
Abstract
Large granular lymphocytic (LGL) leukemia is an incurable chronic disease, characterized by clonal expansion of cytotoxic T- or NK-cells in blood and bone marrow. Cytopenias (anemia, neutropenia) and autoimmune disorders such as rheumatoid arthritis are the most common clinical manifestations of LGL leukemia. Recently, somatic activating STAT3 gene mutations were shown to be specific for LGL leukemia with a prevalence of up to 70%. Analogous mutations in the STAT5b gene were seen in a smaller proportion of patients. These gain-of-function mutations are located in the SH2 domain of STAT3 and affect the phosphotyrosine–SH2 interaction required for dimerization of STAT3. The mutations increase the phosphorylation of STAT3 and STAT5b and enhance the transcriptional activity of the mutated proteins. STAT3 and STAT5b mutations can be used as molecular markers for LGL leukemia diagnostics, and they present novel therapeutic targets for STAT3 and STAT5b inhibitors, which currently are in development for treatment of cancer and autoimmune disorders.
Key messages
Somatic, gain-of-function STAT3 mutations are seen in 11%–70% of LGL leukemia patients and STAT5b mutations in 2% of LGL leukemia patients.
STAT3 and STAT5b mutations can be used as diagnostic markers of LGL leukemia and facilitate the development of targeted therapies.
Introduction
Large granular lymphocytic (LGL) leukemia is a chronic disorder of either mature cytotoxic CD3 + T-cells or CD3neg natural killer (NK)-cells infiltrating blood and bone marrow (Citation1). The median age of onset is 60 years, and the prevalence is estimated to be 2%–5% of chronic lymphoproliferative diseases in North America (Citation2). The diagnosis, according to the World Health Organization (WHO) 2008 classification, is based on persistent peripheral LGL lymphocytosis (> 0.5 × 109/L) and, in the case of T-LGL, clonal rearrangement of T-cell receptor (TCR) (Citation3). Considering NK-LGL leukemia, WHO distinguishes chronic lymphoproliferative disease of NK-cells (CLPD-NK) from more aggressive, often rapidly progressing and chemorefractory NK-LGL leukemia. Typical clinical and hematological characteristics including anemia, neutropenia, and autoimmune disorders (such as rheumatoid arthritis) further support the diagnosis.
Leukemic T-LGLs display the phenotype of mature, terminal-effector memory cells. Normally these cells undergo programmed apoptosis by activation-induced cell death (AICD) (Citation4). It is hypothesized that after the original antigen-driven immune response and expansion of LGLs, the dysregulation of several signaling pathways sustains a constitutively active clonal cell population, which is resistant to Fas-mediated apoptosis (Citation5–8). One of these key proteins in LGL leukemia is signal transducer and activator of transcription 3 (STAT3). Leukemic LGLs have been shown to constitutively express phospho-STAT3, and STAT3 inhibition resulted in apoptosis of leukemic cells (Citation5). The molecular basis for these observations was recently uncovered with the identification of common somatic, gain-of-function STAT3 mutations in 40%–70% of T-LGL and 30% of NK-LGL leukemia patients (Citation5,Citation9,Citation10). Similar mutations in the STAT5b gene are seen in a smaller proportion of LGL leukemia patients. The purpose of this review is to provide an overview of STAT3 and STAT5b in the molecular pathogenesis of LGL leukemia.
STAT3 and STAT5b signaling
Both STAT3 and STAT5b genes are located in the chromosome 17 in adjacent regions. The coded proteins are transcription factors, which reside in the cytoplasm as latent monomers. Their protein structure is similar consisting of coiled-coil, DNA-binding, linker, SRC-like homology 2 (SH2), and transcriptional activation domain (TAD) () (Citation11). STAT3 exists in two major isoforms: STAT3α and a truncated form STAT3β, which lacks the TAD and seems to work in a dominant-negative manner and has also specific functions of its own (Citation12). STAT5b protein does not have isoforms, but it shares a sequence identity of over 90% in protein level with STAT5a (Citation13).
Figure 1. The protein domains of STAT3α (A) and STAT5b (B) with the mutated amino acids detected in cancer are presented in a linear model.
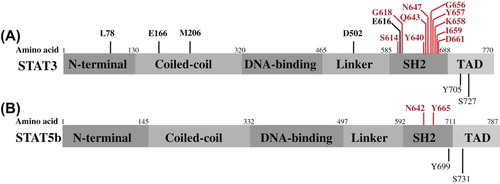
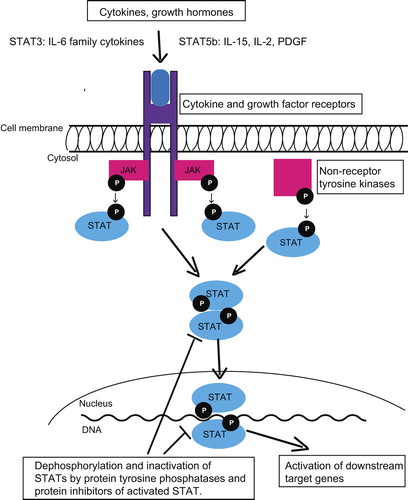
The most important upstream activators of STAT3 and STAT5b are different cytokines, growth factors, and cytoplasmic non-receptor tyrosine kinases, which have either intrinsic or associated tyrosine kinase activity () (Citation14). In a typical activation cascade after cytokine receptor stimuli Janus-activated kinases (JAKs) phosphorylate a single tyrosine residue (Tyr705 of STAT3 and Tyr699 of STAT5b), triggering conformational change required for the dimerization through reciprocal phosphotyrosine–SH2 domain interaction (Citation15). Phosphorylated dimers translocate to the nucleus, bound to the consensus DNA motifs, and activate a number of downstream pathways, which are involved in apoptosis, cell cycle, proliferation, cellular transformation, and inflammation () (Citation16,Citation17). Phosphorylation of Ser727 residue of STAT3 is also likely to affect the transcriptional activity of dimerized STAT3, depending on the cellular context (Citation18). Activated downstream pathways are specific to the cell type, and although upstream regulators of STAT3 and STAT5b are different, based on the ChIPSeq data 90% of STAT5 binding sites are co-occupied with STAT3 in T-cells (Citation19). STAT3 and STAT5b activation is reversed by phosphatase, the suppressor of cytokine signaling (SOCS), and protein inhibitors of STAT (PIAS) protein families (Citation20).
Figure 2. STAT3 and STAT5b signaling cascade.
STAT3 in lymphocytes, cancer, and autoimmunity
STAT3 is essential for development, and total Stat3 ablation leads to early embryonic lethality in mice (Citation21). Targeted disruption of STAT3 function in T-cells results in impaired IL-6-mediated survival and reduced proliferative response when stimulated with IL-2 (Citation22,Citation23). In humans, heterozygous germ-line mutations in DNA-binding and SH2 domain of STAT3 are seen in patients with hyper-IgE syndrome (Citation24,Citation25). Hyper-IgE is characterized by recurrent staphylococcal infections, eczema, cyst-forming pneumonias, and high serum-IgE. The current hypothesis is that these heterozygous mutations affect STAT3 function in a dominant-negative manner: mononuclear cells extracted from hyper-IgE patients respond poorly to IL-6 stimulation, and co-expression of mutated protein with wild-type (wt) STAT3 in different cell lines leads to impaired DNA-binding and transcriptional ability of STAT3 (Citation24,Citation25).
Tyrosine kinases are often overactive in cancer due to genetic or epigenetic alterations, and STAT3 activation is consequently seen in many malignancies as a secondary event, caused mainly by autonomous IL-6 and SRC kinase activation loops. The effects of constitutively active STAT3 depend on the cell type and co-activating proteins: STAT3 activates transcription of downstream anti-apoptotic proteins such as BCL-XL and MCL1, prevents cell cycle arrest through MYC, and down-regulates transcription of the common tumor suppressor TP53 (Citation26). Aberrant STAT3 activation is often observed in head and neck cancer, multiple myeloma, breast cancer, and many hematological malignancies (Citation26–32). However, somatic STAT3 mutations have not been found in these malignancies in genome-scale sequencing of large patient cohorts (Citation33,Citation34).
The oncogenic potential of STAT3 itself was demonstrated in a paper by Bromberg et al., in which a spontaneously dimerizing and constitutively active STAT3-molecule, STAT3-C, was created by substituting two amino acids (A662 and N664) in the SH2 domain of STAT3 with cysteine (Citation35). STAT3-C protein transformed fibroblasts independent of phosphorylation status, and these cells were able to form tumors in nude mice.
In addition to cancer, IL-6-STAT3 activation has been observed in many autoimmune and chronic inflammatory diseases. STAT3 is required for the TH17 immune response, and aberrant function of TH17 helper T-cells is linked with STAT3 overexpression in rheumatoid arthritis, psoriasis, multiple sclerosis, and Crohn's disease (Citation36–40).
Somatic STAT3 mutations in LGL leukemia and other malignancies
Leukemic LGLs express constitutively active STAT3, and STAT3 inhibition leads to decreased MCL-1 expression, restored FAS sensitivity, and apoptosis of leukemic cells (Citation5). The actual cause for constitutive STAT3 activation in LGL leukemia was recently discovered, as somatic STAT3 mutations were found in up to 70% of T-LGL and 30%–40% of NK-LGL leukemia patients () (Citation9,Citation10,Citation41–43). The most common STAT3 mutations observed were Y640F and different missense mutations in the D661 codon. Notably, the reported prevalence of STAT3 mutations in these studies differs significantly. Based on our data, the mutations were more common in patients with one large monoclonal LGL expansion (Citation10). Some patients who meet the diagnostic criteria of LGL leukemia harbor multiple smaller lymphocyte expansions, and thus the discrepancy in mutation frequencies can be related to the differences between patient cohorts and also to the insensitivity of capillary sequencing techniques, which require that at least 20%–30% of the cells in the sample represent the mutated clone.
Table I. Somatic STAT3 mutations reported in different studies.
The mutations detected in LGL leukemia patients are located in exons 20 and 21 in the SH2 domain of STAT3, and are different from the inactivating mutations identified in hyper-IgE syndrome ( and ). Based on functional analyses, mutated STAT3 is constitutively phosphorylated and transcriptionally active when compared to the wt STAT3 (Citation9,Citation10,Citation44). The effects of STAT3 mutation Y640F have also recently been studied in mice. The expression of Y640F-mutated STAT3 led to the development of myeloproliferative neoplasm in a murine retroviral transduction–bone marrow transplantation model, but no abnormalities were found in T- or NK-cell compartments (Citation45).
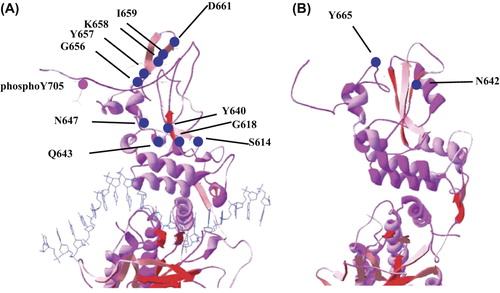
Figure 3. The STAT3 and STAT5b mutations seen in LGL leukemia patients are marked in blue dots in 3D model of STAT3 (A) and STAT5b (B) monomers. The STAT3 monomer is attached to target DNA, which is shown as a blue strand.
IL-6-STAT3 pathway mutations have also been observed in inflammatory hepatocellular adenomas (IHCA): 60% of these tumors have activating somatic IL-6 mutations, and 12% of wt IL-6 patients carry somatic STAT3 mutations (Citation44,Citation46). Two of these mutations are in the same amino acid as in LGL leukemia (K658Y, Y640F). In other tumor types studied thus far, STAT3 mutations have been rarely detected. Sequencing of CD30 + ALK-negative anaplastic large cell lymphomas (ALCL) and CD30 + peripheral T-cell lymphomas (PTCL) revealed STAT3 mutations in 2/18 and 2/8 cases, respectively (Citation42). In another patient series the STAT3 mutation frequency was 2/79 (2.5%) in diffuse large B-cell lymphoma and 4/258 (2%) in different T-cell lymphomas (Citation45). A novel activating STAT3 mutation M206K was detected when 40 patients with diffuse large B-cell lymphoma (DLBCL) were sequenced (Citation47). Screening of a large cohort of cancer patients with different solid tumors and hematological malignancies (n = 2677) excluding LGL leukemia revealed only two non-recurrent STAT3 mutations (G656D in a peripheral T-cell lymphoma case, and C643K in rectal adenocarcinoma) (Citation48). Different mutations found in other cancers are presented in and .
Somatic STAT3 mutations in aplastic anemia and myelodysplastic syndromes
Aplastic anemia (AA) is characterized by failing hematopoiesis and pancytopenia, which are thought to be caused by TH1 and cytotoxic lymphocytes inhibiting pluripotent hematopoietic precursor cells in the bone marrow (BM) (Citation49). Myelodysplastic syndromes (MDS) are a group of BM failure diseases with ineffective hematopoiesis, also originating from a primitive hematopoietic stem cell (Citation50). The pathogenetic events in MDS probably consist of a heterogenous mixture of somatic mutations, epigenetic changes, and deregulation of the immune system and BM stromal cells (Citation50). Interestingly, in a proportion of both AA and MDS patients skewing of T-cell receptor repertoire has been reported, and some of these patients meet the criteria of concomitant LGL leukemia (Citation51). In a recent study, the two most common STAT3 mutations in LGL leukemia (Y640F and D661Y) were screened by ARMS-PCR, and these mutations were detected in AA and MDS patients both with and without concurrent LGL leukemia (Citation52). The frequency of the mutations was 55% and 7% in AA (with and without LGL expansions, respectively), and 38% and 3% in MDS (Citation52). It is intriguing to speculate that these STAT3-mutated clonal lymphocyte expansions could indicate a presence of self-reacting cells targeting hematopoietic precursors in AA and MDS, but further studies are needed to understand the biological significance of the found mutations.
All somatic STAT3 mutations described above in different disease conditions are listed in and .
STAT5b in lymphocytes and cancer
STAT5b is required for the development and normal function of lymphocytes. Stat5b−/− mice are viable but display impaired pituitary growth hormone production, consequently diminished body growth, and diminished NK-cell count with maturation block in bone marrow and impaired NK-cell response to IL-2 and IL-15 (Citation53–55). Similar clinical features and lower number of NK-cells are seen in STAT5b-defective humans (Citation56,Citation57). Depletion of both Stat5a and Stat5b in mice leads to perinatal lethality, severe combined immunodeficiency, and lack of lymphoid cells (Citation58).
Similar to STAT3, constitutive STAT5 activation is also essential in many malignancies, and the mechanism of its activation has been studied in detail for example in chronic myeloid leukemia (CML) and other myeloproliferative neoplasms (Citation59,Citation60). In CML, STAT5 activation is a result of the oncogenic BCR-ABL1 fusion protein, and it confers anti-apoptotic effects through Bcl-XL (Citation61,Citation62). The inhibition of STAT5 induces apoptosis of CML cells (Citation63). In other myeloproliferative diseases (such as polycythemia vera, essential thrombocytosis, and myelofibrosis) STAT5 activation occurs commonly through activating JAK2 V617F mutation (Citation64). The mutation has been observed in over 90% of patients with polycythemia vera and in 30%–50% of cases with essential thrombocytosis or primary myelofibrosis (Citation64,Citation65). JAK2 is an upstream regulator of STAT5, and STAT5 activation is critical for the cellular transformation through V617F-mutated JAK2 (Citation66).
Two constitutively active STAT5 mutants (STAT5-C) have been described, both created by random mutagenesis ex vivo. The first of these had two amino acid substitutions: H299R in DNA-binding domain and S711F in TAD (Citation67). The mutated proteins were phosphorylated, localized in the nucleus of transfected cells, transcriptionally active, and able to sustain growth of IL-3-dependent cells in the absence of IL-3 (Citation67). The second STAT5-C, with one missense mutation N642H, showed similar properties both biochemically and biologically (Citation68). Notably, disruption of Tyr694 abolished all the acquired properties of STAT5-CN642H, indicating that phosphorylation and dimerization are essential for the aberrant function of the STAT5-C (Citation68). The authors also screened 49 patients with acute leukemia, but none harbored mutations in STAT5 genes (Citation69).
STAT5b mutations in LGL leukemia
Interestingly, LGL leukemia patients without STAT3 mutations display phosphorylated STAT3 (pSTAT3), and the comparison between mutated and unmutated patients has demonstrated very similar gene expression patterns (Citation9,Citation10) implying that patients without STAT3 mutations may have other mechanisms inducing STAT3 activation. Exome sequencing of STAT3-mutation-negative LGL leukemia patients led to the discovery of STAT5b mutations in LGL leukemia (Citation70). The two mutations found were N642H and Y665F, and they were located in exon 16 of STAT5b in the SH2 domain ( and ). To date, it seems that the frequency of STAT5b mutations is clearly lower compared to the incidence of STAT3 mutations (4/211, 2%) (Citation70). The Y665F mutation was detected in two untreated patients with CD56 + T-LGL, whereas the N642H mutation was found in two patients suffering from an atypical, aggressive chemorefractory form of LGL leukemia (CD56 + T-LGL and NK-LGL) (Citation70). In vitro, the N642H mutation demonstrated more prominent transcriptional activity in concordance with the clinical phenotype. The biochemical characteristics of the mutations and the phenotype of the patients have raised several questions: are STAT5b mutations typical for CD56 + expressing T- and NK-LGL? Is the N642H mutation associated with the aggressive clinical presentation? Further studies are needed to clarify these issues.
Although STAT5a and STAT5b are closely connected, no STAT5a mutations have been detected in LGL leukemia patients thus far (Citation70). Therefore, the genetic background of LGL leukemia without STAT3 and STAT5 mutations is unknown, and novel mutations affecting the same pathways may still be discovered when more patient samples are sequenced in detail (Citation71).
The origin and the role of STAT3 and STAT5b mutations in LGL leukemia
The initial events leading to the expansion of the mutated LGL clones are not clear. The role of self-antigens or viral infections as triggers of immune activation and clonal expansion has been suspected, but none of the theories have yet been experimentally confirmed. In general, the connection between cancer and certain infectious agents is well established. Epstein–Barr virus (EBV) is often associated with Hodgkin lymphomas and rare aggressive NK-LGL leukemia (also with the nasal form), and human papillomaviruses (HPV) with cervical cancer (Citation72–74). Interestingly, constitutive and aberrant STAT3 activation has been observed in aggressive nasal NK-LGL leukemia patients, and its inhibition led to the apoptosis of MEC04 cells, which are derived from a NK-LGL leukemia patient (Citation75). Similarly, STAT3 expression levels have been shown to increase in HPV16-infected cells when cervical lesions progress to cancer (Citation76). Increased levels of HTLV antibodies have been detected in LGL leukemia, but only in a minority of patients (< 5%) (Citation77). No other viral etiology has clearly been related to LGL leukemia.
In several autoimmune diseases, autoantigens are thought to play a role in disease initiation. The antigen target of LGL leukemia cells is not known, but based on the TCR repertoire no clear preference of the TCR usage has been observed (Citation78). It has been hypothesized that in LGL leukemia the initial polyclonal lymphocyte reaction may evolve first to oligoclonal and then to monoclonal expansion. It is possible that chronic immune activation leading to the amplification of the IL-6-STAT3 activation loop increases the incidence of somatic mutations in this specific pathway. This hypothesis is further supported by the fact that some LGL leukemia patients have multiple mutations in the SH2 domain of STAT3, existing either in the same expanded lymphocyte clone or in multiple clones (Citation43,Citation79).
In a network model of LGL cell survival signaling, IL-15 and platelet-derived growth factor (PDGF) (both upstream regulators of STAT5) were able to sustain leukemic population (Citation8). The role of IL-15 has also been demonstrated in a mouse model in which wt LGL cells cultured with IL-15 and transplanted to mice led to malignant transformation (Citation80). Prolonged exposure to IL-15 caused chromosomal instability through MYC-mediated up-regulation of aurora kinases, and the activation MYC/NF-κBp65/HDAC-signaling cascade repressed micro-RNA miR-29b, resulting in DNMT3 overexpression and DNA hypermethylation (Citation80). The four LGL leukemia patients with STAT5b mutations had either NK-type LGL leukemia, or CD8 + CD56+ T-LGL (Citation70). CD56 is typically a NK-cell surface antigen, but its expression in CD8 + T-cells is linked to more potent cytotoxic activity and NK-like behavior by means of MHC-unrestricted cytotoxicity (Citation81). It is intriguing to speculate that the IL-15 overexpression may induce STAT5b mutations through genomic instability, and also program cells to a NK-like phenotype.
Considering the correlation between the clinical features and STAT3 mutation status, it seemed that the STAT3-mutated patients had more often neutropenia and rheumatoid arthritis in a series of 77 T-LGL leukemia cases (Citation9). In a larger series of LGL leukemia patients consisting both of T- and NK-LGLs, patients with STAT3 mutation had required more lines of therapy during their course of the disease (Citation10). However, no statistically significant differences were observed in the overall survival between STAT3 mutated and non-mutated patients (Citation10).
STAT3 and STAT5b mutations as diagnostic tools in LGL leukemia
The diagnosis of LGL leukemia is sometimes difficult, as no specific molecular markers have existed. The clonality of T-cells can be determined by analyzing the T-cell receptor (TCR) repertoire using either PCR or flow cytometry, but a similar assay is not possible for NK-cells (Citation82,Citation83). The establishment of diagnosis often requires follow-up as reactive poly- or oligoclonal lymphocyte populations can be seen during viral infections, and tyrosine kinase inhibitor therapy (Citation84–86). Furthermore, the TCR repertoire narrows in elderly, and it has been shown that the prevalence of transient LGL-lymphocytosis correlates positively with age (Citation87). STAT3 and STAT5b mutations are the first molecular markers that are highly specific for LGL leukemia and can prove the clonality also in the case of NK-cell proliferations. None of the mutations identified is present in large genome-scale population-variation data sets, such as in the 1000 Genomes (www.1000genomes.org/data). In addition to conventional capillary screening, novel second-generation deep sequencing methods can be used for the screening of STAT3 mutations (Citation43). The sensitivity of amplicon sequencing is even 0.5%–1%, and it is superior to allele-specific PCR methods as it can detect all mutations in the area sequenced.
STAT3 and STAT5b as therapeutic targets in LGL leukemia
LGL leukemia patients are currently treated with lymphotoxic, immunosuppressive agents such as methotrexate, cyclosporine, and cyclophosphamide (Citation2). Although the disease is indolent, the majority of patients require treatment at some time point due to anemia, neutropenia, or autoimmune manifestations. The overall response rate for methotrexate, which is the most common first-line treatment, is approximately 50% in retrospective patient series, and relapses are common if the treatment is discontinued (Citation2).
STAT3 is a potential therapeutic target not only in cancer, but also in autoimmune disorders. STAT3-specific small molecule SH2-inhibitors have shown promising results in preclinical trials. A novel orally administered compound, BP-1-102, inhibited breast and lung cancer cells in cell culture and mouse xenografts by disrupting the STAT3 SH2–phosphotyrosine interaction and the dimerization of STAT3 (Citation88). Another inhibitor, OPB-3112, induced tumor-specific growth inhibition in leukemic cell lines HEL92.1.7, KU812, and TCCy/sr without affecting growth of normal human hematopoietic cells (Citation89). OPB-3112 has entered phase I/II clinical trials. Another way of STAT3 inhibition is the use of decoy oligonucleotides, which target the DNA binding domain and block the nuclear transfer of STAT3 (Citation90). This could be a potentially interesting approach in STAT3-mutated LGL leukemia, but it needs to be tested whether STAT3 mutations in the SH2 domain could affect the binding affinity of small molecule SH2-inhibitors. Some preliminary work has been done with two different cell lines carrying STAT3 mutations Y640F (YT1, NK/T-cell line) and G618R (FEDP, ALK-negative ALCL) (Citation45). STA-21 (a small-molecule STAT3-inhibitor inhibiting the SH2–phosphotyrosine interaction) was able to prevent the growth of both cell lines, suggesting that these mutations do not affect the binding of the drug significantly (Citation45).
STAT5 inhibitors, such as small molecule STAT5-SH2 domain inhibitors, are under development for the treatment of STAT5-dependent malignancies (Citation91,Citation92). However, there are not yet any specific STAT5 inhibitors in the clinical trials. Pimozide is an antipsychotic drug that has been identified as a STAT5 inhibitor in a high-throughput drug screen for STAT5-targeting drugs (Citation63). Its mechanism of action is unknown, but it has been able to induce apoptosis through BCR-ABL1-independent STAT5 inhibition in CML cells and also in FLT3-mutated AML model (Citation63,Citation93). Considering the STAT5b targeting in LGL leukemia, it is notable that the two patients discovered with the STAT5b N642H mutation suffered from an atypical, aggressive, and treatment-resistant form of LGL leukemia (Citation70). If the N642H is the driver mutation in their disease, targeted STAT5 inhibition could have significant therapeutic potential in this type of LGL leukemia.
No clinical studies targeting JAK-STAT pathway with available JAK inhibitors (such as ruxolitinib) have been done in LGL leukemia, although STAT3 activation was already discovered more than 10 years ago in LGL leukemia patients. In vitro, JAK2 inhibitor AG-490 induced apoptosis of leukemic LGLs with a corresponding decrease in STAT3-DNA binding activity, but, in addition to JAK2, AG-490 inhibits also other kinases such as epidermal growth factor receptor kinase (Citation94). Furthermore, STAT3 and STAT5b mutations seem to cause constitutive activation without upstream stimulation needed, and therefore it is possible that the inhibition of JAKs would not have adequate effect in LGL leukemia patients with activating STAT3 or STAT5b mutations (Citation9,Citation68–70).
Conclusion
A large proportion (30%–70%) of LGL leukemia patients have activating somatic mutations in the STAT3 gene, whereas STAT5b mutations are observed only in a minority of patients. STAT3 mutations seem fairly specific for LGL leukemia, although they have recently been found also in other bone marrow failure conditions such as in AA and MDS and in rare cases of other T-cell malignancies. STAT3 and STAT5 mutation status should be included in the diagnostic criteria of LGL leukemia along with other already established hematological and clinical factors. Further studies are needed to understand the initiating events of STAT3 mutations and their functional significance. The inhibition of STAT3 pathway is an attractive treatment target in LGL leukemia.
Declaration of interest: K.P. has received research funding and honoraria from Novartis and Bristol-Myers Squibb. S.M. has received honoraria from Novartis and Bristol-Myers Squibb.
References
- Loughran TP Jr, Kadin ME, Starkebaum G, Abkowitz JL, Clark EA, Disteche C, et al. Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med. 1985; 102:169–75.
- Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011; 117:2764–74.
- Mohan SR, Maciejewski JP. Diagnosis and therapy of neutropenia in large granular lymphocyte leukemia. Curr Opin Hematol. 2009;16: 27–34.
- Zhang J, Xu X, Liu Y. Activation-induced cell death in T cells and autoimmunity. Cell Mol Immunol. 2004;1:186–92.
- Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107:351–62.
- Yang J, Epling-Burnette PK, Painter JS, Zou J, Bai F, Wei S, et al. Antigen activation and impaired Fas-induced death-inducing signaling complex formation in T-large-granular lymphocyte leukemia. Blood. 2008;111:1610–16.
- Leblanc F, Zhang D, Liu X, Loughran TP. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol. 2012;8:787–801.
- Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, et al. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008;105:16308–13.
- Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–13.
- Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120:3048–57.
- Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2:536–50.
- Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401–9.
- Ambrosio R, Fimiani G, Monfregola J, Sanzari E, De Felice N, Salerno MC, et al. The structure of human STAT5A and B genes reveals two regions of nearly identical sequence and an alternative tissue specific STAT5B promoter. Gene. 2002;285:311–18.
- Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314.
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56.
- Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–73.
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
- Wen Z, Darnell JE Jr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–7.
- Kang K, Robinson GW, Hennighausen L. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genomics. 2013;14:4.
- Greenhalgh CJ, Hilton DJ. Negative regulation of cytokine signaling. J Leukoc Biol. 2001;70:348–56.
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–4.
- Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–60.
- Akaishi H, Takeda K, Kaisho T, Shineha R, Satomi S, Takeda J, et al. Defective IL-2-mediated IL-2 receptor alpha chain expression in Stat3-deficient T lymphocytes. Int Immunol. 1998;10:1747–51.
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19.
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62.
- Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105.
- Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19:2489–95.
- Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth In vitro. J Clin Invest. 1998;102:1385–92.
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10: 105–15.
- Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513.
- Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54.
- Kube D, Holtick U, Vockerodt M, Ahmadi T, Haier B, Behrmann I, et al. STAT3 is constitutively activated in Hodgkin cell lines. Blood. 2001;98:762–70.
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7.
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72.
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303.
- Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007; 179:4313–17.
- Gaston JS. Cytokines in arthritis—the ‘big numbers’ move centre stage. Rheumatology (Oxford). 2008;47:8–12.
- Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep. 2007;9:461–7.
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5.
- Schmechel S, Konrad A, Diegelmann J, Glas J, Wetzke M, Paschos E, et al. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL-22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis. 2008;14:204–12.
- Fasan A, Kern W, Grossmann V, Haferlach C, Haferlach T, Schnittger S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia. 2013;27:1598–600.
- Ohgami RS, Ma L, Merker JD, Martinez B, Zehnder JL, Arber DA. STAT3 mutations are frequent in CD30 + T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia. 2013;27:2244–7.
- Rajala HL, Eldfors S, Kuusanmaki H, Andersson EI, van Adrichem AJ, Lagstrom S, et al. Deep sequencing reveals small subclones of STAT3 mutations in large granular lymphocytic leukemia. European Hematology Association, 18th Congress of EHA, June 13–16, 2013, Stockholm, Sweden.
- Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JT, Paradis V, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011;208:1359–66.
- Couronne L, Scourzic L, Pilati C, Valle VD, Duffourd Y, Solary E, et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematologica. 2013;98:1748–52.
- Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200–4.
- Hu G, Witzig TE, Gupta M. A novel missense (M206K) STAT3 mutation in diffuse large B cell lymphoma deregulates STAT3 signaling. PLoS One. 2013;8:e67851.
- Kim MS, Lee SH, Yoo NJ, Lee SH. STAT3 exon 21 mutation is rare in common human cancers. Acta Oncol. 2013;52:1221–2.
- Young NS. Acquired aplastic anemia. Ann Intern Med. 2002;136: 534–46.
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872–85.
- Karadimitris A, Li K, Notaro R, Araten DJ, Nafa K, Thertulien R, et al. Association of clonal T-cell large granular lymphocyte disease and paroxysmal nocturnal haemoglobinuria (PNH): further evidence for a pathogenetic link between T cells, aplastic anaemia and PNH. Br J Haematol. 2001;115:1010–14.
- Jerez A, Clemente MJ, Makishima H, Rajala H, Gomez-Segui I, Olson T, et al. STAT3-mutations indicate the presence of subclinical T cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013;122:2453–9.
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94: 7239–44.
- Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188:2067–74.
- Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. 2011;117:1565–73.
- Hwa V, Camacho-Hubner C, Little BM, David A, Metherell LA, El-Khatib N, et al. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene. Horm Res. 2007;68:218–24.
- Kanai T, Jenks J, Nadeau KC. The STAT5b pathway defect and autoimmunity. Front Immunol. 2012;3:234.
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A. 2006;103:1000–5.
- Fabbro D. BCR-ABL signaling: a new STATus in CML. Nat Chem Biol. 2012;8:228–9.
- Rajala HL, Mustjoki S. STAT5b in LGL leukemia - a novel therapeutic target?. Oncotarget. 2013;4:808–9.
- Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96:2269–76.
- Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–54.
- Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011; 117:3421–9.
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97.
- Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32:2601–13.
- Funakoshi-Tago M, Tago K, Abe M, Sonoda Y, Kasahara T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J Biol Chem. 2010; 285:5296–307.
- Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–9.
- Ariyoshi K, Nosaka T, Yamada K, Onishi M, Oka Y, Miyajima A, et al. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J Biol Chem. 2000;275:24407–13.
- Yamada K, Ariyoshi K, Onishi M, Miyajima A, Hayakawa F, Towatari M, et al. Constitutively active STAT5A and STAT5B in vitro and in vivo: mutation of STAT5 is not a frequent cause of leukemogenesis. Int J Hematol. 2000;71:46–54.
- Rajala HL, Eldfors S, Kuusanmaki H, van Adrichem AJ, Olson T, Lagstrom S, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541–50.
- Andersson EI, Rajala HL, Eldfors S, Ellonen P, Olson T, Jerez A, et al. Novel somatic mutations in large granular lymphocytic leukemia affecting the STAT-pathway and T-cell activation. Blood Cancer J. 2013;3:e168.
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19.
- Saha A, Robertson ES. Epstein-Barr virus-associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res. 2011;17: 3056–63.
- Hart DN, Baker BW, Inglis MJ, Nimmo JC, Starling GC, Deacon E, et al. Epstein-Barr viral DNA in acute large granular lymphocyte (natural killer) leukemic cells. Blood. 1992;79:2116–23.
- Coppo P, Gouilleux-Gruart V, Huang Y, Bouhlal H, Bouamar H, Bouchet S, et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia. 2009;23:1667–78.
- Shukla S, Shishodia G, Mahata S, Hedau S, Pandey A, Bhambhani S, et al. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: implications in high-risk human papillomavirus infection. Mol Cancer. 2010;9:282.
- Thomas A, Perzova R, Abbott L, Benz P, Poiesz MJ, Dube S, et al. LGL leukemia and HTLV. AIDS Res Hum Retroviruses. 2010;26:33–40.
- O’Keefe CL, Plasilova M, Wlodarski M, Risitano AM, Rodriguez AR, Howe E, et al. Molecular analysis of TCR clonotypes in LGL: a clonal model for polyclonal responses. J Immunol. 2004;172:1960–9.
- Rajala HL, Eldfors S, Kuusanmaki H, Andersson EI, van Adrichem AJ, Lagstrom S, et al. Discovery of STAT5b mutations and small subclones of STAT3 mutations in large granular lymphocytic (LGL) leukemia. Blood (ASH Annual Meeting Abstracts). 2012;120(Abstract 871).
- Mishra A, Liu S, Sams GH, Curphey DP, Santhanam R, Rush LJ, et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell. 2012;22:645–55.
- Ohkawa T, Seki S, Dobashi H, Koike Y, Habu Y, Ami K, et al. Systematic characterization of human CD8 + T cells with natural killer cell markers in comparison with natural killer cells and normal CD8 + T cells. Immunology. 2001;103:281–90.
- Langerak AW, Groenen PJ, Bruggemann M, Beldjord K, Bellan C, Bonello L, et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26:2159–71.
- Langerak AW, van Den Beemd R, Wolvers-Tettero IL, Boor PP, van Lochem EG, Hooijkaas H, et al. Molecular and flow cytometric analysis of the Vbeta repertoire for clonality assessment in mature TCRalphabeta T-cell proliferations. Blood. 2001;98:165–73.
- Kreutzman A, Juvonen V, Kairisto V, Ekblom M, Stenke L, Seggewiss R, et al. Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood. 2010;116:772–82.
- Gillespie GM, Wills MR, Appay V, O’Callaghan C, Murphy M, Smith N, et al. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol. 2000;74:8140–50.
- Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia. 2009;23:1398–405.
- Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to “benign monoclonal gammapathy”. J Exp Med. 1994;179:609–18.
- Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–8.
- Hayakawa F, Sugimoto K, Kurahashi S, Sumida T, Naoe T. A novel STAT3 inhibitor OPB-31121 induces tumor-specific growth inhibition in a wide range of hematopoietic malignancies without growth suppression of normal hematopoietic cells. Blood (ASH Annual Meeting Abstracts). 2011;118(Abstract 577).
- Souissi I, Najjar I, Ah-Koon L, Schischmanoff PO, Lesage D, Le Coquil S, et al. A STAT3-decoy oligonucleotide induces cell death in a human colorectal carcinoma cell line by blocking nuclear transfer of STAT3 and STAT3-bound NF-kappaB. BMC Cell Biol. 2011;12:14.
- Page BD, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, et al. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem. 2012;55:1047–55.
- Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12:611–29.
- Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, et al. The STAT5 inhibitor pimozide displays efficacy in models of acute myelogenous leukemia driven by FLT3 mutations. Genes Cancer. 2012;3:503–11.
- Epling-Burnette PK, Catlett-Falcone R, Kothapalli R, Oshiro M, Jove R, Loughran TP. Induction of apoptosis in large granular lymphocyte (Lgl) leukemia by jak tyrosine kinase inhibitor AG-490: role of STAT3-regulated MCL-1. ISEH Annual Meeting: Experimental Hematology. 2000;28:90–1.