Abstract
Sudden cardiac death (SCD) following ventricular tachyarrhythmias constitutes an important clinical cause of mortality; 4% of cases may involve ion channel-mediated cellular excitation in structurally normal hearts. Alterations in such processes could disturb action potential conduction, depolarization/ repolarization gradients, or Ca2+ homeostasis with potential arrhythmogenic consequences. Although SCD may be the first presentation of arrhythmic syndromes, patients may present to the general physician with symptoms of palpitations or hemodynamic compromise, including dizziness, seizure, or syncope, particularly following exertion. In all inherited cardiac death syndromes, first-degree relatives should be referred to a cardiologist and should undergo testing appropriate for the condition. While management of patients at risk of SCD largely centers on risk stratification and, if necessary, insertion of an implantable cardioverter-defibrillator, there are a number of other, pharmacological, treatments being developed. Furthermore, as the genetic basis of these diseases becomes established, genetic testing will form an increasingly important part of diagnosis, and gene-specific therapy is an area under investigation. This article bridges the gap between molecular medicine and clinical practice by reviewing recent developments in the pathophysiological understanding of SCD, and their implications for the management of patients with these complex diseases.
Key messages
Inherited arrhythmic disorders may present as sudden death, as well as syncope, palpitations, hemodynamic compromise, or seizure activity, and have a broad variety of phenotypes and genetic etiologies.
Risk stratification of individuals and family members is important in determining treatment options, in particular the need for insertion of an implantable cardioverter-defibrillator.
Improvements in molecular biology have resulted in progress in the elucidation of the cellular mechanisms of these diseases, with increasing emphasis placed on pharmacological treatments tailored to the specific genetic defect.
Introduction
Sudden cardiac death (SCD) is defined as unexpected death from cardiac causes occurring within 1 hour of the onset of symptoms (Citation1) and is a major cause of morbidity and mortality in the developed world. Arrhythmias account for between 180,000 and 250,000 deaths in the United States (Citation2,Citation3) and up to 70,000 deaths per year in the United Kingdom (Citation1). An assembly of reports from different studies (Citation4–7) is consistent with a global incidence of 4–5 million cases per year. This reflects a slight reduction in incidence from previous suggestions (USA: > 300,000) (Citation8,Citation9) that may reflect improved intervention for coronary artery disease, although increased incidences of diabetes and obesity may result in a resurgence in incidence (Citation3). Survival rates from sudden cardiac arrest (SCA) are very low, at only 2% (Citation1).
In most cases, cardiac arrhythmias result from underlying ischemic heart disease (Citation10). However, autopsy fails to reveal a cause in around 4% of SCD patients, ˜ 3% of all out-of-hospital cardiac arrests (Citation11), and 14% of all resuscitation attempts performed on patients aged < 40 years (Citation12) with much variation in these frequencies between centers. Cardiac causes are thought to account for 56.4% of non-traumatic, sudden death in autopsies from patients between 5 and 35 years old. Of these, arrhythmia is the most common cause at approximately 30%, presenting in patients with little or no structural change and therefore likely reflecting a causative channelopathy (Citation13,Citation14). Furthermore, 60%–80% of deaths in infants less than one year of age are autopsy-negative and thus defined as sudden infant death syndrome (SIDS) (Citation15). Of these, 10%–20% are thought to be due to pathognomonic channelopathies, extensively reviewed by Klaver et al. (Citation16).
Such arrhythmic disorders in the absence of structural abnormalities are termed primary electrical diseases and are exemplified by Brugada syndrome (BrS), long QT syndrome (LQTS), and catecholaminergic polymorphic ventricular tachycardia (CPVT). SCD in structurally normal hearts can often be attributed to one or more mutations in the genes controlling electrical conduction and repolarization through the heart, although other associated regulatory proteins may also be affected. Molecular autopsy studies reported pathogenic mutations in LQTS and CPVT-associated genes in over one-third of cases. Similar post-mortem studies demonstrated mutations in 5%–10% of sudden infant death syndrome cases (Citation17,Citation18). Different mutations in the same gene can variously lead to either gain-of-function or loss-of-function phenotypes, and often the same phenotype may be associated with a wide range of genotypes (e.g. LQTS).
Recent reviews of inherited SCD have focused on genetic and molecular aspects of these diseases. However, for the general physician, their diagnosis and practical management can be confusing. All clinical management should begin with a full history including family history, as well as a preliminary electrocardiographic (ECG) assessment. Investigations should also exclude other causes of similar clinical manifestations such as seizures. In all inherited SCD syndromes, first-degree relatives should be referred to a cardiologist and should undergo testing appropriate for the condition, including genetic testing, if the causative gene in that family has been identified.
Despite the clinical importance, high prevalence, and high associated incidence of mortality of cardiac arrhythmias, there is only limited knowledge of the mechanisms of their initiation, maintenance, and propagation. This compromises effective risk stratification and treatment. However, recent work has characterized their underlying genetic abnormalities thereby leading to clarifications of their associated functional abnormalities. The resulting insights have positively impacted upon their possible clinical diagnosis and treatment. The roles of environmental as well as genetic modifiers have been shown to be important in larger patient cohorts for the determination of arrhythmia susceptibility and severity. Further insights have come from the use of animal models of arrhythmic disease syndromes, whether these involve targeted genetic changes or specific pharmacological interventions, both typically involving the genes and proteins responsible for action potential (AP) generation and propagation. Finally, recent papers have reported that induced pluripotent stem cells derived from carriers of the relevant conditions can be made to replicate expected cellular properties of both LQT2 syndrome (Citation19) and CPVT, offering further valuable avenues for investigating disease mechanisms in vitro, developing new drugs, predicting their toxicity, and optimizing treatment strategies (Citation20,Citation21). This article seeks to bridge the gap between basic molecular physiology and clinical practice by reviewing recent developments in the understanding of SCD in structurally normal hearts, exploring the implications for the risk stratification and management of patients with these complex diseases.
Long QT syndrome
The current population prevalence of LQTS in the US is estimated at around 0.01% (Citation22). A recent Italian study on 44,596 infants suggested a prevalence close to 1:2000; among genotyped infants, disease-causing mutations were found in 12 of 28 with a QTc > 470 ms and in 4 of 14 with a QTc of 461–470 ms (Citation23). Long QT syndromes have a wide range of genetic causes, but all are characterized by increased risks of polymorphic ventricular tachycardia (VT), especially torsades de pointes (TdP) (), and increased action potential duration (APD), resulting from prolonged ventricular repolarization (). The normal cardiac AP classically consists of an initial depolarization (phase 0) mediated by transmembrane Na+ currents, immediately followed by an early recovery (phase 1) mediated by transient outward K+ currents, a plateau period (phase 2) mediated by Ca2+ currents, and a repolarization (phase 3) mediated by K+ currents. It can be directly demonstrated using basic intracellular experimental sharp or patch electrode recording techniques (). However, such studies are clinically invasive, and so the ECG remains the mainstay of diagnostic investigation for cardiac electrophysiological disorders. However, as the ECG is an extracellular recording, it effectively produces a signal that reflects electrical summation of cardiac action potentials from different regions of the heart, with a P wave representing atrial depolarization, a QRS complex representing ventricular depolarization, and a T wave representing ventricular repolarization. When the ECG does not provide a definitive diagnosis it is possible to use a scoring system based on criteria in the ECG findings, clinical history, and family history () (Citation24). Action potential prolongation reflecting a delayed repolarization thus results in an increase in the QT interval (). As QT interval varies with heart rate, it is often corrected (QTc) using an algorithm (e.g. Bazett's formula: QTc = QT/√(RR)) to allow comparisons of results obtained at different heart rates. A recent multivariate analysis of LQT1, LQT2, and LQT3 loci patients (see below) implicated genetic locus as an independent predictor of risk. The QTc emerged as an independent risk predictor in LQT1 and LQT2 patients, and sex as an independent predictor of events in patients with a mutation in the LQT3 locus (Citation25). Thus the greater the degree of QT prolongation, the greater the arrhythmic risk, with the locus of the causative mutation both affecting the clinical course and modulating the effects of QTc and sex.
Figure 1. Mechanisms of arrhythmogenesis in long QT syndrome. A: ECG of patient with LQTS degenerating into torsades de pointes (TdP) and showing classical undulating baseline. B: (i) APDs in control and LQTS human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) showing significant prolongation. (ii) ECG in patient with LQTS demonstrating significant increase in QT duration. C: (i) Early after-depolarizations (EADs) in stem cell-derived ventricular and atrial cells. (ii) EADs resulting in triggered activity in iPSC-CMs. D: 12-lead ECG from a patient with LQT2 showing profoundly bifid T waves. Reproduced with permission from (A, B(ii)) Salama and London, J Physiol 2007;578:43–53 (Citation26); (B(i), C) Itzhaki et al., Nature 2011;471(7337): 225–9 (Citation19); (D) Zhang et al., Circulation 2000;102(23):2849–55 (Citation140).
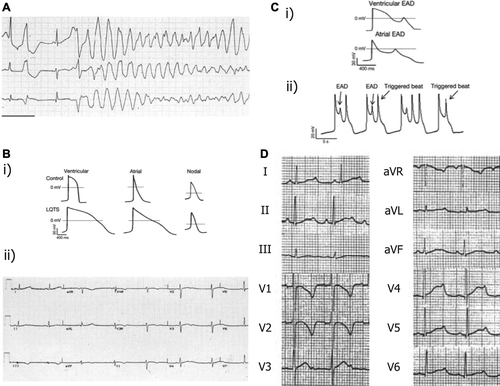
Table I. 2011 LQTS diagnostic criteria.
In general, hereditary LQTS may be caused by abnormally prolonged inward Na+ or Ca2+ currents during the depolarizing or plateau phase, or decreased K+ currents during the repolarizing phase of the cardiac AP. Evidence from several mouse models (Citation26) implicates the prolonged APD in the increased arrhythmic tendency. Arrhythmic substrate may arise from both increased refractory periods creating pockets of functional conduction block and differences in dispersion of repolarization across the myocardium. Both could potentially act as re-entry foci. Extended AP plateau phases may permit L-type Ca2+ channel reactivation and therefore more frequent early after-depolarization phenomena (). Where this involves a sufficient number of cardiac cells, it may result in ectopic beats triggering polymorphic VT in the primed myocardial substrate (Citation27).
To date 13 genes have been implicated in LQTS; all either directly or indirectly concern the ion channels involved in the cardiac AP (). Genetic testing for the known mutations is possible, with identification of an LQTS genetic mutation confirming the diagnosis. However, in common with numerous other Mendelian disorders, the cardiac channelopathies often exhibit incomplete penetrance, variable expressivity, and phenotypic overlap. Genotype-positive individuals within the same genetic lineage can assume markedly differing clinical courses as reflected in their phenotypic outcomes including ECG features as well as the number and type of cardiac events and findings attributable to variable existences of additional, modifier, genes, often including single nucleotide polymorphisms (Citation28–33). However, currently only approximately 50% of LQTS patients have known mutations (Citation34).
Table II. Non-structural disorders associated with ventricular arrhythmia and sudden death.
The most common forms of inherited LQTS are LQT1, 2, and 3, which account for 45%, 45%, and 5% of LQT cases, respectively. In LQT1, 80% of carriers show a broad-based T wave with an indistinct onset (Citation35), representing slowed AP repolarization. Patients typically show events during exercise. Over 170 missense mutations have been characterized in the KCNQ1 (formerly termed KVLQT1) gene. This encodes a protein subunit that mediates the delayed rectifier K+ channel (IKs) involved in phase 3 repolarization, explaining the observed APD prolongation and lengthened QT interval.
LQT2 is characterized by low-amplitude bifid T waves seen in most of the 12 leads in 80% of patients (Citation35). This may represent increased transmural dispersions of repolarization with some APs repolarizing more slowly than others to give the biphasic T wave shape (). Cardiac events in these patients tend to occur during sudden arousal, such as being startled by a loud noise. LQT2 is caused by loss-of-function mutations in the KCNH2 (formerly termed HERG) gene, resulting in reduction of the rapidly activating and rapidly deactivating delayed rectifier K+ current (IKr).
In LQT3, 65% of patients show long isoelectric ST segments (Citation35), caused by a late Na+ current. Patients are at risk during sleep (Citation36). In contrast to the previous conditions, LQT3 results from gain-of-function mutations in the SCN5A gene. Such mutations result in slower inactivation of the channel and faster recovery from inactivation, thereby increasing the late Na+ current and hence the APD and QT interval.
Other forms of LQTS are less common, but all result from prolonged depolarizing or reduced repolarizing currents. LQT4 results from mutations in the ankyrin-B gene, an adapter protein that serves to regulate the activity of several ion channel proteins. LQT5 is caused by mutations in the complex generating the IKs current; its mechanism is hence comparable with that causing LQT1. Mutations in MinK-related protein 1, which assembles with the KCNH2 channel, give rise to LQT6, and hence its arrhythmogenic mechanism is similar to that of LQT2. Further examples include: LQT7 involving the K+ channel protein, Kir 2.1; LQT8 involving the L-type Ca2+ channel; LQT9 involving caveolin 3, a caveolae plasma membrane component protein involved in scaffolding proteins; LQT10 involving the Nav β4, a subunit of the voltage-gated Na+ channel; LQT11 involving the A-kinase anchor protein 9; LQT11 involving alpha-1 syntropin; and LQT13 affecting the B-protein activated inward rectifier K+ channel (Citation37,Citation38).
Due to the high risk of mortality, especially in LQT1 and LQT2 (Citation39), even asymptomatic patients with congenital long QT should be actively managed. As both syndromes are classically associated with either exercise or sudden arousal, traditional management has centered on reduction of heart rate, which may be achieved using β-blockers, typically high-dose propranolol or nadolol, but also atenolol and metoprolol (Citation40). However, β-blockers do not systematically reduce QTc as their effect is mediated by rate control produced through sympathetic blockade and not phenotypic correction via actions on individual ion channel currents. They therefore work only by reducing the precipitating tachycardia (Citation40). β-Blockers are nevertheless effective for the majority of patients, particularly in LQT1, where treatment reduces mortality to < 1% per annum. Lifestyle advice such as avoiding strenuous exercise especially without supervision may also be beneficial.
Patients who remain refractory to β-blocker therapy are regarded as being at high risk, as are those with a QT interval greater than 500 ms, a history of cardiac events including cardiac arrest, syncope or TdP, or Jervell and Lange-Nielsen syndrome; family history is not a strong outcome predictor (Citation41,Citation42). In these patients, the benefits of an implantable cardioverter-defibrillator (ICD) insertion in terms of mortality reduction, from 16% to 1.3% during a mean 8-year follow-up in one study (Citation43), greatly outweigh the risks. Furthermore, ICDs should be programmed to pace at 70–80/min to reduce the risk of pause-dependent TdP, and with prolonged detection times to minimize inappropriate shocks for non-sustained VT precipitating adrenergically triggered VT storm. Left cervicothoracic stellectomy may also be beneficial in uncontrolled, particularly LQT1, patients as a method to reduce further the adrenergic drive. The use of any therapy known to prolong QT should be avoided in all such patients.
Future therapy will likely address the underlying genetic cause, such as the use of class I Na+ channel antagonists in the treatment of LQT3 to reduce the late Na+ current contribution and thus normalize the QTc. However, while this concept is logical, other regulatory pathways may complicate this approach. For example, mexiletine paradoxically facilitates F1473 mutant protein trafficking, resulting in increased Na+ currents and QTc despite channel blockade (Citation44). Modeling and drug testing platforms must hence incorporate these whole-system effects rather than simply focusing on the single channel properties.
Brugada syndrome
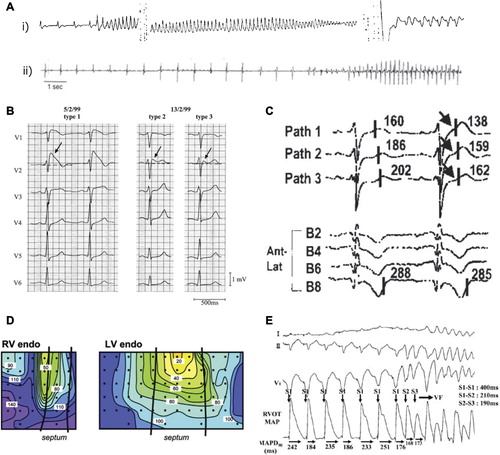
Available evidence implicates BrS in 4%–12% of unexpected sudden deaths, and up to 20% of worldwide deaths in patients with structurally normal hearts, suggesting a population incidence of such SCD of around 0.05% (Citation45). However, its relatively recent identification makes it difficult to ascertain its precise worldwide incidence and distribution. There are also significant worldwide variations in incidence: it is thought to be the commonest cause of sudden death in individuals younger than 50 in South Asia with otherwise anatomically normal hearts. It has a significantly greater male compared to female prevalence (Citation46). Inheritance of the disease is as an autosomal dominant characteristic with incomplete penetrance. Mutations in SCN5A, which encodes the α-subunit of the cardiac voltage-gated Na+ channel, constitute the most common known cause of BrS, accounting for 15%–30% of BrS cases (Citation47); links have been made with almost 300 different mutations in the gene (Citation48,Citation49). Mutations in other genes have also been associated with BrS. These include: GPD1L, encoding glycerol-3-phosphate dehydrogenase 1-like protein (Citation50); CACNA1C (Citation51) and CACNB2 (Citation52,Citation53), which encode the α1 subunit and α2 subunit of the L-type, voltage-gated Ca2+ channel (Cav1.2); SCN1B (Citation54) and SCN3B (Citation55–57), which encode the β1 subunit and β3 subunit of the cardiac Na+ channel; and KCNE3, which encodes the peptide MiRP2, one of five homologous ancillary β subunits (KCNE peptides) of voltage-gated K+ channels (Citation58,Citation59) (). BrS symptoms occur mainly during adulthood, with a mean age of sudden death around 40 years (Citation45). However, a wide range exists, with the youngest patient diagnosed at 2 days old, and the oldest at age 84 years. The disease is much more common in men than in women, probably reflecting gender differences in K+ and Ca2+ channel expression (Citation60,Citation61). Increased testosterone levels in males may lead to a more prominent epicardial Ito-mediated notch which has been implicated in the greater predisposition to develop a BrS phenotype (Citation62). In contrast, females have a greater predisposition for longer APDs and therefore QT intervals, which is potentially protective against BrS (Citation63). The episodes of syncope and SCD are caused by fast polymorphic VT or ventricular fibrillation (VF) (), which often originates from the right ventricular outflow tract (RVOT) (Citation64). BrS is characterized electrocardiographically by right precordial ST elevation, negative T waves, and right ventricular delay; three types (types 1 to 3) of repolarization pattern have been characterized () (Citation65): 1) A diagnostic pattern in the form of coved ST segment elevation ≥ 2 mm followed by a negative T wave; 2) a saddleback appearance resulting in a ST segment elevation of ≥ 2 mm followed by a biphasic or positive T wave; and 3) a saddleback or coved ST elevation of < 1 mm.
Figure 2. Mechanisms of arrhythmogenesis in Brugada syndrome. A: (i) Two different episodes of polymorphic VT, and (ii) ventricular fibrillation documented in BrS patients with ICDs. B: Precordial leads of a resuscitated patient with BrS. There are dynamic ECG changes in the course of a few days, with all three repolarization patterns shown. Arrows denote the ST elevation. The left panel shows a clear type 1 ECG. Between 7–2–99 and 13–2–99, types 2 and 3 are shown. C: Unipolar electrograms from selected electrodes of a pathfinder catheter (Path1–3) positioned in the great cardiac vein to record from the RVOT epicardium, and of a basket catheter (B2-B8) positioned to record from the RVOT endocardium, after a 20 mg pilsicainide injection. The last beat of constant atrial pacing at a cycle length of 600 ms and a subsequent sinus beat are shown. The second beat shows dramatic abbreviation of activation recovery interval (ARI) in pathfinder electrodes (arrows), whereas ARI in the basket electrode did not change, demonstrating increased dispersion of ARI between the epicardium and endocardium of the RVOT. D: Conduction delay in the RV; activation maps are shown for the RV and left ventricular endocardia. In both panels the black lines represent the septal borders, the dots represent the electrode placement, and the colored isochrones indicate 10 ms increments in activation time. E: Monophasic APD alternans recorded in the RV endocardium of a patient with BrS, followed by induction of ventricular fibrillation by extra-stimuli. Reproduced with permission from (A(i)) Brugada et al., Europace 1999;1:156–66 (Citation141); (A(ii)) Priori et al., J Am Coll Cardiol 2012;59(1):37–45 (Citation142); (B) Wilde et al., Circulation 2002;106:2514–9 (Citation65); (C) Shimizu et al., J Cardiovasc Electrophysiol 2001;12(12): 1418–21 (Citation70); (D) Coronel et al., Circulation 2005;112(18):2769–77 (Citation73); (E) Kofune et al., Circ J 2009 Mar;73(3):580–3 (Citation76).
A definite diagnosis of BrS may be made if a type 1 ECG pattern can be demonstrated with one of the following features: 1) Documented history of VF; 2) self-terminating polymorphic VT; 3) family history of SCD; 4) type 1 ECG in family members; 5) induced VT with paced electrical stimulation; 6) syncope; 7) nocturnal agonal respiration.
Some individuals have the typical ECG of right bundle branch block (RBBB) and ST segment elevation in the right precordial leads, and exhibit episodes of VT. Others are asymptomatic and are only diagnosed serendipitously on ECG examination or through screening prompted by a positive family history. Other patients, both symptomatic and asymptomatic, do not exhibit the typical ECG pattern, while in other cases the electrocardiographic manifestations of BrS can be unmasked pharmacologically by administration of Na+ channel blockers, such as flecainide, ajmaline, or procainamide. Other predisposing conditions include pyrexia (Citation66) and exposure to vagotonic agents (Citation67), α-adrenergic agonists, β-adrenergic blockers, tricyclic antidepressants, hyper- and hypokalemia, and hypercalcemia. Genetic testing may also be used to aid the diagnosis, but will only pick up around 30% of patients with the disease, as in 70% of cases the causative mutation is unknown (Citation68).
The cause of the ST elevation in BrS and the reason for its strong linkage to VT/VF remains unresolved. The proposed mechanism which originally appeared to receive the widest support, both from experimental studies using canine pharmacological ventricular preparations (Citation69) and some small clinical studies (Citation70), attributes BrS to a primary repolarization disorder (). The deep phase 1 notch in the epicardial AP, particularly prominent in the right ventricle (RV), makes it susceptible to the effects of Na+ current reduction. This establishes a steep voltage gradient across the RV wall and allows reactivation of the RV epicardium by neighboring regions of myocardium, with longer APs producing functional re-entry, referred to as phase 2 re-entry. An alternative explanation for the ECG signature in BrS is based on conduction delays in the RVOT. Clinical evidence implicating such conduction alterations exists but is mainly based on non-invasive techniques such as echocardiography, signal-averaged ECG, and body surface mapping (Citation71,Citation72). A single ex-vivo study of the heart from a BrS patient has demonstrated local conduction delays in the RVOT () (Citation73), while one in-vivo study has shown both conduction and repolarization disturbances (Citation74). These hypotheses can be related to the increased prevalence of T wave and ST segment alternans, which have recently been associated with BrS patients who are asymptomatic (Citation75), who have been challenged with class 1C agents such as pilsicainide (Citation76), or who are pyrexic (Citation77). ECG alternans has been found to precede induction of VF, potentially via a re-entrant mechanism arising from discordance in APDs () (Citation78).
Recent studies using a haplo-insufficient Scn5a+/− mouse model showing reduced Na+ currents in the RV (Citation79) have shed further light on possible arrhythmogenic mechanisms. The RVs of the mouse hearts demonstrate both depolarization (Citation80) and repolarization (Citation81,Citation82) abnormalities, along with fibrosis and reduced connexin expression associated with ageing (Citation83), which may contribute to the slowed conduction and itself promote repolarization gradients.
The only proven effective treatment for BrS is ICD implantation. However, a recent follow-up extending over 3 years demonstrated that only 2.6% of a cohort of BrS patients received appropriate shocks, whereas 8.9% experienced device-related complications (Citation84). The enhanced knowledge of possible molecular mechanisms underlying arrhythmogenesis has accordingly led to proposals for the introduction of pharmacological therapies. Quinidine has been suggested as an alternative to ICD implantation in view of its inhibitory effect on early activating K+ currents (Ito) (Citation85). However, it cannot be used as first-line therapy in BrS owing to a lack of prospective data. Nevertheless it could be considered for adjunctive therapy in high-risk patients with ICD complications, or in children where ICD implantation is difficult. Recently, a prospective registry has started investigating the use of empirical quinidine therapy for asymptomatic patients with BrS (Citation86). Other drugs, such as β-adrenergic agonists (isoproterenol, orciprenaline) or phosphodiesterase inhibitors (cilostazol), may also have anti-arrhythmic effects, especially during VT storm, but evidence so far is limited (Citation87).
Current management focuses on risk stratification and subsequent ICD implantation. Guidelines published in the report of the Second Consensus Conference on Brugada Syndrome (Citation88) suggest that symptomatic patients displaying a type 1 Brugada ECG presenting with aborted SCD receive an ICD. Similar patients presenting with related symptoms, such as syncope, seizure, or nocturnal agonal respiration should also undergo ICD implantation following exclusion of non-cardiac causes. They also suggest that asymptomatic patients displaying spontaneous type 1 Brugada ECG patterns undergo electrophysiological studies (EPS), followed by ICD implantation if inducible for ventricular arrhythmia; asymptomatic patients with no family history who develop a type 1 ECG only after Na+ channel blockade should be closely followed. However, more recent reports (Citation89,Citation90) could not repeat the initial stratifications of arrhythmic risk based on electrophysiological criteria for inducible ventricular fibrillation that had contributed to such recommendations (Citation91), and both recent multicenter studies (Citation92,Citation93) and meta-analyses (Citation94,Citation95) have questioned the prognostic value of EPS in Brugada syndrome (Citation96). EPS is consequently not in current use in such assessment. In addition there are significant levels of complications from the use of prophylactic implantable cardioverter-defibrillator (ICD) devices among young patients with Brugada syndrome (Citation84).
Catecholaminergic polymorphic ventricular tachycardia
First recognized in 1975, catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly lethal form of inherited cardiac arrhythmogenic disease. It is characterized by potentially fatal spontaneous polymorphic VT following adrenergic challenge. It is thought to contribute to around 15% of the incidence of SCD in the absence of structural heart disease (Citation97,Citation98). Genetic investigation has identified two major disease variants. An autosomal dominant form is associated with mutations in the gene encoding the cardiac ryanodine receptor (RyR2) (Citation99). A recessive form is associated with homozygous mutations in the gene encoding the cardiac isoform of calsequestrin (CASQ2), a calcium-buffering protein of the sarcoplasmic reticulum () (Citation100). Cardiac anatomy is normal, in contrast to the situation in arrhythmogenic right ventricular cardiomyopathy (ARVC), similarly associated with abnormalities in the RyR2 (Citation101,Citation102). Overall, genetic analyses, now being used more widely, can detect a disease-causing mutation in RyR2 or CASQ2 in 50% to 70% of patients with CPVT (Citation103,Citation104); CPVT-like conditions have also been linked to a mutation in the gene encoding ankyrin B, the gene responsible for LQT4 (Citation105).
The resting ECG of patients with CPVT is often unremarkable. CPVT itself is characterized by a uniform pattern of QRS axis alternating by 180° on a beat-to-beat basis, a so-called bidirectional VT (). This can be reproducibly induced during exercise or catecholamine infusion, with arrhythmias usually appearing at a heart rate of 110 to 130 beats per minute.
Figure 3. Mechanisms of arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia. A: Exercise stress test in a patient with polymorphic VT and RyR2 mutation. Ventricular arrhythmias are observed with a progressive worsening during exercise. Typical bidirectional VT develops after 1 minute of exercise with a sinus heart rate of approximately 120 beats per minute. Arrhythmias rapidly recede during recovery. B: AP recordings from control and CPVT stem cell-derived ventricular myocytes. Arrows represent 1 Hz pacing procedure. CPVT shows delayed after-depolarizations (DAD) followed by triggered activity (TA), which is absent in the control. Reproduced with permission from (A) Liu et al., Prog Cardiovasc Dis 2008;51:23–30 (Citation97); (B) Jung et al., EMBO Mol Med 2012;4(3):180–91 (Citation20).
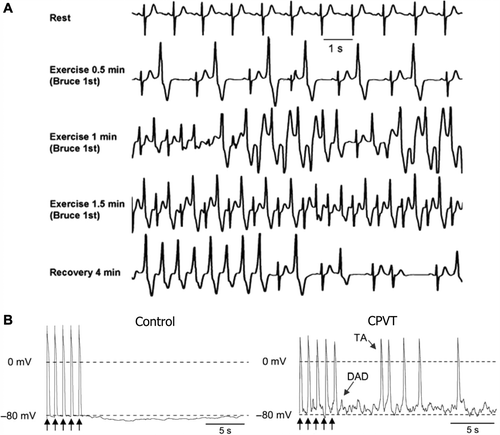
In vitro studies of gain-of-function RyR2 and CASQ2 mutations show calcium leakage from the sarcoplasmic reticulum (Citation106,Citation107), resulting in cytosolic calcium overload. This calcium overload results in increased 3Na+-Ca2+ exchanger activity, which results in delayed after-depolarization (DAD) during phase 4 of the AP. This often results in premature ventricular contractions and could potentially act as an arrhythmogenic trigger in CPVT (). ECG features suggestive of DAD-type phenomena also occur in patients with acute digitalis toxicity. The latter is also associated with increased intracellular Ca2+ (Citation108) resulting from both reduced Na+-K+ ATPase activity and increased RyR2 leakiness. Studies in transgenic mouse models suggest that DADs trigger abnormal beats (Citation109,Citation110).
CPVT is a highly malignant disease, particularly among males with a RyR2 mutation. The mean age of onset of clinical symptoms is between 7 and 9 years (Citation108), and syncope triggered by emotional or physical stress is usually the first clinical manifestation (Citation103,Citation108). A third of patients with CPVT have a family history of premature sudden death or stress-related syncope (Citation108), but sudden death may be the first manifestation of CPVT. β-Blockers are the preferred therapy (Citation108). They may prevent syncope and SCD because adrenergic activation is the main mechanism of DAD-dependent triggered activity in these patients (Citation111). The most commonly used β-blockers are nadolol or propranolol. To maximize control of arrhythmias, it is important that these are prescribed at the maximal tolerated dose. However, recent studies have questioned the efficacy of β-blockers over long-term follow-up, with an ICD required in 30% of patients because of symptomatic recurrence of life-threatening arrhythmia despite such β-blocker therapy (Citation103).
More recent studies have showed that verapamil may be an alternative or adjuvant therapy with β-blockers (Citation112), potentially owing to its effect in reducing cytosolic Ca2+. However, because of the small number of patients studied so far and the limited follow-up, conclusive evidence is lacking. A more novel therapy involving use of the RyR2 blocker flecainide may provide an alternative and more targeted therapy through its effect in preventing Ca2+ leak (Citation113). Lastly and in common with LQTS, in centers where the procedure is available, left cardiac sympathetic denervation (LCSD) (Citation114) may be considered for individuals with intractable arrhythmic storms.
Other syndromes
Whilst most cases of SCD in the absence of structural heart abnormalities are thought to be caused by the three syndromes mentioned above, a number of other syndromes that may also cause sudden death have come to light.
Short QT syndrome is characterized by abnormally short QT intervals, as well as a high incidence of SCD and high prevalence of atrial fibrillation (Citation115). It has been associated with gain-of-function mutations in three genes encoding K+ channels (Citation116–118) and more recently loss-of-function mutations in two genes encoding the cardiac L-type Ca2+ channel. The latter cause a mixed phenotype of short QT syndrome and BrS (Citation53). The first-choice therapy for these conditions is ICD implantation. However, pharmacological treatment with hydroquinidine, which prolongs QT and reduces the inducibility of ventricular arrhythmias, has been suggested for children and possibly elderly asymptomatic patients (Citation119).
Early repolarization syndrome is characterized by ventricular arrhythmias and SCD in the presence of early repolarization (ER) patterns in the ECG (Citation120,Citation121). The ER pattern is relatively common, occurring in approximately 1% to 13% of the general population (Citation122). It was initially thought benign, but patients with documented idiopathic VF and structurally normal hearts show a prevalence of ER of around 31% (Citation122). The ER pattern is identified on the basis of J point elevation manifesting as QRS slurring or notching, but then may be subclassified according to the morphology of the subsequent ST segment, with the horizontal ST segment denoting high risk (Citation123,Citation124). Preliminary evidence from canine studies suggests that increased transmural differences in early phases of the cardiac AP may be responsible (Citation125). However, the exact mechanism for arrhythmogenesis in ER is still unknown. In rare cases, gene mutations causing either decreased inward Ca2+ currents (Citation126) or increased outward K+ currents (Citation127) have been identified. A number of cases of ER are also associated with SCN5A mutations (Citation128). However, mutations have not been identified in most cases. The high frequency of the genetic background underlying the ER pattern in the population suggests that the disease is probably polygenic and influenced by environmental factors. The treatment of choice of patients with VF and ER is ICD implantation. Quinidine may be used as an adjunct to prevent recurrence, and isoproterenol can be used in acute control of arrhythmia.
Even with exhaustive genetic evaluation, all known hereditary causes are excluded in up to 5% of cases of SCD (Citation129). Historically such cases have especially been noted in South East Asian men and are referred to as the sudden unexplained nocturnal death syndrome. However, it has been suggested that a majority of these cases are, in fact, manifestations of BrS (Citation130). The remaining cases are termed idiopathic ventricular fibrillation. Patients often have a history of syncope. However, some of these cases may show ECG repolarization abnormalities of the ER type (Citation122) and therefore may be a variant of this condition. Research is on-going in the remainder of cases to identify possible causative genes (Citation131).
Progressive cardiac conduction disease (PCCD) encompasses a further group of conditions that most commonly manifest as bradycardia but may also cause SCD. The first gene to be associated with PCCD was SCN5A (Citation132). Indeed some SCN5A mutations may be associated with more complex phenotypes combining characteristics of BrS, PCCD, and LQT3, the so-called ‘overlap syndromes’ (Citation133). More recently, PCCD has been associated with altered expression of genes encoding other proteins involved in impulse propagation, including those responsible for Ca2+-activated ion channels and cytoskeletal components (Citation62,Citation134). The resulting phenotype appears to result from a combination of these genetic and environmental factors as well as additional physiological changes including those resulting from ageing. For isolated conduction disease, pacemaker implantation is the treatment of choice, but other strategies may be indicated in the presence of overlap syndromes.
Sick sinus syndrome (SSS) results from a failure of the sino-atrial node (SAN), most frequently due to ageing and fibrosis, which may result in profound bradycardia or conduction block (Citation135). Patients present with syncope, exertional dyspnea, angina, and palpitations. ECG features include bradycardia that does not respond to physiological stresses, sinus pauses, sino-atrial exit block, and arrest. Tachycardia–bradycardia syndrome occurs in over 50%. It may also be seen with other atrial arrhythmias reflecting progressive atrial pathology. While the average age of presentation is 73–76 years (Citation136), reflecting age-related changes such as SAN fibrosis or nodal artery stenosis, an interesting but rare subset of patients present in childhood and have a family history of the disease. Mutations involving two genes have been associated with such cases. Mutations in the SCN5A gene have previously been found in five out of ten children with SSS, from three families (Citation137). As such there may be overlap with other inherited arrhythmias such as BrS. However, not all patients with SCN5A mutations develop SSS. There is also involvement of the HCN4 gene which encodes the α-subunit of the hyperpolarization-activated cyclic nucleotide-gated channel, which carries the pacemaker current, If (Citation138). Such mutations tend to run in a small number of families who may present with symptomatic or asymptomatic SSS. The mainstay of treatment for symptomatic or asymptomatic SSS is the insertion of a pacemaker once reversible causes, such as drugs, have been excluded. Concomitant AF must be assessed and anticoagulated appropriately (Citation139).
Summary
Syncope and risk of SCD caused by ventricular tachyarrhythmia are common manifestations of several inherited disorders. SCD may be the first presentation, but patients may present with symptoms of palpitations or hemodynamic compromise such as dizziness, seizure, or syncope, particularly following exertion, and therefore may undergo initial evaluation by the general hospital physician. In all inherited cardiac death syndromes, first-degree relatives should be referred to a cardiologist, and undergo testing appropriate for the condition, which may include genetic testing. Hence, while the follow-up of patients with known channelopathies often occurs in a tertiary setting, it is vital that the non-specialist has a basic knowledge of the mechanisms and clinical manifestations of these diseases, in order to enable initial diagnosis and onward referral.
Since the mid-1990s there has been a surge of interest in unexplained cardiac arrest occurring in patients with normal autopsy. Recent characterizations of their underlying genetic bases in combination with clarifications of their physiological consequences have led to clarification of their cellular mechanisms. In combination with clinical studies this has improved our understanding of pathogenesis, diagnosis, risk stratification, and management. While many treatment strategies still rely on ICD implantation, these have led to an increased interest in pharmacological treatment options, particularly those tailored to the genetic defect responsible for the arrhythmia generation.
Declaration of interest: Funding was provided by the British Heart Foundation, the Medical Research Council, the Wellcome Trust, and the Biotechnology and Biological Research Council, UK. Claire A. Martin was supported by a Medical Research Council Clinical Research Fellowship and a Sackler Studentship of the University of Cambridge School of Clinical Medicine. Gareth D. K. Matthews was supported by the Stanley-Elmore Scholarship, Gonville and Caius College, University of Cambridge and by the Wellcome Trust Translational Medicine and Therapeutics Program, University of Cambridge. The authors report no conflicts of interest.
References
- National Institute for Health and Clinical Excellence (NICE). Implantable cardioverter defibrillators for arrhythmias, www.nice.org.uk/TA095 . In: Department of Health U, editor. 2006. p. 1–33.
- Stecker EC, Vickers C, Waltz J, Socoteanu C, John BT, Mariani R, et al. Population-based analysis of sudden cardiac death with and without left ventricular systolic dysfunction: two-year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol. 2006; 47:1161–6.
- Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, et al. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–28.
- de Vreede-Swagemakers JJ, Gorgels AP, Dubois-Arbouw WI, van Ree JW, Daemen MJ, Houben LG, et al. Out-of-hospital cardiac arrest in the 1990’s: a population-based study in the Maastricht area on incidence, characteristics and survival. J Am Coll Cardiol. 1997; 30:1500–5.
- Tokashiki T, Muratani A, Kimura Y, Muratani H, Fukiyama K. Sudden death in the general population in Okinawa: incidence and causes of death. Jpn Circ J. 1999;63:37–42.
- Byrne R, Constant O, Smyth Y, Callagy G, Nash P, Daly K, et al. Multiple source surveillance incidence and aetiology of out-of- hospital sudden cardiac death in a rural population in the West of Ireland. Eur Heart J. 2008;29:1418–23.
- Vaillancourt C, Stiell IG. Cardiac arrest care and emergency medical services in Canada. Can J Cardiol. 2004;20:1081–90.
- Kannel WB, Cupples LA, D’Agostino RB. Sudden death risk in overt coronary heart disease: the Framingham Study. Am Heart J. 1987; 113:799–804.
- Willich SN, Levy D, Rocco MB, Tofler GH, Stone PH, Muller JE. Circadian variation in the incidence of sudden cardiac death in the Framingham Heart Study population. Am J Cardiol. 1987;60:801–6.
- Behr E, Wood DA, Wright M, Syrris P, Sheppard MN, Casey A, et al. Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet. 2003;362:1457–9.
- Tung RT, Shen WK, Hammill SC, Gersh BJ. Idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors. Pacing Clin Electrophysiol. 1994;17:1405–12.
- Viskin S, Belhassen B. Idiopathic ventricular fibrillation. Am Heart J. 1990;120:661–71.
- Puranik R, Chow CK, Duflou JA, Kilborn MJ, McGuire MA. Sudden death in the young. Heart Rhythm. 2005;2:1277–82.
- Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49:240–6.
- Arnestad M, Vege A, Rognum TO. Evaluation of diagnostic tools applied in the examination of sudden unexpected deaths in infancy and early childhood. Forensic Sci Int. 2002;125:262–8.
- Klaver EC, Versluijs GM, Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol. 2011;152: 162–70.
- Tester DJ, Ackerman MJ. The role of molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol. 2006;21:166–72.
- Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc. 2012;87:524–39.
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9.
- Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–91.
- Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnaiz-Cot JJ, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28:579–92.
- Vincent GM. Long QT syndrome. Cardiol Clin. 2000;18:309–25.
- Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
- Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181–4.
- Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74.
- Salama G, London B. Mouse models of long QT syndrome. J Physiol. 2007;578(Pt 1):43–53.
- January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2 + current. Circ Res. 1989;64:977–90.
- Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res. 2013;161:1–14.
- Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47.
- Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases?:the intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1120–1.
- Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112: 2602–10.
- Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8.
- Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–41.
- Vincent GM, Timothy K, Fox J, Zhang L. The inherited long QT syndrome: from ion channel to bedside. Cardiol Rev. 1999;7:44–55.
- Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–34.
- Roden DM, Anderson ME. The pause that refreshes, or does it?Mechanisms in torsades de pointes. Heart. 2000;84:235–7.
- Cerrone M, Napolitano C, Priori SG. Genetics of ion-channel disorders. Curr Opin Cardiol. 2012;27:242–52.
- Barsheshet A, Brenyo A, Moss AJ, Goldenberg I. Genetics of sudden cardiac death. Curr Cardiol Rep. 2011;13:364–76.
- Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–5.
- Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–23.
- Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–91.
- Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, et al. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–8.
- Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337–41.
- Ruan Y, Denegri M, Liu N, Bachetti T, Seregni M, Morotti S, et al. Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3. Circ Res. 2010;106:1374–83.
- Antzelevitch C, Brugada P, Brugada J, Brugada R. Brugada syndrome: from cell to bedside. Curr Probl Cardiol. 2005;30:9–54.
- Matsuo K, Akahoshi M, Nakashima E, Suyama A, Seto S, Hayano M, et al. The prevalence, incidence and prognostic value of the Brugada-type electrocardiogram: a population-based study of four decades. J Am Coll Cardiol. 2001;38:765–70.
- Alings M, Wilde A. ”Brugada” syndrome: clinical data and suggested pathophysiological mechanism. Circulation. 1999;99:666–73.
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A- encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46.
- Berne P, Brugada J. Brugada syndrome 2012. Circ J. 2012;76:1563–71.
- London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8.
- Takimoto K, Li D, Nerbonne JM, Levitan ES. Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J Mol Cell Cardiol. 1997;29: 3035–42.
- Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–5.
- Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9.
- Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–8.
- Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–58.
- Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–8.
- Valdivia CR, Medeiros-Domingo A, Ye B, Shen WK, Algiers TJ, Ackerman MJ, et al. Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res. 2010;86:392–400.
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87.
- Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–18.
- Sacher F, Meregalli P, Veltmann C, Field ME, Solnon A, Bru P, et al. Are women with severely symptomatic Brugada syndrome different from men?J Cardiovasc Electrophysiol. 2008;19:1181–5.
- Shimizu W. Gender difference and drug challenge in Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:70–1.
- Fish JM, Antzelevitch C. Cellular and ionic basis for the sex-related difference in the manifestation of the Brugada syndrome and progressive conduction disease phenotypes. J Electrocardiol. 2003;36(Suppl): 173–9.
- Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, et al. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm. 2012;9: 823–32.
- Morita H, Morita ST, Nagase S, Banba K, Nishii N, Tani Y, et al. Ventricular arrhythmia induced by sodium channel blocker in patients with Brugada syndrome. J Am Coll Cardiol. 2003;42:1624–31.
- Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–9.
- Antzelevitch C, Brugada R. Fever and Brugada syndrome. Pacing Clin Electrophysiol. 2002;25:1537–9.
- Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996; 27:1061–70.
- Hedley PL, Jorgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, et al. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat. 2009;30:1256–66.
- Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–6.
- Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol. 2001;12:1418–21.
- Tukkie R, Sogaard P, Vleugels J, de Groot IK, Wilde AA, Tan HL. Delay in right ventricular activation contributes to Brugada syndrome. Circulation. 2004;109:1272–7.
- Eckardt L, Bruns HJ, Paul M, Kirchhof P, Schulze-Bahr E, Wichter T, et al. Body surface area of ST elevation and the presence of late potentials correlate to the inducibility of ventricular tachyarrhythmias in Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:742–9.
- Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112:2769–77.
- Lambiase PD, Ahmed AK, Ciaccio EJ, Brugada R, Lizotte E, Chaubey S, et al. High-density substrate mapping in Brugada syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation. 2009;120:106–17, 1–4.
- Morita H, Zipes DP, Fukushima-Kusano K, Nagase S, Nakamura K, Morita ST, et al. Repolarization heterogeneity in the right ventricular outflow tract: correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm. 2008;5:725–33.
- Kofune M, Watanabe I, Ashino S, Ohkubo K, Okumura Y, Kofune T, et al. Action potential alternans in the right ventricular outflow tract in a patient with asymptomatic Brugada syndrome. Circ J. 2009;73: 580–3.
- Lamelas P, Labadet C, Spernanzoni F, Saubidet CL, Alvarez PA. Brugada electrocardiographic pattern induced by fever. World J Cardiol. 2012;4:84–6.
- Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98: 1244–53.
- Martin CA, Siedlecka U, Kemmerich K, Lawrence J, Cartledge J, Guzadhur L, et al. Reduced Na+ and higher K+ contribute to right ventricular origin of arrhythmias in Scn5a+/2 mice. Open Biol. 2012; 2:120072.
- Martin CA, Guzadhur L, Grace AA, Lei M, Huang CL. Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a++/− mouse model. Am J Physiol Heart Circ Physiol. 2011; 300:H1853–62.
- Martin CA, Grace AA, Huang CL. Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am J Physiol Heart Circ Physiol. 2011;300: H605–16.
- Matthews GD, Martin CA, Grace AA, Zhang Y, Huang CL. Regional variations in action potential alternans in isolated murine Scn5a+/2 hearts during dynamic pacing. Acta Physiol (Oxf). 2010;2:129–46.
- van Veen TA, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, et al. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation. 2005;112:1927–35.
- Sacher F, Probst V, Iesaka Y, Jacon P, Laborderie J, Mizon-Gerard F, et al. Outcome after implantation of a cardioverter-defibrillator in patients with Brugada syndrome: a multicenter study. Circulation. 2006;114:2317–24.
- Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731–7.
- Viskin S, Wilde AA, Tan HL, Antzelevitch C, Shimizu W, Belhassen B. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm. 2009;6:401–4.
- Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm. 2009;6:1335–41.
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–70.
- Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–7.
- Eckardt L, Probst V, Smits JP, Bahr ES, Wolpert C, Schimpf R, et al. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation. 2005;111:257–63.
- Brugada P, Brugada R, Mont L, Rivero M, Geelen P, Brugada J. Natural history of Brugada syndrome: the prognostic value of programmed electrical stimulation of the heart. J Cardiovasc Electrophysiol. 2003; 14:455–7.
- Probst V, Veltmann C, Eckardt L, Meregalli PG, Gaita F, Tan HL, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121:635–43.
- Kamakura S, Ohe T, Nakazawa K, Aizawa Y, Shimizu A, Horie M, et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ Arrhythm Electrophysiol. 2009;2:495–503.
- Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AA, et al. Role of programmed ventricular stimulation in patients with Brugada syndrome: a meta-analysis of worldwide published data. Eur Heart J. 2007;28:2126–33.
- Gehi AK, Duong TD, Metz LD, Gomes JA, Mehta D. Risk stratification of individuals with the Brugada electrocardiogram: a meta-analysis. J Cardiovasc Electrophysiol. 2006;17:577–83.
- Viskin S, Rosso R. Risk of sudden death in asymptomatic Brugada syndrome: not as high as we thought and not as low as we wished. ..but the contrary. J Am Coll Cardiol. 2010;56:1585–8.
- Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51:23–30.
- Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin Proc. 2004;79:1380–4.
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200.
- Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation. 2001;103: 2822–7.
- Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–32.
- Lanner JT. Ryanodine receptor physiology and its role in disease. Adv Exp Med Biol. 2012;740:217–34.
- Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74.
- Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol. 2009;2:6–15.
- Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–42.
- Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–81.
- Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–14.
- Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–9.
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2-R4496C knock-in mouse model. Circ Res. 2006;99:292–8.
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–20.
- Nakajima T, Kaneko Y, Taniguchi Y, Hayashi K, Takizawa T, Suzuki T, et al. The mechanism of catecholaminergic polymorphic ventricular tachycardia may be triggered activity due to delayed afterdepolarization. Eur Heart J. 1997;18:530–1.
- Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149–54.
- Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3.
- Wilde AA, Bhuiyan ZA, Crotti L, Facchini M, De Ferrari GM, Paul T, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358: 2024–9.
- Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965–70.
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–5.
- Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394–7.
- Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800–7.
- Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494–9.
- Mehta M, Jain AC, Mehta A. Early repolarization. Clin Cardiol. 1999; 22:59–65.
- Miyazaki S, Shah AJ, Haissaguerre M. Early repolarization syndrome - a new electrical disorder associated with sudden cardiac death. Circ J. 2010;74:2039–44.
- Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–23.
- Tikkanen JT, Junttila MJ, Anttonen O, Aro AL, Luttinen S, Kerola T, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation. 2011;123:2666–73.
- Rosso R, Halkin A, Viskin S. J waves and early repolarization: do not confuse me with the facts!Heart Rhythm. 2012;9:1603–4.
- Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–9.
- Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, Desai M, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–82.
- Haissaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20:93–8.
- Watanabe H, Nogami A, Ohkubo K, Kawata H, Hayashi Y, Ishikawa T, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–81.
- Survivors of out-of-hospital cardiac arrest with apparently normal heart. Need for definition and standardized clinical evaluation. Consensus Statement of the Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe and of the Idiopathic Ventricular Fibrillation Registry of the United States. Circulation. 1997;95: 265–72.
- Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96: 2595–600.
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MW, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468–76.
- Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–1.
- Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–9.
- Marsman RF, Bardai A, Postma AV, Res JC, Koopmann TT, Beekman L, et al. A complex double deletion in LMNA underlies progressive cardiac conduction disease, atrial arrhythmias, and sudden death. Circ Cardiovasc Genet. 2011;4:280–7.
- Ferrer MI. The sick sinus syndrome in atrial disease. JAMA. 1968; 206:645–6.
- Lamas GA, Lee KL, Sweeney MO, Silverman R, Leon A, Yee R, et al. Ventricular pacing or dual-chamber pacing for sinus-node dysfunction. N Engl J Med. 2002;346:1854–62.
- Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003; 112:1019–28.
- Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–7.
- Andersen HR, Nielsen JC, Thomsen PE, Thuesen L, Mortensen PT, Vesterlund T, et al. Long-term follow-up of patients from a randomised trial of atrial versus ventricular pacing for sick-sinus syndrome. Lancet. 1997;350:1210–6.
- Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849–55.
- Brugada J, Brugada P, Brugada R. The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death—the Brugada syndrome. Europace. 1999;1:156–66.
- Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59:37–45.