Abstract
Background. Hypertrophic cardiomyopathy (HCM) is predominantly caused by a large number of various mutations in the genes encoding sarcomeric proteins. However, two prevalent founder mutations for HCM in the alpha-tropomyosin (TPM1-D175N) and myosin-binding protein C (MYBPC3-Q1061X) genes have previously been identified in eastern Finland. Objective. To assess the prevalence of these founder mutations in a large population of patients with HCM from all over Finland. Patients and methods. We screened for two founder mutations (TPM1-D175N and MYBPC3-Q1061X) in 306 unrelated Finnish patients with HCM from the regions covering a population of ∼4,000,000. Results. The TPM1-D175N mutation was found in 20 patients (6.5%) and the MYBPC3-Q1061X in 35 patients (11.4%). Altogether, the two mutations accounted for 17.9% of the HCM cases. In addition, 61 and 59 relatives of the probands were found to be carriers of TPM1-D175N and MYBPC3-Q1061X, respectively. The mutations showed regional clustering. TPM1-D175N was prevalent in central and western Finland, and MYBPC3-Q1061X in central and eastern Finland. Conclusion. The TPM1-D175N and MYBPC3-Q1061X mutations account for a substantial part of all HCM cases in the Finnish population, indicating that routine genetic screening of these mutations is warranted in Finnish patients with HCM.
Key messages
TPM1-D175N and MYBPC3-Q1061X account for nearly one-fifth of the cases of hypertrophic cardiomyopathy in Finland and should be routinely sought in all Finnish patients with confirmed or suspected HCM.
Introduction
During the two past decades, over 900 different mutations in 23 genes, most of which encode sarcomeric proteins, have been reported to cause hypertrophic cardiomyopathy (HCM) (Citation1,Citation2). The cardiac myosin-binding protein C (MYBPC3) and beta-myosin heavy chain (MYH7) genes are the two most prevalent HCM disease genes worldwide, whereas mutations in other sarcomeric and non-sarcomeric genes are relatively uncommon (Citation3–7).
In contrast to most populations where HCM is caused by a large number of individual mutations in sarcomeric genes, in genetically isolated populations a few founder mutations may account for a substantial part of the disease. There are a few previous reports on HCM-causing founder mutations in the Netherlands (Citation8), Japan (Citation9), and South Africa (Citation10). In Finland, the founder effect has been demonstrated in various genetic diseases, including long QT syndrome (Citation11) and HCM (Citation12). In our previous studies we found two prevalent HCM mutations, the D175N mutation in the alpha-tropomyosin gene (TPM1-D175N) (Citation13,Citation14) and the Q1061X mutation in the myosin-binding protein C gene (MYBPC3-Q1061X) (Citation15), which together accounted for 28% of 35 index HCM cases in the Kuopio University Hospital area in eastern Finland (Citation12). Altogether nine sarcomeric genes were screened in patients with HCM from eastern Finland, but only few other disease-causing mutations with a low frequency were found (Citation12). The two most prevalent mutations, TPM1-D175N and MYBPC3-Q1061X, showed a founder effect in haplotype analysis (Citation12,Citation14,Citation15). Families with TPM1-D175N shared a common haplotype of microsatellite markers HTMαCA, D15S1036, and D15S108 (Citation12,Citation14), whereas MYBPC3-Q1061X co-segregated with a common haplotype of markers D11S4133, D11S1344, D11S1350, and D11S1326 (Citation15). In addition, we have found TPM1-D175N in 37 and MYBPC3-Q1061X in 32 relatives of the aforementioned index patients, respectively (Citation12,Citation14,Citation15; J. Kuusisto, unpublished data, 2011). Furthermore, TPM1-D175N and MYBPC3-Q1061X have been detected in 46 and 34 of 308 unselected Finnish subjects with suspected or confirmed HCM whose DNA was sent to the University of Eastern Finland 2007–2011 for commercial screening of these founder mutations (M. Laakso, unpublished data, 2011). Our previous genetic studies, however, have been confined only to one region of Finland and/or were based on a small patient population. Indeed, so far, there are only few national surveys of the genetics of HCM (Citation16). Therefore, the purpose of this study was to investigate the prevalence of these two founder mutations in a large nationwide Finnish patient population with HCM.
Patients and methods
Between 2001 and April 2010, consecutive unrelated patients aged ≥ 16 years with definite HCM from 12 Finnish university and central hospitals (FinHCM study group; and , ) were recruited for the FinHCM study. All but five patients were of Finnish origin. The regions of the hospitals cover a population of ∼4,000,000, comprising about 80% of the Finnish population. All patients underwent an interview and transthoracic echocardiography. Unrelatedness to other patients of the study was confirmed by the cardiologist responsible for the data collection in each study center. Relatedness up to second degree could be ruled out. Bicycle ergometer stress test and Holter electrocardiography (ECG) registration were performed when possible. The diagnosis of HCM was based on the demonstration of left ventricular hypertrophy (LVH) ≥ 15 mm on two-dimensional echocardiography (or by cardiac magnetic resonance imaging (MRI)) in the absence of other obvious causes for LVH, such as hypertension or aortic stenosis (Citation17).
Table I. Clinical characteristics of the index patients with hypertrophic cardiomyopathy.
Table II. Frequencies of the mutations TPM1-D175N and MYBPC3-Q1061X according to study center.
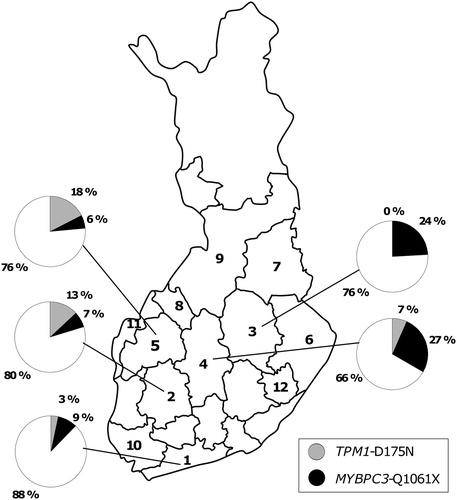
Figure 1. Prevalence of the mutations TPM1 (alpha-tropomyosin)-D175N and MYBPC3 (cardiac myosin-binding protein C)-Q1061X in the five largest study centers (Helsinki, Tampere, Kuopio, Jyväskylä, and Seinäjoki). Hospital districts are numbered (Citation1–12) corresponding to the numbers in .
All genetic analyses were performed at the University of Eastern Finland (previously University of Kuopio). DNA was extracted from peripheral blood leukocytes and amplified using polymerase chain reaction (PCR). Two previously reported mutations, TPM1-D175N (Citation13,Citation14) and MYBPC3-Q1061X (Citation15), were genotyped from DNA samples using either the TaqMan Allelic Discrimination Assay (ABI PRISM 7000 Sequence Detection System, PE Applied Biosystems, Foster City, CA, USA) or direct sequencing (ABI PRISM 3100 Genetic Analyzer, PE Applied Biosystems, Foster City, CA, USA).
In statistical analyses, founder mutation carriers versus non-carriers were compared by using the chi-square or the Mann– Whitney U test, when appropriate. The differences between the three groups (TPM1-D175N versus MYBPC3-Q1061X versus no mutation) were evaluated with the chi-square test or one-way ANOVA, and between the two groups by the Bonferroni post-hoc test. Because of their skewed distribution, all continuous variables except for left ventricular end-diastolic dimension and ejection fraction were logarithmically transformed. SPSS 14.0 (SPSS, Chicago, IL, USA) was used for all analyses. Data are presented as n (%) or mean ± SD. A P < 0.05 was considered statistically significant.
The study was approved by the Ethics Committee of the Kuopio University Hospital. All subjects gave written informed consent.
Results
A total of 381 index patients were initially evaluated; 75 patients were excluded from the study group because they did not meet the inclusion criteria (age ≥ 16 years, maximal LV thickness ≥ 15 mm in the absence of other causes for LVH, unrelated to other FinHCM subjects) or because there were insufficient clinical data available. The final number of unrelated index patients with definite HCM in the FinHCM study was 306.
The clinical characteristics of the index patients (60% men) of the FinHCM study are shown in . The mean age of the study subjects was 53 years. About one-quarter of the patients had a family history of HCM, and one-third reported a history of sudden cardiac death in the family. Exertional dyspnea and syncope or presyncope were the most common cardiac symptoms. Most of the patients had a good or moderate functional class (New York Heart Association (NYHA) 1–2). One-quarter of the patients had paroxysmal or chronic atrial fibrillation. Sustained ventricular tachycardia was reported in ten patients. Fourteen patients had been resuscitated from ventricular fibrillation. Altogether, 30 patients had undergone an implantable cardioverter defibrillator (ICD) implantation for either primary or secondary prevention. The occurrence of congestive heart failure was low, and only seven patients had undergone a surgical myectomy.
Most patients had moderate LVH with a mean wall thickness of 20 mm (range 10–38 mm) on echocardiography. Two of the patients had a maximal LV wall thickness < 15 mm on two-dimensional echocardiography but were included because subsequent cardiac MRI showed LV thickness > 15 mm. Left ventricular dimensions and ejection fraction were within the normal limits in most patients. Maximal flow velocity in the left ventricular outflow tract (LVOT) was slightly elevated. Twenty-three patients had maximal flow velocity ≥ 2.75 m/s in the LVOT.
Bicycle ergometer stress test was performed in 214 patients. Most patients had a good or moderate exercise capacity. A minority of the patients had an abnormal blood pressure response. Holter ECG registration was performed in 249 patients. Short bursts of ventricular tachycardia or atrial fibrillation were found in 20% and 16% of the patients, respectively.
In genetic screening the mutations TPM1-D175N and MYBPC3-Q1061X were found in 20 (6.5%) and 35 (11.4%) of the FinHCM index patients, respectively. Altogether these two mutations accounted for ∼18% of the disease cases. None of the index patients or relatives carried both of the aforementioned mutations. In addition, 61 and 59 relatives of the probands were found to be carriers of TPM1-D175N and MYBPC3-Q1061X, respectively.
Among the 75 probands excluded from the FinHCM study group there was 1 patient who carried TPM1-D175N and 2 patients with MYBPC3-Q1061X. All these patients had LVH < 15 mm on echocardiography.
When comparing the index patients with and without founder mutation, the patients with either TPM1-D175N or MYBPC3-Q1061X more often had a family history of HCM compared to patients with no detected mutation (27 of 55 () versus 56 of 251, respectively, P < 0.001). Mutation carriers had higher maximal thickness of LV wall (21 ± 4 mm versus 19 ± 4 mm, P = 0.006) and lower left ventricular end-diastolic dimension (LVEDD) (44 ± 6 mm versus 47 ± 7 mm, P = 0.033) compared to non-carriers.
In the comparison of the three groups of FinHCM index patients (TPM1-D175N, MYBPC3-Q1061X, no founder mutation), D175N carriers had the highest rate (75%) of family history of HCM (15 of 20 versus 12 of 35 versus 56 of 251, P < 0.001). A history of myectomy was more common in patients with D175N than in patients carrying Q1061X or no detected mutation (2 of 20 versus 0 of 35 versus 5 of 245, respectively, P = 0.048). Among the patients with D175N, Q1061X, and without detected mutation the maximal LV wall thickness was 20 ± 2 mm, 22 ± 5 mm, and 19 ± 4 mm, respectively (P = 0.012). The patients with Q1061X had thicker LV wall than did patients without detected mutation (Ppost-hoc = 0.009). LVEDD differed between the three groups (43 ± 6 mm versus 45 ± 6 mm versus 47 ± 7 mm, respectively, P = 0.046), but there was no statistical difference between the groups in the Bonferroni post-hoc test. Furthermore, the differences were found in maximum load of bicycle ergometry (120 ± 57 W versus 166 ± 65 W versus 166 ± 70 W, respectively, P = 0.042), D175N carriers having lower maximum load than patients with no detected mutation (Ppost-hoc = 0.038).
Among relatives ≥ 16 years of age carrying the TPM1-D175N and MYBPC3-Q1061X mutations, echocardiographic data were available in 31 and 38 subjects, respectively. Twenty-two of 31 relatives with TPM1-D175N had HCM (LVH ≥ 13 mm), giving the overall penetrance of 71% among relatives and 82% (42/51) in all mutation carriers with echocardiographic data. In the carriers of MYBPC3-Q1061X the respective penetrances were 58% (22/38) and 78% (57/73).
In the FinHCM study, TPM1-D175N was common in western Finland, whereas MYBPC3-Q1061X was common in central and eastern Finland (, ). When comparing centers with ten or more FinHCM index cases, the prevalence of TPM1-D175N was the highest in the western Finland district of Sein joki (17.6%), and the prevalance of MYBPC3-Q1061X was the highest in the central Finland district of Jyv skl (26.7%). No carriers of TPM1-D175N were found in five centers (Kuopio, Joensuu, Kajaani, Kokkola, and Turku), whereas MYBPC3-Q1061X was not found in four centers (Joensuu, Kokkola, Vaasa, and Savonlinna). Neither of the two mutations was found in patients from the most eastern district of Joensuu and the western district of Kokkola. In the five largest centers (Helsinki, Tampere, Kuopio, Jyväskylä, and Seinäjoki), the two founder mutations together accounted for 12.3%, 20.4%, 24.2%, 33.3%, and 23.5% of the HCM cases, respectively.
Discussion
We screened for the two previously reported founder mutations TPM1-D175N and MYBPC3-Q1061X in a large sample of Finnish patients with HCM. The two mutations accounted for approximately one-fifth of the HCM cases in our study, indicating that these mutations, especially MYBPC3-Q1061X, are major causes of HCM in Finland.
The high prevalence of TPM1-D175N and MYBPC3-Q1061X in our study is most probably explained by the presence of a founder effect. Although no haplotype analysis was performed in the present study, our previous studies showed the founder effect of these mutations in patients with HCM in eastern Finland (Citation12,Citation14,Citation15). Given the inhabitation history of Finland, it is likely that all carriers of an identical mutation share a common ancestral origin. Recently, HCM-causing founder mutations have also been reported in several other populations. In a French study 13 of 197 (7%) index patients carried an identical MYBPC3 splice acceptor mutation IVS21–2:a13858g (Citation3). In this study a founder effect in some of the families was reported, but no haplotype data were presented. In a study by Alders et al. the 2373InsG (W792fs) mutation of MYBPC3 accounted for 23% of the 259 Dutch HCM patients screened (Citation8). A shared haplotype of the repeat markers and intragenic SNPs indicated that the mutation has arisen from a common origin. In another large Dutch cohort of 1040 patients, most of whom had HCM, an evident founder mutation (R145W) in the troponin I gene was found to cause either HCM or restrictive cardiomyopathy (Citation18). Kubo et al. reported a frameshift mutation (V592fs/8) of MYBPC3 in 15 of 94 (16%) Japanese probands (Citation9). Haplotype analysis with microsatellite markers indicated a common ancestral origin among the mutation carriers. The presence of a founder effect was suggested also in a study by Girolami et al. in which 13 of 88 (15%) of Italian HCM patients screened carried a previously reported E258K mutation of MYBPC3 (Citation6). However, in that study no haplotype analysis was performed. In addition, Moolman et al. reported a founder effect confirmed by haplotype analysis for the three mutations (MYH7-R403W, MYH7-A797T, troponin T R92W) among South African HCM patients (Citation10). Haplotype analysis showed that a MYBPC3- IVS23 + 1G-A defect carried by 18 of 154 (12%) Spanish HCM families was a founder mutation (Citation19).
There are only few national surveys of the genetics of HCM. A national survey of common founder mutations causing HCM has previously been reported in the Netherlands (Citation16). The combined prevalence of three founder mutations (W792fs, R943X, P955fs,) in MYBPC3 was 21% (157 of 735) among all index patients screened in Dutch centers. The W792fs was by far the most common mutation, accounting alone for 17% of the cases.
TPM1-D175N has previously been reported in a few studies from the US, Japan, and Europe (Citation7,Citation13,Citation20,Citation21). In these reports the D175N mutation was found in one to three families, respectively, without any evidence of a founder effect. No mutation carriers of D175N have been found in any of the recent European and American large-scale HCM patient screenings (Citation3–5). MYBPC3-Q1061X was first reported by our group (Citation15). Later, this mutation was also found in 2 of 118 Polish HCM patients (Citation22). Recently, in a Dutch study, the Q1061X mutation was reported in a single patient with HCM and in a family member without signs of HCM (Citation23). Hence, TPM1-D175N and MYBPC3-Q1061X appear to be uncommon in other populations, in contrast to Finland where these mutations have been enriched due to genetic isolation.
As with most missense mutations, TPM1-D175N is likely to result in a mutated protein that acts in a dominant-negative fashion. Bottinelli et al. showed by studying skeletal muscle samples from HCM patients with TPM1-D175N that the D175N-mutated protein was expressed in skeletal muscle and was associated with increased calcium sensitivity compared to controls (Citation24). MYBPC3-Q1061X is predicted to create a premature stop-codon and a truncated protein in which the binding sites for both myosin and titin are missing. There are no functional studies on MYBPC3-Q1061X, but based on previous studies on other MYBPC3 mutations the truncated protein may not be expressed in the sarcomere, and therefore the disease may be mediated by haploinsufficiency. Van Dijk et al. studied the expression of two MYBPC3 frameshift mutations (W792fs and P955fs) in cardiac biopsies from HCM patients carrying either of the two mutations (Citation25). The lack of the truncated proteins and reduced protein expression of myosin-binding protein C in the samples from patients with a frameshift mutation indicated haploinsufficiency. In addition, the samples from the mutation carriers showed abnormal phosphorylation of contractile proteins, reduced maximal force-generating capacity of myocytes, and increased calcium sensitivity compared to controls.
In our study TPM1-D175N was prevalent in western and central Finland, whereas MYBPC3-Q1061X was more common in central and eastern parts of the country. This is explained by the genetic diversity between the eastern and western parts of the country. The existence of genetic sub-isolates in Finland may explain why neither of the founder mutations was found in the most eastern Joensuu area and the most western area of Kokkola. Hence, yet unidentified founder mutations may account for HCM in the regions with a low prevalence of the founder mutations TPM1-D175N and MYBPC3-Q1061X. However, it should be noted that the relatively small number of patients in many of our study centers does not allow reliable estimation of the prevalence of the founder mutations regionally.
Our study group of 306 probands with HCM from 12 university and central hospitals probably reflects the overall Finnish HCM population, allowing a rough estimate of the prevalence of the two founder mutations in Finland. However, by the use of strict diagnostic criteria for HCM (LVH ≥ 15 mm instead of ≥ 13 mm) some mutation carriers with mild LVH are inevitably missed. Hence, the true prevalence of TPM1-D175N and MYBPC3-Q1061X may be even higher than reported in the present study. This notion is supported by our finding that these mutations were found in some probands who were excluded from the present study because the diagnostic criteria for LVH were not fulfilled. On the other hand, by using looser diagnostic criteria of HCM the overall size of the patient population would increase, and, consequently, the prevalence of the mutations TPM1-D175N and MYBPC3-Q1061X might even decrease. In the present study both TPM1-D175N and MYBPC3-Q1061X were associated with an incomplete penetrance (82% and 78%, respectively, in adult carriers). In our previous study in patients from eastern Finland the penetrance of TPM1-D175N and MYBPC3-Q1061X among adult carriers was 91% and 67%, respectively (Citation12).
Screening of the most common disease-causing mutations in all new patients with HCM enables the confirmation of the clinical diagnosis of HCM in many cases. Furthermore, once a mutation has been identified in a proband, the cascade screening of the relatives helps to find those needing clinical evaluation and follow-up. There is also some evidence that HCM patients with an identified sarcomeric myofilament mutation have a more unfavorable clinical course compared to those without sarcomeric protein mutation (Citation26). Accordingly, in our study, carriers of the sarcomeric founder mutations had thicker LV wall compared to non-carriers. In our present and previous studies, the mutations TPM1-D175N and MYBPC3-Q1061X were associated with incomplete penetrance, variable LVH, and relatively favorable prognostic factors (Citation12). However, a longer prospective follow-up of these patients is needed to confirm the benign course of HCM among the patients carrying these mutations.
In conclusion, the two founder mutations TPM1-D175N and MYBPC3-Q1061X accounted for 6.5% and 11.4%, respectively, of the Finnish HCM cases, giving the combined prevalence of ∼18% for these mutations. Altogether, in the present and our previous studies, the founder mutations TPM1-D175N and MYBPC3-Q1061X have been found in 169 and 168 Finnish patients, respectively, indicating that TPM1-D175N and MYBPC3-Q1061X are major HCM-causing mutations in the Finnish population. Therefore, routine screening for these mutations is warranted in Finnish patients with suspected or confirmed HCM.
Acknowledgements
We thank Raija Miettinen, MSc, Teemu Kuulasmaa, MSc, Satu Nenonen, RN, Eila Ruotsalainen, RN, and Sini Weckström, RN, for assistance in data collection.
This study has previously been presented as an abstract in Florence International Course on Advances in Cardiomyopathies, 2008 (J.K.)
Declaration of interest: The authors state no conflict of interest. This study was supported by the Academy of Finland, the Finnish Foundation for Cardiovascular Research and the Kuopio University Hospital (grants to J.K.). T.H. has received grants from the Finnish foundation for Cardiovascular Research, Finnish Medical Foundation, the special governmental subsidy for health sciences research, and the Helsinki University Central Hospital.
References
- Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122:2441–9.
- Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–40.
- Richard P, Charron ,P, Carrier L, Ledeuil C, Cheav T, Pichereau C, . Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–32.
- Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, . Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003;64:339–49.
- Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–51.
- Girolami F, Olivotto I, Passerini I, Zachara E, Nistri S, Re F, . A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown). 2006;7:601–7.
- Andersen PS, Havndrup O, Hougs L, Sørensen KM, Jensen M, Larsen LA, . Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–70.
- Alders M, Jongbloed R, Deelen W, van den Wijngaard A, Doevendans P, Ten Cate F, . The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24:1848–53.
- Kubo T, Kitaoka H, Okawa M, Matsumura Y, Hitomi N, Yamasaki N, . Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac myosin-binding protein C gene among Japanese. J Am Coll Cardiol. 2005;46:1737–43.
- Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA. The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet. 1999;65:1308–20.
- Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, . High prevalence of four long QT syndrome founder mutations in the Finnish population.Ann Med. 2009;41:234–40.
- Jääskeläinen P, Miettinen R, Kärkkäinen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23–32.
- Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, . Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–12.
- Jääskeläinen P, Soranta M, Miettinen R, Saarinen L, Pihlajamäki J, Silvennoinen K, . The cardiac beta-myosin heavy chain gene is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population. J Am Coll Cardiol. 1998;32:1709–16.
- Jääskeläinen P, Kuusisto J, Miettinen R, Kärkkäinen P, Kärkkäinen S, Heikkinen S, . Mutations in the cardiac myosin-binding protein C gene are the predominant cause of familial hypertrophic cardiomyopathy in eastern Finland. J Mol Med. 2002;80:412–22.
- Christiaans I, Nannenberg EA, Dooijes D, Jongbloed RJ, Michels M, Postema PG, . Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth Heart J. 2010;18:248–54.
- Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, . American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–713.
- van den Wijngaard A, Volders P, Van Tintelen JP, Jongbloed JD, van den Berg MP, Lekanne Deprez RH, . Recurrent and founder mutations in the Netherlands: cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth Heart J. 2011;19:344–51.
- Oliva-Sandoval MJ, Ruiz-Espejo F, Monserrat L, Hermida-Prieto M, Sabater M, García-Molina E, . Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart. 2010;96: 1980–4.
- Coviello DA, Maron BJ, Spirito P, Watkins H, Vosberg HP, Thierfelder L, . Clinical features of hypertrophic cardiomyopathy caused by mutation of a “hot spot” in the alpha-tropomyosin gene. J Am Coll Cardiol. 1997;29:635–40.
- Nakajima-Taniguchi C, Matsui H, Nagata S, Kishimoto T, Yamauchi-Takihara K. Novel missense mutation in alpha-tropomyosin gene found in Japanese patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 1995;27:2053–8.
- Rudziński T, Selmaj K, Drozdz J, Krzemińska-Pakuła M. Selected mutations in the myosin binding protein C gene in the Polish population of patients with hypertrophic cardiomyopathy. Kardiol Pol. 2008;66:821–5.
- Michels M, Soliman OI, Phefferkorn J, Hoedemaekers YM, Kofflard MJ, Dooijes D, . Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur Heart J. 2009;30:2593–8.
- Bottinelli R, Coviello DA, Redwood CS, Pellegrino MA, Maron BJ, Spirito P, . A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ Res. 1998;82:106–15.
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, . Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–83.
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, . Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83:630–8.