
Abstract
Sleep parallels brain functioning and mental health. Neuronal activity during wakefulness leads to a subsequent increase in sleep intensity as measured using electroencephalographic slow-wave activity (SWA; index of neuronal synchrony in the low-frequency range). Wakefulness, and particularly prolonged wakefulness, also drives important changes in brain gene expression and changes in protein regulation. The role of these two cellular mechanisms in sleep-wake regulation has typically been studied independently, and their exact contribution to SWA remains poorly defined. In this review, we highlight that many transcriptional pathways driven by sleep deprivation are associated to protein regulation. We first describe the relationship between cytokines, clock genes, and markers of sleep need with an emphasis on transcriptional processes. Observations regarding the role of protein metabolism in sleep-wake regulation are then depicted while presenting interconnections between transcriptional and translational responses driven by sleep loss. Lastly, a manner by which this integrated response can feed back on neuronal network activity to determine sleep intensity is proposed. Overall, the literature supports that a complex cross-talk between transcriptional and translational regulation during prolonged wakefulness drives the changes in sleep intensity as a function of the sleep/wake history.
Key messages
Prolonged wakefulness changes the brain transcriptome via an impact on a variety of transcription regulatory pathways.
Prolonged wakefulness changes protein regulation by affecting pathways involved in protein synthesis, folding, and degradation.
The coordinated transcriptional and translational response to prolonged wakefulness likely regulates the synchrony of neuronal firing in the cerebral cortex by a control of glutamate receptor function.
Introduction
Sleep is perceived as a recovery process, which is easily understandable since sleep is needed to feel refreshed and alert. Although sleep behaviors vary greatly between and also within species, sleep is required and highly conserved across mammalian and many non-mammalian species. The essential role of sleep for brain function and maintenance of mental health is increasingly recognized (Citation1–4), as underlying molecular and cellular mechanisms are being described and identified as overlapping with those implicated in key brain functions such as memory and emotion. Thus, recent research contributed to delineate the manner by which wakefulness quality is intimately linked to sleep.
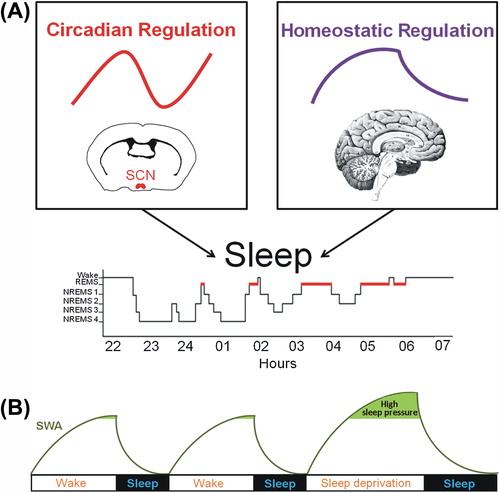
According to a model developed more than three decades ago (Citation5,Citation6), sleep is regulated by two main mechanisms: a circadian and a homeostatic process (). The circadian process controls the propensity for wakefulness with a close to 24 h rhythm and originates from the suprachiasmatic nuclei of the hypothalamus in mammals (Citation7,Citation8). The homeostatic process represents the recovery process according to which the intensity of sleep is determined by previous wakefulness duration and intensity. The relationship between these sleep-regulatory processes determines the consolidation and quality of both wakefulness and sleep in humans (Citation9–11). The present review will focus on the homeostatic aspect of sleep-wake regulation. In particular, this review will emphasize that characteristics of sleep homeostasis are linked to molecular mechanisms involving both gene and protein expression in the brain.
Figure 1. Physiological processes regulating the sleep-wake cycle and indicator of sleep propensity. (A) The sleep-wake cycle is influenced by two main regulatory processes. The circadian process is mainly orchestrated by the master pacemaker located in the suprachiasmatic nuclei (SCN) of the hypothalamus and drives a circadian (about 24 h) rhythm in sleep propensity. The homeostatic process is represented by the build-up of sleep need during wakefulness and its decay in the course of sleep. (B) It is commonly accepted that slow-wave activity (SWA; 0.75–4.5 Hz EEG activity) during non-rapid eye movement sleep (NREMS) is a key marker of the homeostatic process because it increases as a function of wakefulness duration and dissipates during sleep, reflecting the decay in homeostatic sleep need. During sleep deprivation, high sleep pressure leads to increased SWA that returns to baseline level during the recovery sleep. (EEG = electroencephalographic; REMS = rapid-eye-movement sleep).
Markers of the sleep recovery process
From the electroencephalogram
Sleep homeostasis has been studied through the dynamics of its acknowledged markers measured during sleep and wakefulness under normal ‘undisturbed’ condition or under condition of elevated or alleviated sleep need (e.g. sleep deprivation (SD) or nap, respectively). The dynamics of slow-wave activity (SWA; 0.75–4.5 Hz) or delta power (1–4 Hz) during non-rapid eye movement (NREM) sleep measured using electroencephalography (EEG) index the homeostatic sleep need (Citation12–14). Indeed, an increase in the duration of wakefulness, for instance using SD, increases EEG activity in the delta frequency range during subsequent sleep () (Citation15–18). Besides wakefulness duration, increased neuronal activity during waking, such as during learning, has also been shown to enhance SWA (Citation19–22). Such increases have been linked to task performance following sleep (Citation23,Citation24), which indicates that the extent of SWA during sleep may be functionally linked to the efficacy of learning processes in given circumstances.
In the course of sleep, SWA dissipates, reflecting the decay in homeostatic sleep need () (Citation11,Citation13,Citation18). Spindle frequency activity (12–16 Hz) measured during NREM sleep, also referred to as sigma activity, seems to mirror sleep pressure as well (Citation25–27), given that some sigma frequencies increase during sleep and are reduced following SD (Citation11,Citation18,Citation28). EEG activity during wakefulness has also been linked to sleep homeostasis. Notably, spectral power in delta (1–4 Hz), theta (4–8 Hz), and beta (12–25 Hz) frequencies increases with wakefulness duration particularly during SD (Citation29–31).
The manner by which these EEG markers, especially SWA, can inform about the dynamics of sleep homeostasis and about the role of sleep in brain functions remains to be clearly defined, and some authors even questioned the use of SWA as an index of sleep need (Citation32). In particular, the contribution of relative versus absolute changes in defining the recovery capacity of sleep intensity is still controversial. For instance, elevated absolute SWA is often interpreted as a change to sleep homeostasis and considered an indication of better sleep. Nevertheless, absolute SWA importantly differs between individuals and does not provide information about the capacity to recover after a challenge (i.e. the ability to increase SWA after SD). This is rather quantified using the analysis of the increase relative to baseline. As such, the dynamics of sleep need has been modeled using relative changes in SWA or delta power (Citation5,Citation6,Citation33). However, accumulating data show that an absolute difference in NREM sleep SWA between experimental groups associates with differences in cognitive performance (Citation34,Citation35). Thus, the present review will consider both the relative changes in SWA and differences in absolute SWA values as potential evidence of a role for a molecular element in the recovery aspect of sleep-wake regulation.
From a molecular perspective
In parallel, the mRNA expression of many transcripts in the brain has been shown to change reliably with elevated sleep pressure (Citation26,Citation36–38). These molecular elements can therefore be used as indicators of sleep need as well (Citation39–41). Many immediate early genes (IEG; Fos, Arc) increase with SD (Citation42,Citation43), and this is probably related to their well-recognized increase following elevated neuronal activity. Another marker is Homer1a, coding for an intracellular protein that regulates glutamatergic transmission, identified by way of QTL analysis of the SWA response to SD (Citation37,Citation44), which expression is robustly increased by SD (Citation37,Citation39–41, Citation44,Citation45). The expression of several clock genes (e.g. Per2, Dbp) was also shown to track the need for sleep (Citation46) (see (Citation47) for a review). This SD-related change was observed independently of their role in the regulation of internal time, and in areas outside the main circadian clock (Citation38). Although the precise contribution of these transcripts to the SWA increase after SD is still under investigation, their reliable change after sleep loss reveals molecular pathways associated with the sleep recovery process.
In addition, microarray studies portrayed the genome-wide effects of elevated sleep need on brain gene expression (Citation37,Citation39, Citation48–50). These data contributed to current hypotheses regarding the role of sleep in metabolism and energy regulation, synaptic plasticity, and neuroprotection (Citation37,Citation39,Citation51,Citation52). A similar genome-wide response to neuronal activity was observed using an in vitro model, which, in addition to metabolism, also points to regulation of neuronal membrane homeostasis as a function of sleep (Citation53). This function is also supported by recent findings made specifically in oligodendrocytes (Citation54), in which sleep seems to benefit phospholipid synthesis and myelination. Overall, the large effect of SD on the transcriptome (often > 1000 genes) suggests that the modulation of transcriptional regulation is a major pathway that allows the brain, and likely other organs (Citation37,Citation55), to orchestrate an adapted and integrated response to sleep loss.
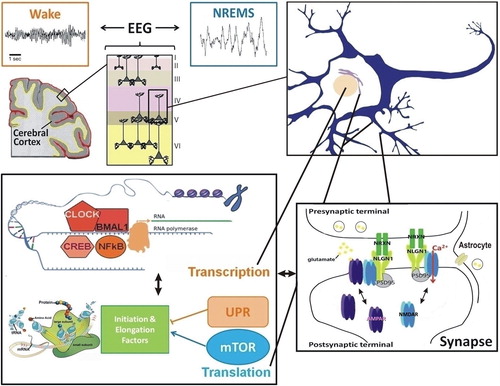
Importantly, microarray studies also revealed that modification in pathways involved in protein regulation and synthesis are additional hallmarks of the response to increased sleep pressure in the brain (Citation37,Citation39,Citation48–50). These studies highlighted, in general, that elevated sleep pressure mostly down-regulates translation and protein transport in the forebrain (Citation39), cerebral cortex (Citation49), and hippocampus (Citation50). Thus, the dynamics of brain molecular processes in response to neuronal activity during wakefulness and sleep are complex and involve the recruitment of a number of cellular functions (). In particular, gene expression and protein biosynthesis are modulated by extended wakefulness showing both high potential to shape neuronal connectivity and cognition, and will be focused on in this review.
Figure 2. Interconnections between molecular elements linked to sleep homeostasis. The EEG reflects the synchronization of neuronal firing in the cerebral cortex to which pyramidal cells, among others, contribute. Changes in neuronal firing synchrony across the sleep-wake cycle, which occur together with changes in neurotransmitters (e.g. glutamate, acetylcholine, dopamine, serotonin), depend on modifications in individual neurons leading to modifications in properties of neuronal communication (e.g. synaptic function). At the transcriptional level, numerous transcription factors seem to respond to neuronal activity associated with prolonged wakefulness and to impact on EEG activity during wakefulness and sleep (e.g. NFκB, CLOCK/BMAL1, CREB). Protein regulation also seems a relevant cellular function linked to sleep, particularly in the context of memory consolidation. Indeed, the mTOR pathway, involved in protein synthesis, and the unfolded protein response (UPR), linked to protein folding and degradation, are usually associated with the sleep and wake states, respectively. Importantly, transcriptional and translational machineries are interacting such that many of these pathways were shown to influence each other directly. In addition, these interactions may feed back on synaptic properties to determine the function of the synapses, notably glutamatergic synapses, as a function of sleep need. This likely involves neurons and non-neuronal cells like astrocytes, and leads to the modulation of numerous synaptic components (e.g. NMDA and AMPA receptors, Neuroligins). Such cross-talk between these molecular and cellular levels of regulation accounts for the changes in EEG synchrony as a function of the sleep/wake history. (EEG = electroencephalogram; mTOR = mammalian target of rapamycin; NLGN1 = Neuroligin-1; NREMS = non-rapid-eye-movement sleep; NRXN = Neurexin; PSD95 = postsynaptic density protein 95; UPR = unfolded protein response).
Humoral factors to clock genes: early emphasis on transcription
Sleep regulatory substances
Early sleep research has focused on the discovery of sleep regulatory substances (SRS) as underlying molecular mechanisms governing the need for sleep (Citation15). To be considered a SRS, a molecule had to meet the following five criteria: 1) should promote sleep when administered; 2) should reduce sleep when inhibited; 3) should vary as a function of sleep propensity; 4) should act on sleep regulatory systems; 5) should be altered in pathological states associated with enhanced sleepiness. One of the first SRS identified was the Factor S that accumulated during SD in the cerebrospinal fluid of animals and humans, and that was shown to induce NREM sleep when infused in the rabbit or rat brain (Citation56,Citation57). Further studies from the same group identified this Factor S as an immunoadjuvant and demonstrated the somnogenic effect of other immunoadjuvants (Citation58). This research led to the identification of the non-negligible role of components of the immune system in sleep-wake regulation. In fact, the relationship between sleep and the immune system seems bidirectional as cytokines influence sleep structure and EEG, and sleep impacts on the immune response. Subsequently, the pro-inflammatory cytokines interleukin-1beta (IL-1β) and tumor necrosis factor alpha (TNFα) were proposed as sleep-promoting substances (Citation59,Citation60). Since the relationship between these cytokines and sleep has been studied in detail (Citation61), and because they are important regulators of gene expression (Citation62), their role will be further addressed in the next section.
Cytokines
Cytokines were first shown to modulate sleep architecture. Central or systemic injection of IL-1β or TNFα increases the duration of NREM sleep in many mammals, and their inhibition decreases spontaneous sleep (for review, see (Citation63)). Moreover, mutant animals for TNFα and/or IL-1β receptors show reduced NREM sleep duration (Citation64–67). The level of IL-1β and TNFα follows the sleep/wake distribution peaking with the onset of sleep, in addition to being increased by SD (Citation68–70). In parallel, infectious diseases increasing sleep, like the African sleeping sickness, are characterized by an increase in pro-inflammatory cytokines such as TNFα (Citation71). Importantly, cytokines and their receptors can change EEG activity during sleep. Indeed, in addition to elevated NREM sleep, delta power is also increased when either IL-1β or TNFα is administered in the cerebral cortex (Citation72,Citation73). Moreover, cortical administration of small interfering RNA targeting TNFα decreased SWA (Citation74). Although genetic inactivation of the receptors of these cytokines did not lead to reduced SWA as would be expected from a positive relationship between cytokines and sleep intensity, mutant animals consistently showed changes in low-frequency EEG activity during NREM sleep. In fact, combined absence of both TNFα receptors, or of Il-1β and TNFα receptors, leads to a reduction of NREM sleep and an increased delta power in the lower frequencies (Citation64,Citation65), as well as to an impaired response to SD with shortened NREM sleep rebound and increased SWA (Citation64).
A mechanism that likely underlies the relationship between cytokines and sleep was proposed (for review, see (Citation63)), which resides in the transduction of the neuronal activity signal into a transcriptional signal. Neuronal activity, such as the one during wakefulness, leads to ATP release (Citation75), to which astrocytes contribute (Citation76). ATP then accumulates in the extracellular space, binds to the purine type 2 P2X7 receptor found on glial cells, and participates to both IL-1β and TNFα release from glia, mainly microglia (Citation77,Citation78). In parallel, ATP is hydrolyzed to adenosine that was shown to accumulate in the course of wakefulness and to modulate NREM sleep SWA (Citation79,Citation80). Both cytokines and adenosine activate a signaling cascade that leads to the activation of the transcription factor nuclear factor kappa B (NFκB) (Citation81,Citation82). In the central nervous system, this activation may predominantly involve astrocytes (Citation83,Citation84). NFκB drives the transcription of many genes, some of them being implicated in synaptic plasticity (e.g. Bdnf) (Citation85). It thus seems that the implication of cytokines, which are released following neuronal activity, may depend on a transcriptional response involving different types of brain cells.
Another way by which cytokines may shape gene expression in a sleep-relevant way resides in their relationship with clock genes. Clock genes independently govern circadian rhythms and sense homeostatic sleep need (Citation40,Citation47) through a transcription-translation feedback loop that controls the activity of a wide range of transcription factors (see next section). IL-1β and TNFα were shown to repress the transcriptional activity of the core clock elements CLOCK/BMAL1 and thereby decrease the expression of clock genes and clock-controlled genes under their control (Citation86). This effect was shown to depend on calcium and MAP kinase signaling (Citation87), which suggests another pathway, besides NFκB, by which cytokines can detect synaptic activity and trigger a transcriptional response. Importantly, cytokines can feed back on synaptic activity which could represent a way by which they affect EEG activity. Indeed, TNFα seems to participate in the control of NMDA receptor presence at the synaptic membrane as well as in the modulation of associated excitatory postsynaptic currents (Citation88).
Clock genes: transcriptional regulators
Clock genes govern endogenous circadian rhythms via their molecular oscillations in various organs. However, in areas outside the hypothalamic suprachiasmatic nuclei in which the master circadian pacemaker resides, many clock genes are expressed in response to neuronal activity, similar to IEG, and may help adapting cellular functions to metabolic demand (for review, see (Citation89)). The clock-independent role of clock genes in sleep homeostasis has been addressed elsewhere (Citation47,Citation90). Here, we will emphasize the impact of the relationship between clock genes and sleep homeostasis on the transcriptional response associated with elevated sleep need.
On the one hand, evidence for the role of clock genes in sleep homeostasis comes from studies in knock-out mice, in which the absence of one or two clock genes was shown to alter the distribution of wakefulness and sleep, the EEG activity during sleep, or the response to SD. For instance, mice lacking Bmal1 expressed an increase in total sleep time, sleep fragmentation, and EEG delta power under baseline conditions (Citation91). Npas2****/**** mice showed a blunted response to SD indexed by both low delta frequencies and Per2 expression (Citation26). Mice lacking both Cry1 and Cry2 also showed a blunted delta power rebound after SD, which accompanied a higher general level of delta during NREM sleep (Citation36). Interestingly, these animals also show elevated expression of TNFα likely due to enhanced activation of NFκB (Citation92), which supports, in addition to data presented above, a bidirectional molecular regulation between cytokines and clock elements. Of note, a Per3 polymorphism in humans has also been linked to changes in delta power during sleep (Citation35). On the other hand, wakefulness and sleep duration impact clock genes expression. Indeed, SD was repeatedly shown to increase the expression of Per genes in the mouse brain (Citation26,Citation37–41). Moreover, these changes depended on the duration of enforced wakefulness and were observed in various mouse strains (Citation46) highlighting their robustness. However, we showed that some of these changes (e.g. Per1, Per3) were mostly driven by the corticosterone surge produced by SD (Citation39). The clock-controlled gene Dbp has also consistently been observed to decrease with SD (Citation39,Citation40). Our observation that the binding of the core clock element CLOCK (or NPAS2) and BMAL1 to specific clock genes (i.e. Per2, Dbp) in the mouse cortex is altered by SD supports the involvement of these transcriptional regulators in the response to sleep loss (Citation93).
It is not surprising that clock genes respond to neuronal activity because of their capacity to adapt the transcriptome to cellular conditions via their function of transcriptional regulators (reviewed in (Citation94)). Indeed, the core feedback loop of this self-sustained molecular oscillation involves DNA-binding basic helix-loop-helix Per-Arnt-Sim (PAS)-domain transcription factors (CLOCK and homolog NPAS2; BMAL1 and homolog BMAL2). CLOCK/NPAS2 and BMAL1/BMAL2 dimerize to bind specific elements on DNA (E-boxes) and activate transcription. These dimers activate the transcription of Per, Cry, Rev-Erb, and Ror genes, but also the expression of a variety of clock-controlled genes (Citation89,Citation94) with among them numerous transcription factors, such as Dbp and Ppar (Citation94,Citation95), affecting themselves the expression of the genome and thus producing an amplification of the effect on gene expression. The protein products of Per and Cry feed back on the CLOCK:BMAL1 complex to inhibit its transcriptional activity, while Rev-Erb and Ror products, also giving rise to transcription factors, generally repress and activate gene expression (Citation82). Thus, the activation of CLOCK and BMAL1 likely serves to amplify and coordinate the cellular response by way of affecting networks of transcriptional effectors. An additional manner by which clock components affect gene expression is their role in shaping chromatin structure. Indeed, CLOCK histone acetyl transferase activity was reported to be essential to the expression of some clock genes independent of its DNA-binding activity (Citation96,Citation97). This type of activity, which regulates the compaction of DNA, shows a unique potential to affect the expression of a substantial number of genes.
Even though their role in transcriptional regulation is established, the involvement of clock genes in shaping the EEG is not well understood. In order for clock elements to control EEG activity and the delta power response to sleep loss, they should directly or indirectly regulate the expression of cellular components affecting neuronal or glial activity, such as neuro- and gliotransmitter receptors, and synaptic proteins. A direct effect is indeed supported by recent observations. For instance, the presence of E-boxes was reported in the gene coding GluR1, an AMPA receptor (Citation98), and in the Neuroligin-1 gene (Citation41), which regulates glutamatergic transmission. Moreover, we observed that SD decreased the binding of CLOCK to the Neuroligin-1 gene (Citation41), providing a direct mechanism by which clock elements can affect neuronal communication as a function of wakefulness and sleep.
Protein synthesis and regulation
Protein synthesis and regulation has been proposed as an important mechanism underlying the role of sleep in memory consolidation (Citation99). The regulation of proteins, which are effectors of cellular function and widespread cell components, is intimately linked to brain plasticity and learning (Citation100). In addition, microarray studies identified pathways of protein regulation as responsive to extended wakefulness and sleep (see above). Protein regulation may thus represent an additional required pathway underlying the sleep recovery process and its associated EEG dynamics. The first papers interested in protein regulation with regard to sleep were published in the early 1970s and authors either blocked protein synthesis with inhibitors or reported an increased incorporation of radioactive amino acids into newly formed proteins during sleep (Citation101,Citation102). More recently, reports showing that, in the mammalian brain, the rate of protein synthesis associated with the occurrence of NREM sleep (Citation103,Citation104) set the pace for studies linking sleep to protein synthesis. A summary of findings with regard to sleep and protein synthesis inhibitors, protein synthesis effectors (mTOR), and protein folding will be presented here.
Protein synthesis inhibitors and sleep
Blockage of protein synthesis with anisomycin impairs memory consolidation (Citation105). This observation is reminiscent of the effect of SD, because total SD also impairs long-term potentiation (LTP) in the hippocampus and cerebral cortex (Citation106–109). In these same two brain areas, SD was also shown to decrease the protein level of plasticity players such as BDNF and CaMKII (Citation110). However, the administration of protein synthesis inhibitors has led to conflicting results regarding sleep, probably because the route and especially the topography of administration modulate the effect. First, intraperitoneal administration of anisomycin was shown to reduce deep NREM sleep and/or REM sleep (Citation111), while intracerebroventricular injection induced a fast and transient decrease of wakefulness and a reduction of NREM sleep followed by a long-lasting increase in REM sleep (Citation112). More recently, anisomycin administration in a NREM sleep-promoting region, the lateral preoptic area, increased NREM sleep and delta power (Citation113), and similar findings were made with intracerebroventricular infusion of another protein synthesis inhibitor, salubrinal (Citation114). Together, these last two studies point to a role for protein synthesis in the regulation of EEG activity during sleep.
Role of the mTOR pathway
A potential mechanism mediating the action of protein synthesis inhibitors on sleep resides in the mTOR (mammalian Target Of Rapamycin) pathway. This pathway involves key protein kinases (mTORC1, mTORC2) for the initiation of translation because they regulate the translation initiation complex eIF4F and favor the translation of mRNA coding for elongation proteins (for review, see (Citation115)). One microarray study pictured many characteristics of the relationship between this pathway and sleep in the hippocampus (Citation50). First, authors reported that SD decreased mTOR transcription. Second, they observed that SD also decreased the gene expression of other components of the translation machinery, such as translation initiation factors (e.g. eIF2a, eIF3s6ip, eIF4el3, eIF5). Last, they highlighted that SD decreased mTOR protein level, and especially phosphorylated mTOR, which is the active form. These observations strongly suggest that this molecular pathway underlies, at least in part, the adverse effects of sleep loss on memory, since the mTOR pathway seems required for several forms of brain plasticity (Citation100,Citation115).
This mechanism is also relevant to the cerebral cortex, where SD was shown to increase phosphorylation of eukaryotic elongation factor 2 (Citation116) and eukaryotic initiation factor 2 alpha (Citation117), which both importantly regulate translation. In the visual cortex, early sleep also promoted phosphorylation of eukaryotic elongation factor 2, and inhibition of mTOR during sleep blocked sleep-dependent plasticity following monocular deprivation (Citation118). These authors proposed that while wakefulness may be associated with increased transcription of plasticity-related transcripts (i.e. Arc, Bdnf), the subsequent first 2 h of sleep would rather show increased translation. Interestingly, the elements affecting transcriptional regulation, described above for their role in the sleep recovery process, have also been linked to the mTOR pathway. Indeed, cytokines, and particularly TNFα, activate the Akt/mTORC1 pathway (Citation119,Citation120). In addition, mTOR has been shown to regulate clock proteins in both the fruit fly (Citation121) and mammals (Citation122). Furthermore, rhythmic activity of mTOR may suggest regulation by clock elements (Citation123). These observations support that molecular routes activated by prolonged wakefulness drive both transcriptional and translational responses and that this synergy is likely necessary to adapt sleep intensity and brain plasticity.
However, although the role of the mTOR pathway in sleep- dependent brain plasticity seems well supported (Citation118), the manner by which these changes may regulate the sleep EEG as a function of neuronal activity remains to be defined. To understand the contribution of elements of the mTOR pathway to delta power and sleep need, translational effectors will need to be turned off in the course of SD or at the beginning of recovery sleep through inhibitor administration or genetic engineering. Nevertheless, the fact that protein synthesis mediated by the mTOR pathway can occur in close proximity to spines in dendrites (Citation124) provides a mechanism by which this pathway may tune synaptic activity and thus overall neuronal synchrony. Interestingly, the mTOR pathway has been shown to regulate central elements of synaptic transmission, like glutamate receptor activity and adhesion molecules (Citation125,Citation126). The importance of these synaptic elements in the EEG response to sleep loss and their relationship to the mTOR pathway will be reviewed in a subsequent section.
Unfolded protein response and sleep homeostasis
Another manner by which protein regulation may be involved in the regulation of sleep need is via the unfolded protein response (UPR). This response, which engages the endoplasmic reticulum stress response, helps to restore normal endoplasmic reticulum function by up-regulating the expression of chaperones to increase endoplasmic reticulum capacity for protein folding, or to promote degradation of misfolded proteins through the process of endoplasmic reticulum-associated degradation (Citation127). It is closely linked to protein synthesis because it freezes protein synthesis via interaction with the mTOR pathway (Citation128). Prolonged wakefulness was repeatedly shown to activate the UPR as it increases the expression of heat-shock proteins (e.g. Hspa5, Hspa8, Dnajb5) and various chaperones (e.g. Cirpb, Bip) (Citation37,Citation39,Citation49,Citation50,Citation129). Moreover, in the fruit fly, overexpression of the chaperone BiP led to extended recovery sleep after SD (Citation129). Interestingly, the up-regulation of the UPR after SD seems also present in non-neuronal tissues (Citation130). Hence, during prolonged wakefulness, decreased protein synthesis and elevated UPR may contribute to the negative impact of sleep loss on cognition. Their potential functional links to the SD-dependent EEG response will be highlighted hereafter.
Mechanisms of plasticity at the junction of transcription and translation
Relationship between synaptic plasticity and sleep
The extensive changes in EEG synchrony as a function of sleep and wakefulness, including those observed for delta activity during NREM sleep, necessarily depend on mechanisms controlling overall neuronal communication. Therefore, elements changing synaptic activity need to be involved in the sleep recovery process. One of the first papers tentatively describing the effect of sleep on neuronal plasticity was published in 1899 (Citation131). It proposed an effect of the different phases of cerebral activity and sleep on the amoeboid movement (e.g. structural plasticity) of the protoplasmic prolongations (e.g. dendrites and axons) of neurons. At that time, the author presented a controversy regarding the occurrence of plasticity. Nowadays, the controversy is no longer about the occurrence of plasticity as a function of sleep and wakefulness but rather about the mechanisms behind it.
The synaptic homeostasis hypothesis presented in detail by Tononi and Cirelli in 2006 (Citation132) highlighted a theory describing the function of sleep. According to this hypothesis, wakefulness, characterized by high neuronal activity, would potentiate synapses and provide a source of high sleep intensity in subsequent sleep as measured by SWA. Then, SWA would enable synaptic down- regulation in parallel with declining sleep intensity. This hypothesis is supported with extensive and robust data sets that have been reviewed more recently (Citation133). For instance, during wakefulness (or SD), the expression of plasticity markers (e.g. Arc, Fos, Bdnf) is increased, as is the case for hallmarks of synaptic potentiation (e.g. AMPA receptor subunit level and their phosphorylation). Nevertheless, synaptic depression and potentiation are not strictly features of the sleep and wake states, respectively, and have been observed in both states and state-like models (Citation134,Citation135). Also, different types of plasticity may be independently favored by wakefulness or sleep (Citation134,Citation136). The observation that both wakefulness and sleep may trigger synaptic potentiation suggests that elevated synaptic strength in itself may not be the sole mechanism underlying the build-up of SWA during wakefulness and its decay during sleep. The integrated transcriptional and translational response to prolonged high neuronal activity (i.e. elevated sleep need) shows a unique potential to impact many synaptic pathways and thus to regulate SWA dynamics.
Importance of glutamatergic receptors in shaping the EEG
Glutamate is the main excitatory neurotransmitter in the brain and the most well-known source of synaptic plasticity through activation of NMDA and AMPA receptors (Citation137). On the one hand, the activity of these receptors, and particularly that of NMDA receptors due to their property of calcium channels, was shown to impact both gene and protein regulation (Citation138,Citation139). This is due to the effect of NMDA receptors on multiple intracellular signaling pathways (MAPK, CREB, NFκB) and underscores their role in the modulation of synaptic connectivity and associated learning. Since, in the cerebral cortex, glutamate level is high during wakefulness and low during NREM sleep (Citation140), the glutamate receptor-dependent effect on gene and protein expression will vary accordingly. Of particular interest, glutamate, NMDA or AMPA receptors can activate clock gene expression (Citation141,Citation142), TNFα production (Citation143,Citation144), the mTOR pathway (Citation145), and the UPR (Citation146), providing mechanisms by which neuronal activity associated with wakefulness can produce large changes in gene as well as protein expression as a function of wakefulness and sleep ().
On the other hand, the expression and membrane targeting of glutamate receptors is regulated by pathways involving the same key molecular elements, thus providing a cellular feedback loop of neuronal excitability. For instance, TNFα and IL-1β increase synaptic AMPA and NMDA receptors (Citation147,Citation148), the UPR controls the mobilization of AMPA receptors (Citation149), the circadian expression of NMDA receptors suggests a regulation by clock components (Citation141), and mTOR modulates AMPA receptor function (Citation150). Furthermore, changes in NMDA and AMPA receptor functioning may represent a mechanism by which neuronal activity shapes the EEG dynamics. Indeed, studies indicate that the relative contribution of NMDA and AMPA receptors could determine the degree of synchrony between cortical neurons (Citation151,Citation152). High NMDA receptor contribution would favor neuronal desynchrony reminiscent of wakefulness due to their slower dynamics because of their need to be activated as a function of membrane potential and their faster inactivation following neuronal activity (Citation151,Citation153); whereas higher relative contribution of AMPA receptors would favor neuronal synchrony as the one observed during deep sleep because of their faster dynamics (Citation151,Citation153,Citation154). In fact, SD decreases NMDA receptor-dependent forms of plasticity and directly decreases NMDA receptor function and membrane targeting (Citation107,Citation108). Besides, blocking NMDA receptors using various antagonists induces deep sleep, increases SWA during sleep, and prevents the proper dissipation of SWA (Citation155,Citation156). Similarly, anesthesia achieved via NMDA receptor blockade using ketamine generates a hyper-synchronized state reminiscent of deep sleep (Citation157). Although the physiological relevance of these changes in the context of sleep homeostasis is not clearly established, these data strongly support a role for NMDA receptors in shaping the EEG. During sleep, low glutamate transmission could allow recovery of NMDA receptor function required for the increased NMDA to AMPA receptor contribution and SWA dissipation. To support this, NMDA receptor function has been observed to be potentiated during a sleep-like state in an in vitro system (Citation158), in which a down-regulation of AMPA receptor activity was also reported (Citation159).
This neuronal activity feedback loop involving neuronal excitation by glutamate receptors and subsequent regulation of the function of these receptors may thus represent a mechanism underlying changes in cortical synchronization in function of the sleep/wake history. However, this oversimplified model does not incorporate the role of the delicate balance between excitation and inhibition in shaping cortical synchrony (Citation160). Indeed, GABAergic transmission has also been shown to contribute to SWA during NREM sleep (Citation161). In addition, other wake-promoting peptides (orexin, dopamine, acetylcholine) are undoubtedly contributing to the transcriptional and translational patterns observed during wakefulness and sleep as well as after extended wakefulness.
Adhesion molecules: a component of the relationship between the synapse and sleep
As highlighted above, glutamatergic synapses, and others types of synapses, are finely regulated as a function of neuronal activity. This is done by the interaction of complex pre- and postsynaptic machineries. In the fruit fly, synaptic proteins were shown to follow the distribution of sleep and wakefulness and to respond to SD (Citation162). In mammals, one synaptic adhesion system, the Neurexin/Neuroligin system, is required for plasticity because it regulates trans-synaptic and calcium signaling, neurotransmitter release, and receptor recruitment (Citation163,Citation164). Within this system, we recently focused on Neuroligin-1 because it is specifically required for proper NMDA receptor functioning as its down-regulation impairs the NMDA receptor-mediated excitatory postsynaptic current (Citation165,Citation166) and associative fear memory and spatial learning (Citation167).
We observed that elevated sleep need decreased the forebrain expression of Neuroligin-1 containing an insert in splice site B in three different inbred mouse strains (Citation41), providing a potential route by which SD impairs NMDA receptor function. This SD-dependent decrease was also observed in the cerebral cortex specifically (Citation168) and linked to changes in Neuroligin-1 protein level at the synapse (Citation41). Most importantly, Neuroligin-1-deficient mice showed a reduced capacity to sustain wakefulness accompanied with an amplified delta power response to sleep loss, which supports a role for NMDA receptor full functionality in maintaining adequate wakefulness and normal EEG activity. Although we identified a pathway that may underlie the SD-dependent decrease in Neuroligin-1 mRNA expression that likely involves transcriptional regulation by clock genes (Citation41), the translation of Neuroligin-1 was recently reported to depend on the mTOR pathway (Citation125), which, as highlighted, is equally relevant to sleep and wakefulness. In brief, our recent data point to a role of synaptic adhesion molecules in plastic events occurring across the sleep-wake cycle. A better understanding of molecular changes at the synapse should help to reconstruct the time course of those plastic events and clarify the bidirectional relationship between sleep and synaptic activity.
Conclusion
In this review, we provide evidence supporting the importance of two main cellular mechanisms in the recovery process of sleep regulation: transcription and translation. Cytokines, clock genes, and components of the mTOR pathway and UPR are, among others, responsive to wakefulness and sleep and habilitated to impact on complex molecular networks capable of shaping synaptic activity. This cross-talk between transcription and translation regulation renders sleep a state particularly suited to control the passage of short-term to long-term memory (Citation115). Our recent finding that genome-wide transcriptional changes with SD may originate from modification of the epigenome (Citation168) suggests an additional manner by which sleep loss can impact neuropsychiatric state and brain development.
Beyond neurons, these cellular processes operate in other brain cells where their relevance to sleep-wake regulation is first demonstrated by the SD-dependent transcriptional changes recently reported specifically in oligodendrocytes (Citation54). In addition, astrocytes are active components of the synapse that release gliotransmitters such as glutamate (Citation169,Citation170), which release was shown to be regulated by TNFα (Citation171). Astrocytes not only express the transcription factor NFκB (see above) but also release D-serine, a NMDA receptor co-activator, and participate in synaptic plasticity (Citation172). Indeed, astrocytes shape neuronal activity by way of modulating NMDA receptor function (Citation173,Citation174) and were accordingly shown to define EEG synchrony (Citation173,Citation175). Importantly, the impact of SD on the expression of astrocyte-specific genes regulating glycogen and of genes regulating lactate in astrocytes also supports the role of this cell type in adjusting brain metabolism as a function of wakefulness and sleep (Citation176–178). Although the exact contribution of astrocytes to the integrated transcriptional and translational response driven by elevated sleep need remains to be determined, their role in sleep homeostasis is no longer questionable () (Citation172,Citation179).
In summary, we highlighted that, during wakefulness, NMDA receptor stimulation, among others, activate several mechanisms regulating both transcription and translation, and that these likely modulate, directly or indirectly via for instance Neuroligins, the functioning of NMDA and AMPA receptors. These transcriptional and translational mechanisms, taking place in both neurons and non-neuronal brain cells, provide a coordinated route for the effect of neuronal activity during wakefulness on EEG synchrony during sleep as well as for the role of sleep in brain function and cognition.
Acknowledgements
We thank Erika Bélanger-Nelson and Dr Derk-Jan Dijk for useful comments on the manuscript, Dr Paul Franken for help regarding the revision, and Gaétan Tremblay for technical help with the figures.
Funding
V.M. is supported by a salary award from the Fonds de la recherche du Québec – Santé and by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada.
Declaration of interest: The authors report no conflicts of interest.
References
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26.
- Ohlmann KK, O’Sullivan MI. The costs of short sleep. AAOHN J. 2009;57:381–5.
- Palagini L, Rosenlicht N. Sleep, dreaming, and mental health: a review of historical and neurobiological perspectives. Sleep Med Rev. 2011; 15:179–86.
- Jagannath A, Peirson SN, Foster RG. Sleep and circadian rhythm disruption in neuropsychiatric illness. Curr Opin Neurobiol. 2013;23:888–94.
- Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204.
- Daan S, Beersma DG, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246(2 Pt 2):R161–83.
- Dunlap JC. Molecular biology of circadian pacemaker systems. In: Dunlap JC, Loros JJ, DeCoursey PJ, editors. Chronobiology: biological timekeeping. Sunderland: Sinauer Associates; 2004. p. 213–53.
- Ralph MR, Foster RG, Davis FC, Menaker M. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247:975–8.
- Dijk DJ, Duffy JF, Czeisler CA. Circadian and sleep/wake dependent aspects of subjective alertness and cognitive performance. J Sleep Res. 1992;1:112–17.
- Dijk DJ, Czeisler CA. Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neurosci Lett. 1994;166:63–8.
- Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15(5 Pt 1):3526–38.
- Achermann P, Dijk DJ, Brunner DP, Borbély AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull. 1993;31:97–113.
- Beersma DG. Models of human sleep regulation. Sleep Med Rev. 1998;2:31–43.
- Borbély AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–68.
- Pappenheimer JR, Koski G, Fencl V, Karnovsky ML, Krueger J. Extraction of sleep-promoting factor S from cerebrospinal fluid and from brains of sleep-deprived animals. J Neurophysiol. 1975;38:1299–311.
- Borbély AA, Baumann F, Brandeis D, Strauch I, Lehmann D. Sleep deprivation: effect on sleep stages and EEG power density in man. Electroencephalogr Clin Neurophysiol. 1981;51:483–95.
- Dijk DJ, Beersma DG, Daan S. EEG power density during nap sleep: reflection of an hourglass measuring the duration of prior wakefulness. J Biol Rhythms. 1987;2:207–19.
- Dijk DJ, Hayes B, Czeisler CA. Dynamics of electroencephalographic sleep spindles and slow wave activity in men: effect of sleep deprivation. Brain Res. 1993;626:190–9.
- Kattler H, Dijk DJ, Borbély AA. Effect of unilateral somatosensory stimulation prior to sleep on the sleep EEG in humans. J Sleep Res. 1994;3:159–64.
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81.
- Vyazovskiy VV, Tobler I. Handedness leads to interhemispheric EEG asymmetry during sleep in the rat. J Neurophysiol. 2008;99:969–75.
- Hanlon EC, Faraguna U, Vyazovskiy VV, Tononi G, Cirelli C. Effects of skilled training on sleep slow wave activity and cortical gene expression in the rat. Sleep. 2009;32:719–29.
- Holz J, Piosczyk H, Feige B, Spiegelhalder K, Baglioni C, Riemann D, et al. EEG Σ and slow-wave activity during NREM sleep correlate with overnight declarative and procedural memory consolidation. J Sleep Res. 2012;21:612–19.
- Mascetti L, Muto V, Matarazzo L, Foret A, Ziegler E, Albouy G, et al. The impact of visual perceptual learning on sleep and local slow-wave initiation. J Neurosci. 2013;33:3323–31.
- Landolt HP, Rétey JV, Tönz K, Gottselig JM, Khatami R, Buckelmüller I, et al. Caffeine attenuates waking and sleep electroencephalographic markers of sleep homeostasis in humans. Neuropsychopharmacology. 2004;29:1933–9.
- Franken P, Dudley CA, Estill SJ, Barakat M, Thomason R, O’Hara BF, McKnight SL. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci U S A. 2006;103:7118–23.
- Tinguely G, Finelli LA, Landolt HP, Borbély AA, Achermann P. Functional EEG topography in sleep and waking: state-dependent and state-independent features. Neuroimage. 2006;32:283–92.
- Knoblauch V, Kräuchi K, Renz C, Wirz-Justice A, Cajochen C. Homeostatic control of slow-wave and spindle frequency activity during human sleep: effect of differential sleep pressure and brain topography. Cereb Cortex. 2002;12:1092–100.
- Cajochen C, Brunner DP, Kräuchi K, Graw P, Wirz-Justice A. Power density in theta/alpha frequencies of the waking EEG progressively increases during sustained wakefulness. Sleep. 1995;18:890–4.
- Dumont M, Macchi MM, Carrier J, Lafrance C, Hébert M. Time course of narrow frequency bands in the waking EEG during sleep deprivation. Neuroreport. 1999;10:403–7.
- Finelli LA, Baumann H, Borbély AA, Achermann P. Dual electroencephalogram markers of human sleep homeostasis: correlation between theta activity in waking and slow-wave activity in sleep. Neuroscience. 2000;101:523–9.
- Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM. EEG delta wave power: an independent sleep phenotype or epiphenomenon?J Clin Sleep Med. 2011;7:S16–18.
- Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–21.
- Bachmann V, Klein C, Bodenmann S, Schäfer N, Berger W, Brugger P, et al. The BDNF Val66Met polymorphism modulates sleep intensity: EEG frequency- and state-specificity. Sleep. 2012;35:335–44.
- Viola AU, Archer SN, James LM, Groeger JA, Lo JC, Skene DJ, et al. PER3 polymorphism predicts sleep structure and waking performance. Curr Biol. 2007;17:613–18.
- Wisor JP, O’Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, et al. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002; 3:20.
- Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, et al. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–5.
- Thompson CL, Wisor JP, Lee CK, Pathak SD, Gerashchenko D, Smith KA, et al. Molecular and anatomical signatures of sleep deprivation in the mouse brain. Front Neurosci. 2010;4:165.
- Mongrain V, Hernandez SA, Pradervand S, Dorsaz S, Curie T, Hagiwara G, et al. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep. 2010;33:1147–57.
- Curie T, Mongrain V, Dorsaz S, Mang G, Emmenegger Y, Franken P. Homeostatic and circadian contributions to EEG and molecular state variables of sleep regulation. Sleep. 2013;36:311–23.
- El Helou J, Bélanger-Nelson E, Freyburger M, Dorsaz S, Curie T, La Spada F, et al. Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci U S A. 2013;110:9974–9.
- Pompeiano M, Cirelli C, Tononi G. Immediate-early genes in spontaneous wakefulness and sleep: expression of c-fos and NGFI-A mRNA and protein. J Sleep Res. 1994;3:80–96.
- Terao A, Greco MA, Davis RW, Heller HC, Kilduff TS. Region-specific changes in immediate early gene expression in response to sleep deprivation and recovery sleep in the mouse brain. Neuroscience. 2003; 120:1115–24.
- Mackiewicz M, Paigen B, Naidoo N, Pack AI. Analysis of the QTL for sleep homeostasis in mice: Homer1a is a likely candidate. Physiol Genomics. 2008;33:91–9.
- Nelson SE, Duricka DL, Campbell K, Churchill L, Krueger JM. Homer1a and 1bc levels in the rat somatosensory cortex vary with the time of day and sleep loss. Neurosci Lett. 2004;367:105–8.
- Franken P, Thomason R, Heller HC, O’Hara BF. A non-circadian role for clock-genes in sleep homeostasis: a strain comparison. BMC Neurosci. 2007;8:87.
- Franken P, Dijk DJ. Circadian clock genes and sleep homeostasis. Eur J Neurosci. 2009;29:1820–9.
- Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41: 35–43.
- Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, et al. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics. 2007;31:441–57.
- Vecsey CG, Peixoto L, Choi JH, Wimmer M, Jaganath D, Hernandez PJ, et al. Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol Genomics. 2012; 44:981–91.
- Mackiewicz M, Zimmerman JE, Shockley KR, Churchill GA, Pack AI. What are microarrays teaching us about sleep?Trends Mol Med. 2009; 15:79–87.
- Wang H, Liu Y, Briesemann M, Yan J. Computational analysis of gene regulation in animal sleep deprivation. Physiol Genomics. 2010;42: 427–36.
- Hinard V, Mikhail C, Pradervand S, Curie T, Houtkooper RH, Auwerx J, et al. Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J Neurosci. 2012;32:12506–17.
- Bellesi M, Pfister-Genskow M, Maret S, Keles S, Tononi G, Cirelli C. Effects of sleep and wake on oligodendrocytes and their precursors. J Neurosci. 2013;33:14288–300.
- Möller-Levet CS, Archer SN, Bucca G, Laing EE, Slak A, Kabiljo R, et al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc Natl Acad Sci U S A. 2013;110:E1132–41.
- Krueger JM, Pappenheimer JR, Karnovsky ML. Sleep-promoting factor S: purification and properties. Proc Natl Acad Sci U S A. 1978;75:5235–8.
- Krueger JM, Bacsik J, García-Arrarás J. Sleep-promoting material from human urine and its relation to factor S from brain. Am J Physiol. 1980;238:E116–23.
- Krueger JM, Pappenheimer JR, Karnovsky ML. Sleep-promoting effects of muramyl peptides. Proc Natl Acad Sci U S A. 1982; 79:6102–6.
- Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L. Sleep- promoting effects of endogenous pyrogen (interleukin-1). Am J Physiol. 1984;246(6 Pt 2):R994–9.
- Shoham S, Davenne D, Cady AB, Dinarello CA, Krueger JM. Recombinant tumor necrosis factor and interleukin 1 enhance slow-wave sleep. Am J Physiol. 1987;253(1 Pt 2):R142–9.
- Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, et al. Involvement of cytokines in slow wave sleep. Prog Brain Res. 2011;193:39–47.
- Taishi P, Churchill L, De A, Obal F Jr, Krueger JM. Cytokine mRNA induction by interleukin-1beta or tumor necrosis factor alpha in vitro and in vivo. Brain Res. 2008;1226:89–98.
- Krueger JM. The role of cytokines in sleep regulation. Curr Pharm Des. 2008;14:3408–16.
- Baracchi F, Opp MR. Sleep-wake behavior and responses to sleep deprivation of mice lacking both interleukin-1 beta receptor 1 and tumor necrosis factor-alpha receptor 1. Brain Behav Immun. 2008;22:982–93.
- Kapás L, Bohnet SG, Traynor TR, Majde JA, Szentirmai E, Magrath P, et al. Spontaneous and influenza virus-induced sleep are altered in TNF-alpha double-receptor deficient mice. J Appl Physiol. 2008;105: 1187–98.
- Fang J, Wang Y, Krueger JM. Effects of interleukin-1 beta on sleep are mediated by the type I receptor. Am J Physiol. 1998;274(3 Pt 2): R655–60.
- Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNF alpha treatment. J Neurosci. 1997;17: 5949–55.
- Hu J, Chen Z, Gorczynski CP, Gorczynski LY, Kai Y, Lee L, et al. Sleep-deprived mice show altered cytokine production manifest by perturbations in serum IL-1ra, TNFa, and IL-6 levels. Brain Behav Immun. 2003;17:498–504.
- Patel SR, Zhu X, Storfer-Isser A, Mehra R, Jenny NS, Tracy R, et al. Sleep duration and biomarkers of inflammation. Sleep. 2009; 32:200–4.
- Darko DF, Miller JC, Gallen C, White J, Koziol J, Brown SJ, et al. Sleep electroencephalogram delta-frequency amplitude, night plasma levels of tumor necrosis factor alpha, and human immunodeficiency virus infection. Proc Natl Acad Sci U S A. 1995;92:12080–4.
- Reincke M, Arlt W, Heppner C, Petzke F, Chrousos GP, Allolio B. Neuroendocrine dysfunction in African trypanosomiasis. The role of cytokines. Ann N Y Acad Sci. 1998;840:809–21.
- Yoshida H, Peterfi Z, García-García F, Kirkpatrick R, Yasuda T, Krueger JM. State-specific asymmetries in EEG slow wave activity induced by local application of TNFalpha. Brain Res. 2004;1009: 129–36.
- Yasuda T, Yoshida H, Garcia-Garcia F, Kay D, Krueger JM.Interleukin-1beta has a role in cerebral cortical state-dependent electroencephalographic slow-wave activity. Sleep. 2005;28:177–84.
- Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X, et al. TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res. 2007;1156:125–32.
- Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797.
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–16.
- Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. 2000;75:965–72.
- Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24:1–7.
- Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Sleep and brain energy levels: ATP changes during sleep. J Neurosci. 2010; 30:9007–16.
- Kalinchuk AV, McCarley RW, Porkka-Heiskanen T, Basheer R. The time course of adenosine, nitric oxide (NO) and inducible NO synthase changes in the brain with sleep loss and their role in the non-rapid eye movement sleep homeostatic cascade. J Neurochem. 2011;116:260–72.
- Mercurio F, Manning AM. Multiple signals converging on NF-kappaB. Curr Opin Cell Biol. 1999;11:226–32.
- Milne GR, Palmer TM. Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. ScientificWorldJournal. 2011;11:320–39.
- Choi S, Friedman WJ. Inflammatory cytokines IL-1β and TNF-α regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN Neuro. 2009;1(2).
- Ghosh M, Yang Y, Rothstein JD, Robinson MB. Nuclear factor-κB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J Neurosci. 2011;31:9159–69.
- Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1:212–22.
- Cavadini G, Petrzilka S, Kohler P, Jud C, Tobler I, Birchler T, et al. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc Natl Acad Sci U S A. 2007;104:12843–8.
- Petrzilka S, Taraborrelli C, Cavadini G, Fontana A, Birchler T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J Biol Rhythms. 2009;24:283–94.
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–90.
- Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci. 2010;123(Pt 22):3837–48.
- Franken P. A role for clock genes in sleep homeostasis. Curr Opin Neurobiol. 2013;23:864–72.
- Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409.
- Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, Verma IM. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc Natl Acad Sci U S A. 2012;109: 12662–7.
- Mongrain V, La Spada F, Curie T, Franken P. Sleep loss reduces the DNA-binding activity of BMAL1, CLOCK and NPAS2 to specific clock genes in the mouse cerebral cortex. PLoS One. 2011;6:e26622.
- Lowrey PL, Takahashi JS. Genetics of circadian rhythms in mammalian model organisms. Adv Genet. 2011;74:175–230.
- Guillaumond F, Becquet D, Boyer B, Bosler O, Delaunay F, Franc JL, et al. DNA microarray analysis and functional profile of pituitary transcriptome under core-clock protein BMAL1 control. Chronobiol Int. 2012;29:103–30.
- Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125:497–508.
- Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. 2010;17:1414–21.
- Lin CH, Lee EH. JNK1 inhibits GluR1 expression and GluR1-mediated calcium influx through phosphorylation and stabilization of Hes-1. J Neurosci. 2012;32:1826–46.
- Seibt J, Frank MG. Translation regulation in sleep: making experience last. Commun Integr Biol. 2012;5:491–5.
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75.
- Bobillier P, Sakai F, Seguin S, Jouvet M. Deprivation of paradoxical sleep and in vitro cerebral protein synthesis in the rat. Life Sci II. 1971;10:1349–57.
- Rojas-Ram–rez JA, Aguilar-Jim–nez E, Posadas-Andrews A, Bernal-Pedraza JG, Drucker-Col–n RR. The effects of various protein synthesis inhibitors on the sleep-wake cycle of rats. Psychopharmacology (Berl). 1977;53:147–50.
- Ramm P, Smith CT. Rates of cerebral protein synthesis are linked to slow wave sleep in the rat. Physiol Behav. 1990;48:749–53.
- Nakanishi H, Sun Y, Nakamura RK, Mori K, Ito M, Suda S. Positive correlations between cerebral protein synthesis rates and deep sleep in Macaca mulatta. Eur J Neurosci. 1997;9:271–9.
- Agnihotri NT, Hawkins RD, Kandel ER, Kentros C. The long-term stability of new hippocampal place fields requires new protein synthesis. Proc Natl Acad Sci U S A. 2004;101:3656–61.
- Campbell IG, Guinan MJ, Horowitz JM. Sleep deprivation impairs long-term potentiation in rat hippocampal slices. J Neurophysiol. 2002;88:1073–6.
- Kopp C, Longordo F, Nicholson JR, Lüthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci. 2006;26:12456–65.
- McDermott CM, Hardy MN, Bazan NG, Magee JC. Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J Physiol. 2006;570(Pt 3):553–65.
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8.
- Zhang L, Zhang HQ, Liang XY, Zhang HF, Zhang T, Liu FE. Melatonin ameliorates cognitive impairment induced by sleep deprivation in rats: role of oxidative stress, BDNF and CaMKII. Behav Brain Res. 2013;256C:72–81.
- Uezu E, Sano A, Matsumoto J. Effects of inhibitor of protein synthesis on sleep in rats. Tokushima J Exp Med. 1981;28:9–16.
- Stern WC, Morgane PJ, Panksepp J, Zolovick AJ, Jalowiec JE. Elevation of REM sleep following inhibition of protein synthesis. Brain Res. 1972;47:254–8.
- Methippara MM, Alam MN, Kumar S, Bashir T, Szymusiak R, McGinty D. Administration of the protein synthesis inhibitor, anisomycin, has distinct sleep-promoting effects in lateral preoptic and perifornical hypothalamic sites in rats. Neuroscience. 2008;151:1–11.
- Methippara MM, Bashir T, Kumar S, Alam N, Szymusiak R, McGinty D. Salubrinal, an inhibitor of protein synthesis, promotes deep slow wave sleep. Am J Physiol Regul Integr Comp Physiol. 2009;296:R178–84.
- Costa-Mattioli M, Sonenberg N. Translational control of gene expression: a molecular switch for memory storage. Prog Brain Res. 2008;169:81–95.
- Grønli J, Dagestad G, Milde AM, Murison R, Bramham CR. Post-transcriptional effects and interactions between chronic mild stress and acute sleep deprivation: regulation of translation factor and cytoplasmic polyadenylation element-binding protein phosphorylation. Behav Brain Res. 2012;235:251–62.
- Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92:1150–7.
- Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, et al. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol. 2012;22:676–82.
- Plaisance I, Morandi C, Murigande C, Brink M. TNF-alpha increases protein content in C2C12 and primary myotubes by enhancing protein translation via the TNF-R1, PI3K, and MEK. Am J Physiol Endocrinol Metab. 2008;294:E241–50.
- Wang CH, Cao GF, Jiang Q, Yao J. TNF-α promotes human retinal pigment epithelial (RPE) cell migration by inducing matrix metallopeptidase 9 (MMP-9) expression through activation of Akt/mTORC1 signaling. Biochem Biophys Res Commun. 2012;425:33–8.
- Zheng X, Sehgal A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr Biol. 2010;20:1203–8.
- Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, et al. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron. 2013;79: 712–24.
- Cao R, Anderson FE, Jung YJ, Dziema H, Obrietan K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience. 2011;181:79–88.
- Martin KC, Zukin RS. RNA trafficking and local protein synthesis in dendrites: an overview. J Neurosci. 2006;26:7131–4.
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–7.
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR- dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64.
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–99.
- Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol. 2012; 22:R622–6.
- Naidoo N, Casiano V, Cater J, Zimmerman J, Pack AI. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep. 2007;30:557–65.
- Anafi RC, Pellegrino R, Shockley KR, Romer M, Tufik S, Pack AI. Sleep is not just for the brain: transcriptional responses to sleep in peripheral tissues. BMC Genomics. 2013;14:362.
- A new theory of cerebral activity and sleep. Br Med J. 1899;1:93–5.
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62.
- Tononi G, Cirelli C. Time to be SHY? Some comments on sleep and synaptic homeostasis. Neural Plast. 2012;2012:415250.
- Frank MG. Erasing synapses in sleep: is it time to be SHY?Neural Plast. 2012;2012:264378.
- Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012; 75:1105–13.
- Donlea JM, Thimgan MS, Suzuki Y, Gottschalk L, Shaw PJ. Inducing sleep by remote control facilitates memory consolidation in Drosophila. Science. 2011;332:1571–6.
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26.
- Karpova A, Bär J, Kreutz MR. Long-distance signaling from synapse to nucleus via protein messengers. Adv Exp Med Biol. 2012;970:355–76.
- Bengtson CP, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. 2012;970:377–405.
- Dash MB, Douglas CL, Vyazovskiy VV, Cirelli C, Tononi G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J Neurosci. 2009;29:620–9.
- Bendová Z, Sládek M, Svobodová I. The expression of NR2B subunit of NMDA receptor in the suprachiasmatic nucleus of Wistar rats and its role in glutamate-induced CREB and ERK1/2 phosphorylation. Neurochem Int. 2012;61:43–7.
- Paul KN, Fukuhara C, Karom M, Tosini G, Albers HE. AMPA/kainate receptor antagonist DNQX blocks the acute increase of Per2 mRNA levels in most but not all areas of the SCN. Brain Res Mol Brain Res. 2005;139:129–36.
- De A, Krueger JM, Simasko SM. Glutamate induces the expression and release of tumor necrosis factor-alpha in cultured hypothalamic cells. Brain Res. 2005;1053:54–61.
- Vladychenskaya E, Tyulina O, Urano S, Boldyrev A. Rat lymphocytes express NMDA receptors that take part in regulation of cytokine production. Cell Biochem Funct. 2011;29:527–33.
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–15.
- Choe ES, Ahn SM, Yang JH, Go BS, Wang JQ. Linking cocaine to endoplasmic reticulum in striatal neurons: role of glutamate receptors. Basal Ganglia. 2011;1:59–63.
- Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–70.
- Lai AY, Swayze RD, El-Husseini A, Song C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 2006;175:97–106.
- Shim J, Umemura T, Nothstein E, Rongo C. The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol Biol Cell. 2004;15:4818–28.
- Ran I, Gkogkas CG, Vasuta C, Tartas M, Khoutorsky A, Laplante I, et al. Selective regulation of GluA subunit synthesis and AMPA receptor-mediated synaptic function and plasticity by the translation repressor 4E-BP2 in hippocampal pyramidal cells. J Neurosci. 2013;33: 1872–86.
- Golomb D, Shedmi A, Curtu R, Ermentrout GB. Persistent synchronized bursting activity in cortical tissues with low magnesium concentration: a modeling study. J Neurophysiol. 2006;95:1049–67.
- Harsch A, Robinson HP. Postsynaptic variability of firing in rat cortical neurons: the roles of input synchronization and synaptic NMDA receptor conductance. J Neurosci. 2000;20:6181–92.
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47.
- Fuchs EC, Doheny H, Faulkner H, Caputi A, Traub RD, Bibbig A, et al. Genetically altered AMPA-type glutamate receptor kinetics in interneurons disrupt long-range synchrony of gamma oscillation. Proc Natl Acad Sci U S A. 2001;98:3571–6.
- Campbell IG, Feinberg I. NREM delta stimulation following MK-801 is a response of sleep systems. J Neurophysiol. 1996;76:3714–20.
- Campbell IG, Feinberg I. Noncompetitive NMDA channel blockade during waking intensely stimulates NREM delta. J Pharmacol Exp Ther. 1996;276:737–42.
- Chauvette S, Crochet S, Volgushev M, Timofeev I. Properties of slow oscillation during slow-wave sleep and anesthesia in cats. J Neurosci. 2011;31:14998–5008.
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574(Pt 1):283–90.
- Czarnecki A, Birtoli B, Ulrich D. Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J Physiol. 2007;578(Pt 2):471–9.
- Tahvildari B, Wölfel M, Duque A, McCormick DA. Selective functional interactions between excitatory and inhibitory cortical neurons and differential contribution to persistent activity of the slow oscillation. J Neurosci. 2012;32:12165–79.
- Winsky-Sommerer R. Role of GABAA receptors in the physiology and pharmacology of sleep. Eur J Neurosci. 2009;29:1779–94.
- Gilestro GF, Tononi G, Cirelli C. Widespread changes in synaptic markers as a function of sleep and wakefulness in Drosophila. Science. 2009;324:109–12.
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52.
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–20.
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–31.
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A. 2008;105:20947–52.
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30:2115–29.
- Massart R, Freyburger M, Suderman M, Paquet J, El Helou J, Bélanger-Nelson E, et al. The genome-wide landscape of DNA methylation and hydroxymethylation in response to sleep deprivation impacts on synaptic plasticity genes. Transl Psychiatry. accepted 2013/11/10.
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369: 744–7.
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci U S A. 2004;101:9441–6.
- Santello M, Bezzi P, Volterra A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001.
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–55.
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, et al. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A. 2009;106:15037–42.
- Deng Q, Terunuma M, Fellin T, Moss SJ, Haydon PG. Astrocytic activation of A1 receptors regulates the surface expression of NMDA receptors through a Src kinase dependent pathway. Glia. 2011;59:1084–93.
- Amzica F, Massimini M. Glial and neuronal interactions during slow wave and paroxysmal activities in the neocortex. Cereb Cortex. 2002;12:1101–13.
- Petit JM, Tobler I, Allaman I, Borbély AA, Magistretti PJ. Sleep deprivation modulates brain mRNAs encoding genes of glycogen metabolism. Eur J Neurosci. 2002;16:1163–7.
- Petit JM, Tobler I, Kopp C, Morgenthaler F, Borbély AA, Magistretti PJ. Metabolic response of the cerebral cortex following gentle sleep deprivation and modafinil administration. Sleep. 2010;33:901–8.
- Petit JM, Gyger J, Burlet-Godinot S, Fiumelli H, Martin JL, Magistretti PJ. Genes involved in the astrocyte-neuron lactate shuttle (ANLS) are specifically regulated in cortical astrocytes following sleep deprivation in mice. Sleep. 2013;36:1445–58.
- Frank MG. Astroglial regulation of sleep homeostasis. Curr Opin Neurobiol. 2013;23:812–18.