairment of bulbar musculature (Citation44). Language abnormalities in non-demented ALS also occur but less frequently. Difficulties have been described on naming and comprehension tests, and several reports have been published of ALS patients with spelling and writing errors (Citation45–48). The spectrum of frontotemporal syndromes in ALS further includes mild neurobehavioral impairment, which may occur independent of the cognitive dysfunction (Citation49). Patients with ALS have been reported with increased frequency of a mild to moderate degree of apathy that was mood-independent, with disinhibition, lack of emotional concern or stereotypic behavior (Citation50,Citation51). Imaging studies in non-demented ALS patients have correlated the presence of cognitive or behavioral impairment with dysfunction in the frontal and temporal cortices (Citation49,Citation52–54). When considering a clinical continuum between ALS and FTLD, it is noteworthy that so far no clear progression of minor cognitive or behavioral impairment to FTLD-ALS has been documented in longitudinal studies (Citation55,Citation56). Recently, consensus criteria have been proposed for the diagnosis of frontotemporal dysfunction in ALS, which include the classes FTLD-ALS, ALSci (ALS with mild cognitive impairment), and ALSbi (ALS with minor behavioral impairment) (Citation43).
Table I. Diagnostic categories of the FTLD-ALS disease spectrum.
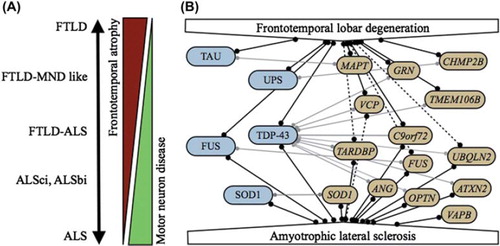
Figure 1. A: FTLD and ALS form a clinical disease continuum. B: Molecular relationships between FTLD and ALS. Pathological proteins are indicated in blue, causal genes in yellow. Full lines indicate strong correlations; dotted lines represent putative correlations. Correlations between genes and pathological proteins are indicated by light gray lines.
The reverse phenomenon of subtle and pre-symptomatic motor abnormalities in FTLD has also been investigated, although less extensively. One prospective study reported that while 14% of FTLD patients met the criteria for clinical definite ALS, another 36% had possible features of ALS such as occasional fasciculations, evidence for neurogenic changes in one limb on EMG, or troubles with swallowing (Citation31). A similar rate of minor motor system dysfunction in FTLD was reported in a second study (Citation32).
Molecular pathology of FTLD and ALS
The histopathological characteristics of FTLD and ALS include, as for most neurodegenerative diseases, abnormal accumulation of protein aggregations in the affected parts of the nervous system. In contrast to the near-uniform nature of the aggregates in case of, for example, AD (amyloid-β plaques and intracellular tau neurofibrillary tangles), the molecular pathology of FTLD and ALS is notably heterogeneous (). Nevertheless, similar molecular inclusions are being observed in FTLD and ALS, which suggests that they share common pathogenic mechanisms leading to neurodegeneration and aggregation of the specific inclusion proteins.
Approximately 35% of the FTLD patients who come to autopsy show intraneuronal and glial aggregations of the microtubule-associated protein tau (indicated as FTLD-tau) (Citation57,Citation58). The prototype of FTLD-tau is Pick's disease, where round inclusions of tau filaments are observed in ballooned neurons, which can be visualized by silver staining (Pick bodies).
In most FTLD brains, however, tau deposits cannot be detected. In the majority of these cases, neuronal intracytoplasmatic and intranuclear inclusions are present that are immunoreactive to ubiquitin (FTLD-U) (Citation59). Similar ubiquitin-positive inclusions were also observed in the degenerating motor neurons of ALS patients (Citation60). Moreover, further research demonstrated that a significant fraction of ALS patients, including those without overt cognitive changes, also have extra motor neuron ubiquitin pathology, which is mostly located in the limbic and frontotemporal brain regions reminiscent of FTLD (Citation61). In 2006, the protein TDP-43 was identified as the main component of the ubiquitinated inclusions in both FTLD-U () (renamed FTLD-TDP) and in the majority (> 90%) of ALS (ALS-TDP) (Citation62,Citation63). Following this discovery, it was proposed that FTLD and ALS form a clinico-pathological spectrum of TDP-43 proteinopathies (Citation64).
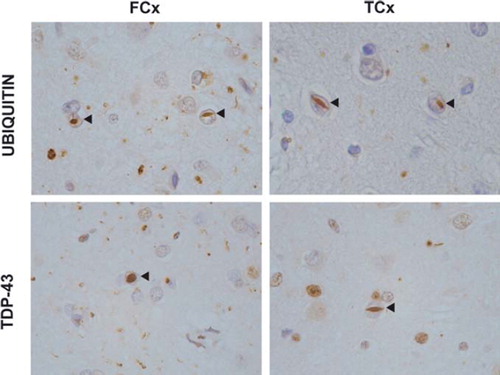
Figure 2. TDP-43 proteinopathy in FTLD. Ubiquitin and TDP-43 immunohistochemistry of sections from the superficial frontal cortex (FCx) and temporal cortex (TCx) of a patient with a VCP mutation (Citation176). Frequent intranuclear inclusions (arrow-heads) are seen that stain positive for ubiquitin and TDP-43.
In 2009, aggregations of the FUS protein were demonstrated in rare ALS patients with a mutation in the FUS gene (ALS-FUS) (Citation65,Citation66). These FUS inclusions were negative for TDP-43, which implies the involvement of mutually distinct pathways that lead to neurodegeneration. For FTLD, a small number of FTLD-U cases remained (10%–20%) that could not be explained by TDP-43 (Citation67,Citation68). When analyzed, almost all these atypical FTLD-U (aFTLD-U) brains appeared to harbor FUS inclusions (Citation69–72). In addition, abundant FUS pathology has been demonstrated in two related rare neurodegenerative disorders: basophilic inclusion body disease (BIBD) (Citation73) and neuronal inclusion filament disease (NIFID) (Citation74). Current pathological nomenclature considers aFTLD-U, NIFID, and BIBD as the three distinct forms of FTLD-FUS (Citation75). Together, ALS-FUS and FTLD-FUS form a new class of FUS proteinopathies (Citation64).
In rare FTLD patients that have been linked to the CHMP2B gene, cellular inclusions are present that are ubiquitin-positive but TDP-43- and FUS-negative (FTLD-UPS) (Citation71). Likewise, in ALS, a third molecular class exists without clear TDP-43 or FUS pathology, ALS-SOD, which is observed in ALS patients with a SOD1 mutation (Citation76).
Notably, TDP-43 and FUS are not specific to FTLD and ALS. Concomitant TDP-43 pathology is also found in several other forms of neurodegenerative diseases, such as AD (20%–26% of patients) (Citation77,Citation78), dementia with Lewy bodies (DLB) (18%–45% of patients) (Citation79,Citation80), or corticobasal degeneration (CBD) (15% of patients) (Citation78) (for review see (Citation81)). The relevance of this phenomenon of concomitant TDP-43 pathology remains unclear, and attempts to correlate the concurrent TDP-43 pathology with the clinical phenotype has given mixed results (Citation78,Citation82). In neurologically healthy elderly, a limited number of studies indicated that TDP-43 pathology is rare in normal aging (3% or less) (Citation83). As in the case of TDP-43, concomitant FUS inclusions have been described in several other diseases, including disorders with expanded polyglutamine stretches such as Huntington's disease (Citation84) and several forms of spinocerebellar ataxia (Citation85).
FTLD-ALS mutation spectrum
FTLD is known to be a principally familial disorder, with the incidence of FTLD-ALS spectrum disorders in a patient's relative being 38%–47% (Citation86–88). Similarly, a positive familial history has been found in 36%–59% of patients with FTLD-ALS (Citation86–88). In ALS, however, most patients are isolated cases, but 5%–10% have a familial history of the disease (Citation89). The notion of a shared genetic susceptibility between FTLD and ALS is also supported by findings of epidemiological studies. First-degree and second-degree relatives of ALS patients have a 2-fold increased risk for dementia (Citation90,Citation91).
Major progress has been made in the last 20 years in unraveling the genetic basis of FTLD and/or ALS. Together, FTLD-ALS disorders have been linked to at least 14 different genes. While some genetic causes are largely specific for either FTLD or ALS, others contribute to different phenotypes of the FTLD-ALS disease spectrum (, ).
Table II. Genes associated with FTLD and ALS.
Autosomal dominant mutations in two genes, MAPT and GRN, account for about 10%–20% of all FTLD patients (Citation87, Citation92–94). ALS has only very rarely been part of the clinical spectrum within MAPT or GRN segregating FTLD families (Citation95–99). Other rare genetic causes of FTLD include mutations in the CHMP2B gene, in which pathogenic nonsense mutations have so far been identified in one large Danish FTLD pedigree (Citation100) and one familial Belgian FTLD patient (Citation101). A small number of CHMP2B missense mutations have also been described in FTLD and ALS patients; however, the pathogenicity of this type of CHMP2B mutations still remains uncertain (Citation101–104). Mutations affecting the gene VCP were previously known to cause a rare multi-system disorder that includes FTLD (Citation105). Recently, the phenotypic spectrum associated with VCP has been extended also to include ALS.
Mutations in the SOD1 gene are being found in about 20% of familial and 3% of sporadic ALS patients (Citation106,Citation107). There are very few reports of dementia in association with SOD1 mutations (Citation108). Further, neuropsychological assessment in series of SOD1 mutation carriers failed to detect significant cognitive impairment (Citation109). Mutations have been detected in the genes that encode the key pathological proteins of the FTLD-ALS spectrum, FUS and TDP-43 (Citation65,Citation66,Citation110,Citation111). Unexpectedly, however, mutations in the respective genes, FUS and TARDBP, appear to occur mostly at the ALS end of the disease spectrum. Occasionally, mutations in these genes have also been reported in FTLD-ALS or FTLD patients. While expansions of a polyglutamine tract in ATXN2 have been associated with ALS (Citation112), screening of this gene in FTLD and FTLD-ALS cohorts has so far been negative (Citation113,Citation114). Lastly, rare genetic causes of ALS include mutations in the ANG, OPTN, UBQLN2, and VAPB genes () (Citation115–118). In UBQLN2 families, the phenotype was mostly ALS, with a few patients reported as FTLD-ALS (Citation118). The ANG gene has also been associated to FTLD-ALS, but this was based on the observation of a mutation in a single FTLD-ALS pedigree (Citation119).
The strongest evidence for a shared genetic etiology between FTLD and ALS came from multiple linkage and genome-wide association (GWA) studies that established a shared disease locus on chromosome 9p (Citation120–131). Most recently, three research groups have independently identified pathological expansions of a hexanucleotide repeat (GGGGCC) in the gene C9orf72 as the underlying genetic cause of chromosome 9p-linked FTLD and ALS (Citation132–134).
A pathological repeat expansion in C9orf72 results in both FTLD and ALS
Since 2006, several large families in which FTLD and ALS co-segregated were reported with linkage to a region on chromosome 9p (Citation120–127). The shared chromosomal region between the linked families was 3.6 Mb in size and was located at 9p21.2-p21.1. The same chromosome 9p region was independently identified in several large population-based GWA studies in cohorts of unrelated ALS patients and control individuals. A trend for association with the same region was also observed in a FTLD GWA study, in a cohort of patients enriched for TDP-43 neuropathology (Citation128–131). Fine mapping in the GWA studies delineated the association signal to a 106.5 Kb sized region at 9p21.2. In spite of extensive genetic analyses of all known and predicted coding and RNA genes within the candidate region, no simple sequence mutation or copy number variation could be identified explaining the established linkage and associations (Citation125–127).
The clinical phenotype both between and within the chromosome 9p-linked families was variable (); 31% of all patients presented with bvFTD only, 35% ALS only, and 34% a combination of both disorders. Additional clinical features included frequent hallucinations and delusions (Citation120,Citation122,Citation124,Citation126,Citation127). Further, Parkinsonism was common in these families, and a CBS phenotype was reported in one patient (Citation122,Citation125–127). One patient had prominent cerebellar ataxia (Citation127). The mean disease onset age in these families age was 53 years, ranging from 31 to 84 years, with some risk haplotype carriers remaining disease-free until late in life (> 75 years) (Citation120,Citation122,Citation125). The disease duration was 5 years on average, with shorter survival for ALS patients (3 years) than for FTLD-ALS (4 years) or bvFTD patients (7 years).
Table III. Characteristics of chromosome 9p-linked FTLD-ALS families.
The neuropathology of the chromosome 9p-linked FTLD-ALS patients was, in case of FTLD, characterized by TDP-43 inclusions, showing a specific pattern of abundant neuronal cytoplasmatic inclusions with few neurites and intranuclear inclusions (a subtype of FTLD with TDP-43 pathology denoted as FTLD-TDP type B) (Citation135). This FTLD-TDP subtype is typical for FTLD-ALS and has also been observed in familial FTLD-ALS patients not linked to chromosome 9p and in sporadic FTLD-ALS patients (Citation136–138). The ALS pathology has been described as classical ALS-TDP. Often, chromosome 9p-linked patients with exclusively clinical FTLD or ALS showed a combined pathology at autopsy.
Most recently, others and we independently demonstrated that a pathological expansion of a GGGGCC hexanucleotide repeat within the promoter region of C9orf72 is at the basis of chromosome 9p-linked FTLD and ALS (Citation132–134). The expanded repeat was shown to co-segregate with disease in several of the linked families. Further screening for GGGGCC repeat expansions in FTLD and ALS patient cohorts resulted in high mutation frequencies. Depending on the study, in patients with familial disease, 80% of FTLD-ALS, 25%–50% of ALS patients, and 10%–30% of FTLD patients had a pathological repeat expansion. The majority of the autopsy-confirmed mutation carrier patients had TDP-43 inclusions in brain or spinal cord. However, also ubiquitin- positive and TDP-43-negative inclusions were observed, suggesting that proteins other than TDP-43 were accumulating (Citation132,Citation139). The identification of a pathological repeat expansion in C9orf72 as a major cause of both FTLD and ALS provided the ultimate evidence that these two diseases are indeed biologically related.
Genotype–phenotype correlations in other FTLD-ALS spectrum genes
TARDBP
Sequencing of the TARDBP gene in FTLD and ALS patients was motivated by the discovery of underlying TDP-43 pathology. By early 2008, the first mutations in TARDBP were reported in ALS, providing conclusive evidence that TDP-43 itself can trigger TDP-43-associated neurodegeneration (Citation110,Citation111). To date, over 40 mutations have been identified in TARDBP, 7 of which were found in FTLD or FTLD-ALS patients (http://www.molgen.vib-ua.be/FTDMutations The majority of the mutations are missense and are located in exon 6 of the gene, encoding the C-terminal glycine-rich domain, which is thought to be important for protein–protein interactions.
Mutations in TARDBP are usually associated with classic adult onset ALS. They account for 3%–4% of familial and around 1% of sporadic ALS. The site of onset varies, and some patients presented with a predominantly lower motor neuron disorder. In 74 index patients carrying 31 different TARDBP mutations, the disease age of onset varied from 30 to 74 years, with an average of 55 years (ALSOD database, http://alsod.iop.kcl.ac.uk/). Several TARDBP families showed evidence for incomplete penetrance. For example, in an Italian founder family segregating the p.A382T mutation, the penetrance was estimated at 60% by the age of 70 years (Citation140).
TARDBP mutations may also be a rare but notable cause of FTLD-ALS and FTLD. Evidence is relatively strong in the case of FTLD-ALS. Several reports have been published of TARDBP mutations in FTLD-ALS patients (Citation141–144). For example, one study that included a large number (n = 149) of FTLD-ALS patients found that two (1%) of those patients carried a TARDBP mutation (Citation144). TARDBP missense mutations have to date been described in two pure FTLD patients with sporadic onset (Citation145,Citation146). One of these mutations has also been found in one ALS patient, adding evidence for its pathogenic character. Further, one TARDBP mutation has been described in a FTLD patient with additional features of supranuclear palsy and chorea, who showed TDP-43 pathology at autopsy (Citation147). On the other hand, negative findings from other large cohorts of FTLD patients (together, n = 674) indicated that TARDBP mutations in pure FTLD are very rare (Citation148–151).
ALS patients with a TARDBP mutation that come to autopsy show ALS-TDP pathology that is indistinguishable from the TDP-43 pathology observed in sporadic ALS (Citation152–156). Although reports are limited, TDP-43 inclusions are found more widespread than the actual motor neuron degeneration (Citation152,Citation153).
FUS
Shortly following the identification of mutations in TARDBP in ALS patients, pathogenic mutations in FUS were detected in autosomal dominant ALS families linked to chromosome 16 (Citation65,Citation66). Over 40 FUS mutations have now been reported in ALS and 3 mutations in FTLD-ALS or FTLD (http://www.molgen.vib-ua.be/FTDMutations). Most were found in the C-terminal end of FUS encoded by exons 14 and 15. These may be missense or nonsense mutations, splice site or frameshift mutations that result in the production of a C-terminal truncated protein. Interestingly, the C-terminal part of the FUS protein holds a nuclear localization signal (NLS), a sequence necessary for transport of the protein into the nucleus of the cell (Citation157–159). A second cluster of mutations is observed in the Gly-rich region of FUS, encoded by exons 4 to 6, which are mostly found in sporadic patients.
Mutations in FUS are also mostly associated with a classic ALS phenotype. In several FUS mutation carriers, the signs of the disease were restricted to LMN damage, with little or no evidence for UMN disease (Citation160–162). An unusual presentation affecting predominantly the axial musculature has been described in a number of ALS patients with the p.R521C mutation (Citation163–165). The frequency of FUS mutations is estimated at 4% in familial ALS and 1% in sporadic ALS.
FUS mutation carriers have been reported with a disease onset for ALS from as early as 13 years old to over 70 years of age. A correlation was observed between onset age and the degree in which the mutation affects the NLS of FUS. Truncating mutations, which completely remove the NLS or missense mutations strongly affecting the NLS, such as the p.P525L mutation, occur in patients with earlier disease onset (before the age of 30 years) and more rapid decline (Citation158,Citation166–168). However, even within families segregating the same FUS mutation a broad spread in onset age has been observed, indicating that other genetic and/or environmental factors also influence the disease manifestation (Citation160,Citation169).
Few FUS mutations have been identified in patients with an FTLD or FTLD-ALS phenotype. We have described a missense mutation in a bvFTD patient without signs of motor neuron dysfunction (Citation170). The mutation affects a highly conserved amino-acid residue and was absent in a large group of control individuals, suggesting a pathogenic role. However, family history was negative for FTLD or ALS, and there was no autopsied brain material available to investigate the associated brain pathology. Other investigators have described FUS mutations in FTLD-ALS patients (Citation143,Citation165,Citation171). Some reports also mentioned the presence of FTLD or dementia in relatives of FUS mutation carries (Citation160,Citation168). However, two further studies of FUS in large FTLD cohorts (together, n = 465) observed no mutations (Citation72,Citation171), suggesting that FUS has only a limited role in the genetic etiology of FTLD.
FUS mutation carriers typically show inclusions of FUS but not of TDP-43 in affected motor neurons (Citation66,Citation160,Citation163,Citation172). Some reports indicate that the degeneration can be limited to the LMN, with minimal damage of the UMN (Citation163,Citation173). FUS staining is, however, more widespread and shows pathological inclusions in glial cells and neurons outside the motor system, including the striatum, basal ganglia, and frontal and temporal cortex. Some researchers also noticed basophilic inclusions in the degenerating motor neurons (Citation160,Citation163).
FUS has also been established as a major inclusion protein in three rare pathological phenotypes related to FTLD, being BIBD, NIFID, and aFTLD-U (Citation75). BIBD can present clinically as ALS, FTLD-ALS, or FTLD (Citation73). In patients with BIBD pathology and juvenile onset ALS, FUS mutations have been identified (Citation166,Citation167). The FUS mutations in these patients result in a strong disruption of the NLS of FUS, which reflects the early onset age of the disease. Intriguingly, not all patients with juvenile or adult onset ALS and FUS-positive BIBD can be explained by a FUS mutation (Citation167). Further, no reports exist to date of FUS mutation carriers with BIBD and an FTLD-ALS or FTLD phenotype. FUS pathology was also demonstrated in NIFID (Citation74). NIFID is a rare disorder and is seen in patients with bvFTD with or without ALS. Mutation screening of FUS in a small number of NIFID patients was negative. Finally, FUS pathology also characterizes the rare FTLD pathology subtype aFTLD-U, in which the ubiquitinated inclusions are FUS-positive but TDP-43-negative (Citation69–72). Patients with aFTLD-U have typical clinical characteristics in common such as early disease onset (around 40 years), negative family history, bvFTD, little motor dysfunction, and pronounced atrophy of the caudate nucleus of the brain (Citation67,Citation68). Atrophy of the caudate nucleus on MRI has been proposed as a useful marker to predict aFTLD-U pathology (Citation70,Citation174). Also, in this subgroup with FUS pathology no mutations were found in the FUS gene (Citation69,Citation71,Citation72).
VCP
The gene VCP has also been linked to different phenotypes of the FTLD-ALS disease spectrum. To date, about 20 different VCP mutations have been identified in 50 unrelated families (http://www.molgen.vib-ua.be/FTDMutations). All are missense mutations, and in about 75% of VCP-linked families the mutation is located in exon 5 of the gene.
VCP mutations were known to cause a rare autosomal dominant disorder with inclusion body myopathy (IBM) associated with Paget's disease of the bone (PDB) and FTLD, denoted as IBMPFD (Citation105). The clinical presentation of IBMPFD is markedly variable. In a review of 20 IBMPFD families, approximately 90% of affected persons had IBM, 45% had PDB, and FTLD was seen in 38% of patients (Citation175). We have described two unrelated families segregating the same p.R159H mutation in which pure FTLD was the most frequent clinical manifestation (Citation176). Onset age of FTLD in these two families was on average 55 years (range 44–63 years) (Citation176).
The clinical spectrum associated with VCP mutations has recently been extended to include also ALS (Citation177). Through a whole-exome sequencing approach a p.R191Q mutation in VCP was identified in a family with autosomal dominant ALS (Citation177). Screening of additional 210 familial and 78 autopsy-confirmed ALS patients revealed 4 more VCP missense mutation carriers (1%–2% of familial ALS). The identified VCP mutation carriers had been diagnosed with clinical definite ALS (mean age at onset of 49 years, range 37–53 years), supported by the typical findings of ALS on EMG testing, and one VCP mutation carrier had a pathologically confirmed diagnosis of ALS-TDP. Of note, four patients of the described VCP families had been diagnosed with FTLD-ALS. The autopsy-confirmed ALS patient was part of a family with documented IBMPFD, but otherwise signs of PDB or IBM were mild or absent in the majority of these VCP families. In FTLD, VCP mutations lead to a signature TDP-43 pathology with numerous intranuclear inclusions, referred to as FTLD-TDP type D () (Citation135,Citation178). Since then, other studies have confirmed a contribution of VCP to the etiology of ALS (Citation179,Citation180).
Risk factors for TDP-43 proteinopathy
The inherent molecular heterogeneity of FTLD and the relative infrequency of the disease initially hampered the set-up of a successful GWA study on FTLD. With the recognition of TDP-43 pathology in FTLD an international GWA study was undertaken which focused on a homogeneous population of autopsy-confirmed FTLD-TDP patients. Significant association was identified with DNA variants located within the TMEM106B gene on chromosome 7p21 (Citation131). Results of a number of follow-up studies seem to indicate that TMEM106B is a true risk factor for FTLD (Citation181–184), but further confirmation is needed (Citation185).
Because of the similar TDP-43 pathology, the disease- modifying effect of TMEM106B was evaluated in a cohort of ALS patients (Citation186). No association between TMEM106B and ALS was observed, which was in line with results from previous ALS GWA studies that did not detect an association with the TMEM106B locus (Citation128,Citation130). However, the TMEM106B risk allele did appear to be associated with cognitive dysfunction in the ALS patients. Correlation with cognitive performance was strong as measured by the phonemic verbal fluency test but could not be confirmed by other cognitive tests. These findings are interesting but need to be replicated in other and larger ALS cohorts with more thorough neuropsychological evaluation. If confirmed, this would indicate that genetic risk factors for FTLD act as modifiers of the clinical presentation in ALS and underlie the risk for associated cognitive impairment.
Conclusions
Substantial overlap exists between FTLD and ALS at the clinical level. In a small group of patients, the concurrence of both disorders is obvious. However, there exists also a large group of patients in which signs of co-morbidity are more subtle, e.g. the ALS patients with mild cognitive impairment or behavior changes. Recent advances in the pathology and genetics of FTLD and ALS have demonstrated that these disorders are also tightly connected at the molecular level. Whilst on the one hand providing evidence for overlap, these discoveries have also highlighted that FTLD and ALS are etiologically diverse. TDP-43 and FUS pathology seems to be largely mutually exclusive, which suggests the involvement of different pathways that lead to neurodegeneration. The relevance of this heterogeneity is supported by the strong genetic–pathological correlations and, to a lesser degree, also by correlations with clinical sub-phenotypes. While some of the identified genes contribute to only one extreme of the clinical continuum, others have been implicated in different phenotypes of the FTLD-ALS spectrum. The exciting recent identification of pathological repeat expansions in C9orf72 in FTLD-ALS spectrum diseases, in particular, provided firm evidence for a biological link between these diseases. Future studies might provide insights into why some carriers have FTLD whereas others develop ALS and what the biological mechanisms could be by which the expanded repeat in C9orf72 contributes to neurodegeneration. TARDBP and FUS mutations have most often been associated with a classical ALS phenotype. Recent findings have suggested that these genes also have a limited role in FTLD pathogenesis. VCP mutations occur in the setting of a rare and heterogeneous multi-systemic syndrome that includes both FTLD and ALS as well as a bone (PDB) and a peripheral muscle disease (IBM). Genetic variants at the TMEM106B gene have been associated with increased risk for the more common sporadic forms of FTLD. An interesting hypothesis is that the susceptibility genes for FTLD-TDP, e.g. TMEM106B, could act as modifiers of cognitive performance in ALS. Finding relationships at the molecular level between two seemingly separate disorders has not been unique to FTLD and ALS. In fact, modern high-throughput genomic and proteomic analyses have discovered links between many diseases, which has culminated in the concept of a human diseasome that maps relationships between diseases and genes associated with them (Citation187). The recognition that FTLD and ALS are closely related could have important implications not only for research but also for the management of FTLD-ALS patients. With the identification of new genes within the spectrum, screening of all FTLD-ALS phenotypes is justified. And in the event of emerging therapeutic techniques, approaches aimed at preventing or removing TDP-43 or FUS aggregations should be investigated in the different phenotypes associated with TDP-43 or FUS proteinopathy, as they will likely benefit patients on both ends of the FTLD-ALS spectrum.
Acknowledgements
The research in the authors’ group was funded in part by the international consortium of Centers of Excellence in Neurodegenerative Brain Diseases, the Interuniversity Attraction Poles program P6/43 of the Belgian Federal Science Policy Office; the Foundation for Alzheimer Research (SAO/FRMA); the Medical Foundation Queen Elisabeth; the Methusalem excellence program of the Flemish Government; the Research Foundation Flanders (FWO); the Agency for Innovation by Science and Technology Flanders (IWT); and the Special Research Fund of the University of Antwerp, Belgium. The IWT provided a PhD fellowship to T.V.L. and the FWO a postdoctoral fellowship to J.v.d.Z.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–90.
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094–7.
- Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatr. 2003;74:1206–9.
- Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004;62:506–8.
- Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology. 2008;71:1496–9.
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–21.
- Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, . Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(Pt 9):2016–22.
- Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, . Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–30.
- Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, . Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208.
- Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, . Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006;129(Pt 6):1385–98.
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, . Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54.
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, . Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77.
- Rascovsky K, Salmon DP, Ho GJ, Galasko D, Peavy GM, Hansen LA, . Cognitive profiles differ in autopsy-confirmed frontotemporal dementia and AD. Neurology. 2002;58:1801–8.
- Rosen HJ, Hartikainen KM, Jagust W, Kramer JH, Reed BR, Cummings JL, . Utility of clinical criteria in differentiating frontotemporal lobar degeneration (FTLD) from AD. Neurology. 2002;58:1608–15.
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, . Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–14.
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005.
- Kertesz A, Munoz D. Relationship between frontotemporal dementia and corticobasal degeneration/progressive supranuclear palsy. Dement Geriatr Cogn Disord. 2004;17:282–6.
- Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003;61:349–54.
- Rascovsky K, Salmon DP, Lipton AM, Leverenz JB, DeCarli C, Jagust WJ, . Rate of progression differs in frontotemporal dementia and Alzheimer disease. Neurology. 2005;65:397–403.
- Vossel KA, Miller BL. New approaches to the treatment of frontotemporal lobar degeneration. Curr Opin Neurol. 2008;21:708–16.
- Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3–9.
- Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. 2009;72:725–31.
- Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78:629–31.
- Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, . Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252–8.
- Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, . The natural history of primary lateral sclerosis. Neurology. 2006;66: 647–53.
- del Aguila MA, Longstreth WT Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–9.
- Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2007;(1):CD001447.
- Bak TH, Hodges JR. Motor neurone disease, dementia and aphasia: coincidence, co-occurrence or continuum? J Neurol. 2001;248:260–70.
- Van Bogaert L. Les troubles mentaux dans la sclerose laterale amyotrophique. L'encephale. 1925;20:27–47.
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002; 59:1077–9.
- Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(Pt 9):2582–94.
- Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatr. 1990;53:23–32.
- Lillo P, Garcin B, Hornberger M, Bak TH, Hodges JR. Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol. 2010;67:826–30.
- Larner AJ. Delusion of pregnancy in frontotemporal lobar degeneration with motor neurone disease (FTLD/MND). Behav Neurol. 2008;19:199–200.
- Mendez MF, Shapira JS, Woods RJ, Licht EA, Saul RE. Psychotic symptoms in frontotemporal dementia: prevalence and review. Dement Geriatr Cogn Disord. 2008;25:206–11.
- Ostberg P, Bogdanovic N. Semantic dementia with lower motor neuron disease showing FTLD-TDP type 3 pathology (sensu Mackenzie). Neuropathology. 2011;31:271–9.
- Caselli RJ, Windebank AJ, Petersen RC, Komori T, Parisi JE, Okazaki H, . Rapidly progressive aphasic dementia and motor neuron disease. Ann Neurol. 1993;33:200–7.
- Catani M, Piccirilli M, Geloso MC, Cherubini A, Finali G, Pelliccioli G, . Rapidly progressive aphasic dementia with motor neuron disease: a distinctive clinical entity. Dement Geriatr Cogn Disord. 2004;17:21–8.
- Josephs KA, Knopman DS, Whitwell JL, Boeve BF, Parisi JE, Petersen RC, . Survival in two variants of tau-negative frontotemporal lobar degeneration: FTLD-U vs FTLD-MND. Neurology. 2005;65:645–7.
- Hu WT, Seelaar H, Josephs KA, Knopman DS, Boeve BF, Sorenson EJ, . Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol. 2009;66:1359–64.
- Massman PJ, Sims J, Cooke N, Haverkamp LJ, Appel V, Appel SH. Prevalence and correlates of neuropsychological deficits in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatr. 1996;61:450–5.
- Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, . Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131–46.
- Abrahams S, Goldstein LH, Al-Chalabi A, Pickering A, Morris RG, Passingham RE, . Relation between cognitive dysfunction and pseudobulbar palsy in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatr. 1997;62:464–72.
- Bak TH, Hodges JR. The effects of motor neurone disease on language: further evidence. Brain Lang. 2004;89:354–61.
- Rakowicz WP, Hodges JR. Dementia and aphasia in motor neuron disease: an underrecognised association? J Neurol Neurosurg Psychiatr. 1998;65:881–9.
- Tsuji-Akimoto S, Hamada S, Yabe I, Tamura I, Otsuki M, Kobashi S, . Writing errors as a result of frontal dysfunction in Japanese patients with amyotrophic lateral sclerosis. J Neurol. 2010;257: 2071–7.
- Ichikawa H, Koyama S, Ohno H, Ishihara K, Nagumo K, Kawamura M. Writing errors and anosognosia in amyotrophic lateral sclerosis with dementia. Behav Neurol. 2008;19:107–16.
- Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–4.
- Gibbons ZC, Richardson A, Neary D, Snowden JS. Behaviour in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:67–74.
- Lillo P, Mioshi E, Zoing MC, Kiernan MC, Hodges JR. How common are behavioural changes in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 2011;12:45–51.
- Abrahams S, Leigh PN, Kew JJ, Goldstein LH, Lloyd CM, Brooks DJ. A positron emission tomography study of frontal lobe function (verbal fluency) in amyotrophic lateral sclerosis. J Neurol Sci. 1995;129 Suppl:44–6.
- Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, . Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand. 1992;85:81–9.
- Kew JJ, Leigh PN, Playford ED, Passingham RE, Goldstein LH, Frackowiak RS, . Cortical function in amyotrophic lateral sclerosis. A positron emission tomography study. Brain. 1993;116(Pt 3):655–80.
- Abrahams S, Leigh PN, Goldstein LH. Cognitive change in ALS: a prospective study. Neurology. 2005;64:1222–6.
- Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology. 1999;53:1665–70.
- Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VM-Y, Hatanpaa KJ, . Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22.
- Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, . Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122:137–53.
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, . Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol. 2001;58:1803–9.
- Leigh PN, Anderton BH, Dodson A, Gallo JM, Swash M, Power DM. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci Lett. 1988;93:197–203.
- Mackenzie IRA, Feldman H. The relationship between extramotor ubiquitin-immunoreactive neuronal inclusions and dementia in motor neuron disease. Acta Neuropathol. 2003;105:98–102.
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, . TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, . Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
- Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007.
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, . Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8.
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, . Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11.
- Mackenzie IRA, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131(Pt 5):1282–93.
- Roeber S, Mackenzie IRA, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 2008;116:147–57.
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922–31.
- Seelaar H, Klijnsma KY, de Koning I, van der Lugt A, Chiu WZ, Azmani A, . Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol. 2010;257:747–53.
- Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, . FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120: 33–41.
- Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, . The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 2011;122:99–110.
- Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, . FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–27.
- Neumann M, Roeber S, Kretzschmar H, Rademakers R, Baker M, Mackenzie IRA. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118(Pt 11):2922–31.
- Mackenzie IRA, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, . Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121:207–18.
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, . Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–4.
- Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, . TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–45.
- Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, . Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67:555–64.
- Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, . Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94.
- Yokota O, Davidson Y, Arai T, Hasegawa M, Akiyama H, Ishizu H, . Effect of topographical distribution of α-synuclein pathology on TDP-43 accumulation in Lewy body disease. Acta Neuropathol. 2010;120:789–801.
- Geser F, Martinez-Lage M, Kwong LK, Lee VM-Y, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256:1205–14.
- Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, . Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70(Pt 2):1850–7.
- Wilson AC, Dugger BN, Dickson DW, Wang D-S. TDP-43 in aging and Alzheimer's disease—a review. Int J Clin Exp Pathol. 2011;4:147–55.
- Doi H, Okamura K, Bauer PO, Furukawa Y, Shimizu H, Kurosawa M, . RNA-binding protein TLS is a major nuclear aggregate- interacting protein in huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem. 2008;283:6489–500.
- Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res. 2010;66:131–3.
- Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, . Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65:1817–9.
- Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, . The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–6.
- Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, . Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71:1220–6.
- Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet. 2009;10: 769–82.
- Majoor-Krakauer D, Ottman R, Johnson WG, Rowland LP. Familial aggregation of amyotrophic lateral sclerosis, dementia, and Parkinson's disease: evidence of shared genetic susceptibility. Neurology. 1994;44:1872–7.
- Fallis BA, Hardiman O. Aggregation of neurodegenerative disease in ALS kindreds. Amyotroph Lateral Scler. 2009;10:95–8.
- Cruts M, Gijselinck I, Van Der Zee J, Engelborghs S, Wils H, Pirici D, . Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006; 442:920–4.
- Baker M, Mackenzie IRA, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, . Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9.
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, . Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
- Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF, . Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology. 1994;44:1878–84.
- Zarranz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atarés B, . A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology. 2005;64:1578–85.
- Wilhelmsen KC, Forman MS, Rosen HJ, Alving LI, Goldman J, Feiger J, . 17q-linked frontotemporal dementia-amyotrophic lateral sclerosis without tau mutations with tau and alpha-synuclein inclusions. Arch Neurol. 2004;61:398–406.
- Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P, . Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatr. 2007;78:754–6.
- Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PWJ, . Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008;71:253–9.
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, . Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37: 806–8.
- van der Zee J, Urwin H, Engelborghs S, Bruyland M, Vandenberghe R, Dermaut B, . CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet. 2008;17:313–22.
- Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, . ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67:1074–7.
- Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, . Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One. 2010;5:e9872.
- Cannon A, Baker M, Boeve B, Josephs K, Knopman D, Petersen R, . CHMP2B mutations are not a common cause of frontotemporal lobar degeneration. Neurosci Lett. 2006;398:83–4.
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, . Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, . Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
- Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46.
- Masè G, Ros S, Gemma A, Bonfigli L, Carraro N, Cazzato G, . ALS with variable phenotypes in a six-generation family caused by leu144phe mutation in the SOD1 gene. J Neurol Sci. 2001;191:11–18.
- Wicks P, Abrahams S, Papps B, Al-Chalabi A, Shaw CE, Leigh PN, . SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J Neurol. 2009;256:234–41.
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, . TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, . TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4.
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, . Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75.
- Ross OA, Rutherford NJ, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, . Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20:3207–12.
- Van Langenhove T, van der Zee J, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Ataxin-2 polyQ expansions in FTLD-ALS spectrum disorders in Flanders-Belgian cohorts. Neurobiol Aging. 2011 Oct 27. [Epub ahead of print].
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, . ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–3.
- Nishimura AL, Mitne-Neto M, Silva HCA, Richieri-Costa A, Middleton S, Cascio D, . A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–31.
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, . Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–6.
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, . Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–5.
- van Es MA, Diekstra FP, Veldink JH, Baas F, Bourque PR, Schelhaas HJ, . A case of ALS-FTD in a large FALS pedigree with a K17I ANG mutation. Neurology. 2009;72:287–8.
- Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, . Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2 - 21.3. Brain. 2006;129(Pt 4):868–76.
- Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, . A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–44.
- Le Ber I, Camuzat A, Berger E, Hannequin D, Laquerrière A, Golfier V, . Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–76.
- Valdmanis PN, Dupre N, Bouchard J-P, Camu W, Salachas F, Meininger V, . Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–5.
- Luty AA, Kwok JBJ, Thompson EM, Blumbergs P, Brooks WS, Loy CT, . Pedigree with frontotemporal lobar degeneration—motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32.
- Gijselinck I, Engelborghs S, Maes G, Cuijt I, Peeters K, Mattheijssens M, . Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–16.
- Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, . Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011;82:196–203.
- Pearson JP, Williams NM, Majounie E, Waite A, Stott J, Newsway V, . Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258:647–55.
- Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, . Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–94.
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai S-L, Myllykangas L, . Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–85.
- van Es MA, Veldink JH, Saris CGJ, Blauw HM, van Vught PWJ, Birve A, . Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–7.
- Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, . Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–9.
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, . A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, . Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56.
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, . A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68.
- Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, . A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–3.
- Seelaar H, Schelhaas HJ, Azmani A, Küsters B, Rosso S, Majoor-Krakauer D, . TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain. 2007;130(Pt 5):1375–85.
- Snowden J, Neary D, Mann DMA. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 2007; 114:31–8.
- Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118:349–58.
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, . p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702.
- Orru S, Manolakos E, Orru N, Kokotas H, Mascia V, Carcassi C, . High frequency of the TARDBP p.Ala382Thr mutation in Sardinian patients with amyotrophic lateral sclerosis. Clin Genet. 2012;81:172–8.
- Chiò A, Calvo A, Moglia C, Restagno G, Ossola I, Brunetti M, . Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch Neurol. 2010;67:1002–9.
- Chio A, Borghero G, Pugliatti M, Ticca A, Calvo A, Moglia C, . Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol. 2011;68:594–8.
- Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, . SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet. 2010;47:554–60.
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, . TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–3.
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, . Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30:E974–83.
- Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, . TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13: 509–17.
- Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, . TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–7.
- Gallone S, Giordana MT, Scarpini E, Rainero I, Rubino E, Fenoglio P, . Absence of TARDBP gene mutations in an italian series of patients with frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2009;28:239–43.
- Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin J-J, Vandenberghe R, . Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2009;30:1329–31.
- Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DMA, Pickering-Brown SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419:1–4.
- Origone P, Caponnetto C, Bandettini Di Poggio M, Ghiglione E, Bellone E, Ferrandes G, . Enlarging clinical spectrum of FALS with TARDBP gene mutations: S393L variant in an Italian family showing phenotypic variability and relevance for genetic counselling. Amyotroph Lateral Scler. 2010;11:223–7.
- Yokoseki A, Shiga A, Tan C-F, Tagawa A, Kaneko H, Koyama A, . TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–42.
- Cairns NJ, Perrin RJ, Schmidt RE, Gru A, Green KG, Carter D, . TDP-43 proteinopathy in familial motor neurone disease with TARDBP A315T mutation: a case report. Neuropathol Appl Neurobiol. 2010;36:673–9.
- Tamaoka A, Arai M, Itokawa M, Arai T, Hasegawa M, Tsuchiya K, . TDP-43 M337V mutation in familial amyotrophic lateral sclerosis in Japan. Intern Med. 2010;49:331–4.
- Pamphlett R, Luquin N, McLean C, Jew SK, Adams L. TDP-43 neuropathology is similar in sporadic amyotrophic lateral sclerosis with or without TDP-43 mutations. Neuropathol Appl Neurobiol. 2009;35:222–5.
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, . TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–16.
- Fushimi K, Long C, Jayaram N, Chen X, Li L, Wu JY. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell. 2011;2:141–9.
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, . ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57.
- Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, . Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011;39:2781–98.
- Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, . FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2010;81:639–45.
- Groen EJN, van Es MA, van Vught PWJ, Spliet WGM, van Engelen-Lee J, de Visser M, . FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol. 2010;67:224–30.
- Drepper C, Herrmann T, Wessig C, Beck M, Sendtner M. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging. 2009;32:548.e1–4.
- Rademakers R, Stewart H, Dejesus-Hernandez M, Krieger C, Graff-Radford N, Fabros M, . Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. 2010;42:170–6.
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, . Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet. 2010;47:190–4.
- Ticozzi N, Silani V, Leclerc AL, Keagle P, Gellera C, Ratti A, . Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73:1180–5.
- Bäumer D, Hilton D, Paine SML, Turner MR, Lowe J, Talbot K, . Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010;75:611–8.
- Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, . Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010;20:1069–76.
- Yan J, Deng H-X, Siddique N, Fecto F, Chen W, Yang Y, . Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010;75:807–14.
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, . Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75.
- Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, . Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–71.
- Broustal O, Camuzat A, Guillot-Noël L, Guy N, Millecamps S, Deffond D, . FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;22:765–9.
- Suzuki N, Aoki M, Warita H, Kato M, Mizuno H, Shimakura N, . FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J Hum Genet. 2010;55:252–4.
- Hewitt C, Kirby J, Highley JR, Hartley JA, Hibberd R, Hollinger HC, . Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2010;67:455–61.
- Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack CR, Jr, . Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol. 2010;17:969–75.
- Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008;1782:744–8.
- van der Zee J, Pirici D, Van Langenhove T, Engelborghs S, Vandenberghe R, Hoffmann M, . Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. 2009;73:626–32.
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, . Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64.
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, . TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–7.
- Koppers M, van Blitterswijk MM, Vlam L, Rowicka PA, van Vught PW, Groen EJ, . VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:837.e7–13.
- DeJesus-Hernandez M, Desaro P, Johnston A, Ross OA, Wszolek ZK, Ertekin-Taner N, . Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology. 2011;77: 1102–3.
- van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, . TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134(Pt 3):808–15.
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, . TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–74.
- Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, . Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68:581–6.
- Rollinson S, Mead S, Snowden J, Richardson A, Rohrer J, Halliwell N, . Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32:758.e1–7.
- van der Zee J, Van Broeckhoven C. TMEM106B a novel risk factor for frontotemporal lobar degeneration. J Mol Neurosci. 2011;45:516–21.
- Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, . Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121:373–80.
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci USA. 2007;104: 8685–90.