
Abstract
Rheumatoid arthritis (RA) is a destructive autoimmune disease that mainly affects synovial joints. RA patients can be subdivided in two distinct disease subsets based on the presence of anti-citrullinated protein antibodies (ACPA). These two disease phenotypes are associated with different environmental and genetic risk factors and clinical parameters. The HLA class II locus is the most important risk factor for ACPA-positive RA (ACPA+ RA). ACPA can be found up to 10 years before diagnosis and can be used as a predictive biomarker. During progression from breaking tolerance to a citrullinated protein to ACPA+ RA, the ACPA response matures. Recent work implicates the HLA class II locus as a risk factor in the progression from ACPA positivity to ACPA+ RA. We now propose that this locus directly influences the maturation of the ACPA response, most likely via antigen-specific T-cells providing help to ACPA-producing B-cells allowing for maturation of the citrullinated protein-specific autoantibody response. We present and discuss several models and underlying data, including antibody cross-reactivity, molecular mimicry, and neo-antigen formation, that could explain the HLA–RA connection.
Key messages
Rheumatoid arthritis (RA) can be subdivided in two distinct disease entities based on the presence of anti-citrullinated protein antibodies (ACPA).
Recent work suggests a role for CD4 + T-cells in the maturation of the ACPA response, which precedes the onset of ACPA + RA.
To understand disease pathogenesis and to establish novel therapeutics, identification of those CD4 + T-cell populations critically involved in the development of ACPA + RA is required.
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease characterized by extensive inflammation of the synovial joints. About 1% of the Western population is affected by this disease, resulting in a large disease burden. In the past decades seminal work was performed on the drivers of the inflammatory process, resulting in the development of a whole range of therapeutics that have greatly enhanced the quality of life of RA patients. Despite the identification of crucial pathways and the implementation of novel therapeutics, little is known about the factors driving the initiation of RA and about why the inflammation does not spontaneously resolve (Citation1).
RA is considered an autoimmune disease. Autoantibodies are an important hallmark of RA, and several classes of autoantibodies have been described that precede the development of RA. These autoantibodies include rheumatoid factor, anti-citrullinated protein antibodies (ACPA), and the recently identified anti- carbamylated protein antibodies (Citation2,Citation3).
ACPA are of particular interest as these autoantibodies are highly specific for RA and can be found in about 50% of early RA patients. This makes ACPA an important early clinical biomarker, and ACPA status was therefore recently added to the new criteria used for the classification of RA patients (Citation4). In addition, ACPA+ RA patients present with a faster rate of joint destruction. ACPA can therefore also be used as a biomarker for patients with a more severe disease phenotype and could be used to select those patients eligible for a more aggressive treatment (Citation5).
Upon comparing ACPA-positive with ACPA-negative RA patients, it was noted that these patient groups differ not only in clinical phenotype but also in genetic and environmental risk factors (Citation6,Citation7). It is therefore believed that these are two distinct disease subsets with a different underlying pathogenesis. In this review we will focus on the ACPA-positive form of RA (ACPA+ RA) as we will discuss the characteristics of the ACPA response and the influence of genetic risk factors.
ACPA, not just a biomarker
Pathogenic potential of ACPA
Because of the clear association between the rate of joint destruction and ACPA positivity, it has been suggested that ACPA may directly contribute to synovial inflammation. ACPA-producing B-cells are enriched in the synovial fluid, which suggests local production of ACPA in the synovial fluid and a direct role for ACPA and/or ACPA-producing B-cells in synovial inflammation (Citation8,Citation9).
The number of different ACPA isotypes is a predictor of radiographic damage (Citation10). As different isotypes can recruit different immune effector mechanisms, this suggests that ACPA can contribute to damage using multiple different effector pathways. For example, stimulation of osteoclast precursors, cells involved in the degradation of bone, with ACPA has been reported to result in increased osteoclastogenesis (Citation11). Supporting these reports, subclinical joint damage is already present in ACPA+ healthy subjects (Citation12). Enhanced osteoclastogenesis could provide a direct link between the presence of ACPA and more severe joint destruction. This was also confirmed in mice since injecting mice with experimental arthritis with ACPA aggravates arthritis severity (Citation13).
Many antibody effector mechanisms are implicated in the recruitment and activation of inflammatory cells. For instance, ACPA can form immune complexes with citrullinated proteins, which can activate inflammatory cells and induce the production of TNF-alpha (Citation14,Citation15). Also, it was reported that ACPA are of low avidity when compared to other antibodies, and the presence of low-avidity ACPA is correlated with a faster rate of joint progression. These low-avidity ACPA are particularly good in activating the complement system, and complement-mediated recruitment of inflammatory cells could therefore be an important effector mechanism of ACPA (Citation16–18).
Together, these data support the notion that ACPA are more than a biomarker for selecting patients with a more severe disease phenotype, as they could be directly involved in the inflammatory process. It is therefore important to understand the evolution and characteristics of the ACPA response.
Citrullination
In 1964, it was first shown that RA patients harbour very specific antibodies, which were called anti-perinuclear factor or anti-keratin antibodies (Citation19–21). In a search for synovial targets to which these autoantibodies bind, fibrinogen and vimentin were identified (Citation22,Citation23). Proteins can undergo many types of post-translational modifications (PTMs). In 1998, it was demonstrated that the described autoantibodies only target citrullinated antigens, hence their name anti-citrullinated protein antibodies (ACPA) (Citation24–26).
Citrullination is a physiological process catalysed by a family of enzymes called peptidylarginine deiminases (PADI1-4). These enzymes convert the positively charged amino-acid (aa) arginine to a novel uncharged aa called citrulline. This process is linked to several aspects of cell biology including apoptosis, necrosis, netosis, and histone modifications. PADI enzymes require relatively high concentrations of calcium for their activity allowing citrullination of the extracellular matrix when they are released from (dying) cells (Citation27,Citation28).
Several data indicate an important role for citrullination in ACPA + RA. Genome-wide association studies identified SNPs in the region encoding the PADI4 gene that are associated with an increased risk to develop ACPA + RA (Citation29). Also, smoking, the most important environmental risk factor, is described to associate with an increased PADI2 expression in bronchoalveolar lavage cells and bronchial mucosal biopsy sections (Citation30). Finally, it was recently shown that RA patients can carry autoantibodies specifically targeting PADI enzymes, which can enhance the activity of these enzymes allowing them to function at lower calcium concentrations enabling enhanced citrullination of the extracellular matrix when they are released from (dying) cells (Citation31).
The antigens targeted by ACPA
Since the identification of the molecular nature of the antigens targeted, much work has been performed on identifying potential citrullinated proteins recognized by ACPA. Unfortunately, this is hampered by the characteristics of the ACPA response. The ACPA repertoire is highly diverse as it displays reactivity against many different citrullinated proteins. This is explained both by the polyclonality of the ACPA response and by several reports that showed that ACPA molecules can directly cross-react between different citrullinated antigens (Citation9,Citation32–34).
An increasing list of citrullinated proteins identified within the synovial compartment and recognized by ACPA is now known, including proteins like collagen, vimentin, fibrinogen, enolase, fibronectin, vinculin, and histones (Citation22,Citation35–38). It is therefore highly likely that a plethora of citrullinated proteins are recognized by ACPA in the synovial compartment that could all conceivably contribute to synovial inflammation. However, due to the high degree of ACPA cross-reactivity, it is difficult to identify those citrullinated antigens that play a critical role in the initiation or development of ACPA+ RA.
Characteristics of the ACPA response
The presence of ACPA can precede RA onset by up to 10 years, without any clinical signs of arthritis (Citation39,Citation40). Interestingly, by comparing the characteristics of ACPA before and after disease onset, it has become clear that the ACPA response is, on average, not the same between these subjects. In RA patients, ACPA are increased in level, use more isotypes, display a different glycosylation pattern, and are more cross-reactive towards different citrullinated epitopes. Interestingly, the ‘maturation’ of the ACPA response takes place before disease onset. Prior to the start of clinical symptoms a sharp rise in ACPA levels, isotype usage, and epitope recognition profile is observed (Citation41).
These findings are in line with the notion that before disease onset the ACPA response undergoes a ‘second hit’, which suggests a role for a matured ACPA response in the onset and the pathogenesis of ACPA + RA. As an ‘immature’ ACPA response can be identified in serum up to 10 years before disease onset, this provides a potentially interesting therapeutic window for interventions preventing maturation of the ACPA response. In a search for such interventions, it is important to understand the progression from ACPA positivity without disease in healthy subjects to ACPA + RA.
A role for CD4 + T-cells
Genetic risk
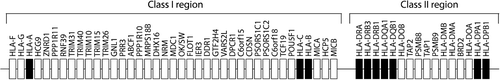
The most important genetic risk factor for RA is the human leukocyte antigen (HLA) class II locus. Importantly, it was shown that the HLA class II locus is strongly associated with ACPA+ RA, not with ACPA– RA (Citation6). The HLA locus is associated with most autoimmune diseases. This locus, located in chromosomal position 6p21, contains many different genes and is particularly well known for its strong linkage disequilibrium (LD) () (Citation42).
Figure 1. Schematic representation of the HLA locus. Scheme shows the different genes located in the class-I and class-II region of the HLA locus. Genes that encode classical HLA class I and class II molecules are indicated in black.
The shared epitope hypothesis
In 1976, it was first shown that the presence of HLA-DRB1*04 strongly predisposes to RA (Citation43). The HLA-DRB1 gene is in LD with genes encoding the alpha and the beta chain of HLA-DQ (HLA-DQA1 and HLA-DQB1), and these genes are inherited together in haplotypes. In 1986, the shared epitope (SE) hypothesis was postulated (Citation44). This hypothesis assumes that the HLA association is explained by polymorphisms in the HLA-DRB1 chain. Upon comparing different HLA-DRB1*04 alleles it was observed that these alleles differ mostly in aa position 70-74 (Citation45). Haplotypes encoding predisposing HLA-DRB1 alleles share the sequences QRRAA, QKRAA, and RRRAA at these positions and hence are collectively called HLA-SE alleles. It was postulated that this sequence directly influences peptide presentation or T-cell recognition (Citation44,Citation46).
Recently, the association of individual amino-acids with ACPA + RA was revisited by using a novel statistical approach leading to the formation of an ‘HLA-SE version 2.0’ hypothesis. Variations encoding amino-acids 11 and 13 of the HLA-DRB1 chain explain, statistically, most of the genetic risk followed by ‘SE aa’ 71 and 74. Based on these four amino-acid positions, RA patients can be subdivided in 16 groups, each with their own genetic risk profile (Citation47). The three groups with the highest odds ratios still harbour HLA-DR-DQ haplotypes encoding the SE alleles derived from HLA-DRB1*01, HLA-DRB1*04, and HLA-DRB1*10 alleles.
Notably, HLA-DRB1*04-positive RA patients are also HLA-DQ3-positive, and it has also been proposed that the predisposing effect of HLA-DR4 can be explained by its LD with genes encoding for HLA-DQ3 (Citation48). The HLA-DR/HLA-DQ linkage makes it difficult to identify which of the two molecules is responsible for the association with RA. Therefore, it is still unclear whether the HLA-DQ genes in linkage with the HLA-DR-encoding SE genes can be excluded from the association with ACPA + RA.
CD4 + T-cells
HLA class II molecules are involved in presenting peptides to CD4 + T-cells. HLA class II molecules contain a peptide-binding groove that can accommodate a wide range of peptide lengths. The part of the peptide that interacts with the binding groove is on average nine aa long. The aa side chains at positions 1, 4, 6, 7, and 9 interact with the corresponding pocket 1, pocket 4, pocket 6, pocket 7, and pocket 9 in the peptide binding groove. The aa shaping the pockets are most polymorphic. Therefore, each allele has a certain preference for accommodating particular aa in each of the pockets resulting in large differences the peptide ligands bound to different HLA class II molecules. Positions 2, 3, 5, and 8 of the peptide generally have a more upward configuration and can interact with the T-cell receptor (TCR) (Citation49).
HLA involvement in ACPA positivity or in ACPA-positive disease?
As mentioned above, the presence of ACPA can be found up to 10 years before disease onset. This suggests that the progression to ACPA + RA involves two ‘hits’. Initially individuals are healthy and ACPA-negative. After the ‘first hit’ they become positive for ACPA. Then, presumably after a ‘second hit’, individuals progress from ACPA positivity to ACPA+ RA, which is accompanied by a maturation of the ACPA response.
The HLA class II locus is strongly associated with ACPA+ RA, and understanding this association would provide important insights in disease pathogenesis. The association suggests an involvement of antigen-specific CD4 + T-cells. This is strengthened by genome-wide association studies that identified many genes involved in the regulation of adaptive immune responses (Citation50). In understanding the HLA class II association it is important to understand if this locus contributes to either the first or the second ‘hit’ thought to accompany ACPA positivity or ACPA-positive disease, respectively.
We know that CD4 + T-cells are important for maturation of antibody responses. Maturation of antibody responses occurs in complex structures in lymph nodes called germinal centres (GCs). The formation of a GC critically depends on the interaction between B- and activated T-cells (Citation51,Citation52). Within a GC, antigen-experienced T-cells produce cytokines that induce the proliferation and differentiation of stimulated B-cells. Also, during proliferation somatic hypermutation occurs. During this process, mutations are introduced in the variable region of immunoglobulin molecules resulting in different B-cell clones. These clones are selected based on the interaction with follicular dendritic cells retaining antigens. The selection of ‘mutated’ clones results in affinity maturation as the clones with the highest avidity for the antigen are selected. The low affinity of the ACPA response therefore suggests that ACPA-producing germinal centre B-cells do not undergo a stiff selection observed for pathogen-directed B-cells, conceivably as a consequence of the relative abundance of citrullinated antigens, thereby allowing low-affinity B-cells to survive selection. Cytokines produced by activated T-cells are also required for class-switch recombination potentially leading to isotype-switching and a different isotype usage (Citation53,Citation54).
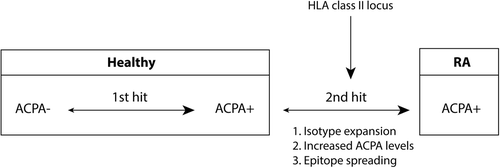
As antigen-experienced CD4 + T-cells are critically important for the generation of mature antibody responses, it seems likely that activated antigen-experienced (self-reactive) CD4 + T-cells are involved in the maturation of the ACPA response. Importantly, recent evidence suggests that the HLA class II locus is not associated with the risk to become ACPA+, but with the risk to progress from ACPA+ to ACPA+ RA (Citation55,Citation56). This supports the hypothesis that the HLA class II locus is not directly involved in the formation of ACPA responses, but rather in its maturation, possibly via T-cells providing help to ACPA-producing B-cells before the precipitation of disease ().
Figure 2. Schematic representation of the progression from healthy to ACPA+ RA. The HLA class II locus associates with the progression from ACPA positivity to ACPA+ RA. During this ‘second hit’ the ACPA response matures as shown by isotype expansion, increased ACPA levels, and epitope spreading. Together it supports a role for the HLA class II locus in the maturation of ACPA.
Towards a molecular basis for the HLA–RA association
T-cell selection and tolerance
Developing thymocytes undergo several rounds of selection resulting in the removal of a large part of the self-reactive T-cell repertoire.
After selection, T-cells leave the thymus and migrate to lymph nodes awaiting their specific antigen, which is required for their activation. Despite selection, a small subset of self-reactive T-cells that are predominantly thought to be directed to low- affinity self-antigens can escape negative selection and will become part of the peripheral T-cell repertoire (Citation57).
The activation of naive T-cells is tightly regulated to prevent activation of potentially autoreactive T-cells. For proper activation, T-cells need to recognize their peptide–HLA complex and receive co-stimulatory signals from APCs. The expression of co-stimulatory molecules by APCs is tightly regulated and enhanced in response to pathogen-derived danger signals (Citation58).
In RA, although other possibilities cannot be excluded, B-cell tolerance to self is apparently lost as ACPA are reactive to a wide variety of citrullinated self-proteins. However, it is unclear to what extent T-cell tolerance is lost. In several mechanisms are shown that could potentially explain the loss of B-cell tolerance in ACPA + RA. These mechanisms are discussed in the next paragraphs.
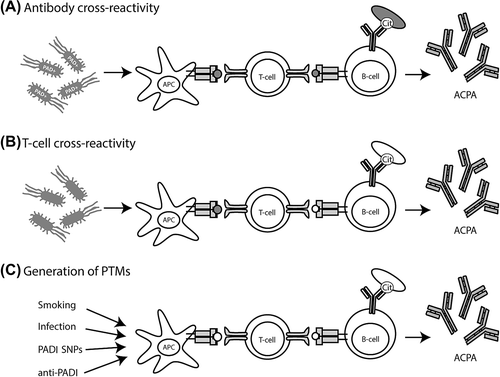
Figure 3. Models explaining loss of tolerance in RA. A: Infection and subsequent citrullination of microbial proteins results in the activation of microbe-directed T-cells. These microbe-directed T-cells can provide help to B-cells specific for a citrullinated microbial protein resulting in the production of ACPA cross-reactive to citrullinated self-proteins. Microbial proteins could become citrullinated by human or by bacterial PADI enzymes. B: Infection with microbes that have proteins with a high degree of similarities with self-protein could activate self-reactive T-cells. These self-reactive T-cells can subsequently provide help to B-cells specific for citrullinated self-proteins resulting in the production of ACPA. C: The amount of citrullinated proteins could be enhanced by smoking, infection with PADI-containing bacteria, polymorphisms within the PADI locus, or by anti-PADI antibodies. Enhanced citrullination of self-proteins occurs by several causes. Presentation of citrullinated neo-antigens by HLA-SE molecules activates citrulline-directed CD4 + T-cells that can help ACPA-producing B-cells. In each of the models antigens indicated in white are self-derived and antigens indicated in grey are pathogen-derived.
Model 1: ACPA-producing B-cells did not receive help from self-reactive CD4 + T-cells
As described, ACPA are highly cross-reactive both at a polyclonal and at a monoclonal level towards different citrullinated self- antigens. Likewise, we know that infection and subsequent apoptosis and necrosis can enhance the extracellular activity of PADI enzymes. Therefore, during infection, pathogen-derived proteins can conceivably become citrullinated by human PADI enzymes. In addition, bacteria can carry enzymes with PADI activity and can citrullinate their own proteins (Citation59–61).
It is therefore plausible that B-cells recognizing pathogen- derived citrullinated proteins are initially helped by CD4 + T-cells targeting (citrullinated) pathogen-derived epitopes. The self- reactive nature of ACPA could result from subsequent epitope spreading from foreign- to self-proteins that can also become citrullinated. In this model, the HLA class II restricted T-cells are conceivably not directed to self, but rather to foreign citrullinated epitopes, and T-cell tolerance was thus never breached ().
A well-known example in line with this model is found in celiac disease. Celiac disease patients often have autoantibodies targeting tissue transglutaminase. The pathogenic T-cells, however, do not target a self-protein, but are directed against food-derived gluten peptides that are deamidated by tissue transglutaminase (Citation62). Thus, in this case, the autoreactive B-cells attract T-cell help through the recruitment of T-cells recognizing non-self-proteins. In RA, several lines of evidence are in support of such a model. Some reports have suggested a role for Porphyromonas gingivalis, an oral bacterium that contains a PADI enzyme. ACPA can cross-react between epitopes derived for bacterial and human citrullinated alpha enolase, and it is conceivable in case enolase is involved in the breach of B-cell tolerance that the T-cells supporting ‘citrullinated enolase’-directed B-cell responses are directed against the bacterial protein (Citation63). Also, treatment of Epstein–Barr virus (EBV)-transformed lymphoblastoid cell lines with calcium ionophores induces the activation of PADI enzymes leading to the citrullination of EBV-derived proteins including the EBV antigen EBNA-1. Citrullinated EBNA-1 was shown to be a target of ACPA that are cross-reactive to citrullinated self-antigens (Citation64). Likewise, EBNA-1-specific CD4 + T-cells are well described and likely to be present in many individuals (Citation65,Citation66). Nonetheless, as mentioned, it is unclear if ACPA were initially generated to bacterial/viral proteins, and pathogen-directed T-cells that recognize (citrullinated) foreign proteins helping ACPA-producing B-cells have not been reported.
Model 2: Molecular mimicry-induced loss of tolerance
The production of ACPA could also result from provision of T-cell help by CD4 + T-cells reactive to self-peptides. In this case, T-cell tolerance would be lost. Naive T-cells, as compared to memory or effector T-cells, require higher concentrations of antigen to become properly activated (Citation67). Infection by pathogens induces the expression of co-stimulatory molecules and thereby lowers the strength of TCR signalling required to induce activation of naive T-cells. Molecular mimicry was therefore proposed as an important mechanism to activate naive T-cells expressing a T-cell receptor that could also recognize autoantigens (Citation68). Molecular mimicry occurs when a self-peptide closely resembles a pathogen-derived peptide. Infection by pathogens can then induce the activation of low-affinity self-reactive T-cells that are cross-reactive to self- peptides () (Citation69). These self-reactive T-cells could provide help to B-cells targeting (citrullinated) self-proteins that present an epitope from the (citrullinated) self-protein after BCR-mediated antigen uptake.
Several pathogens have been implicated in triggering autoimmune diseases. In multiple sclerosis it was first shown that human TCRs targeting the autoantigen myelin basic protein can cross-react with epitopes derived from different pathogens including influenza A virus, EBV, herpes simplex virus, adenovirus, and Pseudomonas aeruginosa. Also in RA, several pathogens have been proposed to trigger disease onset, including EBV, Proteus mirabilis, and Mycoplasma (Citation70). For some of these pathogens sequence homology with human proteins has been proposed to be important. Thus far, however, human T-cells cross-reactive between citrullinated joint antigens and viral or bacterial antigens have not been described.
Model 3: Neo-antigen formation
Another possible mechanism that could explain loss of tolerance is the peripheral introduction of protein post-translation modifications (PTMs) that do not occur in the thymus (). During selection, T-cells targeting the native protein are negatively selected, but T-cells recognizing PTMs possibly escape the thymus and can potentially become activated in the periphery.
Since ACPA are directed to citrullinated proteins, the presence of citrulline-reactive T-cells restricted to HLA-SE alleles has been proposed. The occurrence of citrullination in the thymus has been poorly studied. However, due to the tight association between citrullination and apoptosis, it is conceivable that citrullination occurs in the thymus since thymic selection and apoptosis are strongly linked.
As mentioned, susceptibility is associated with the HLA haplotypes encoding HLA-DR molecules that harbour a shared epitope motif at position 70-74 of the HLA-DRB1 beta chain. It was suggested that this motif is directly involved in antigen presentation (Citation44). Amino-acids 70 and 71 of the SE motif are particularly interesting because they are part of the p4 pocket and are therefore important for peptide presentation to CD4 + T-cells. It has been proposed that SE alleles predispose to RA because they have a more positively charged p4 pocket than non-HLA-SE alleles. The nature of the pocket would therefore prevent the accommodation of positively charged aa residues like arginine residues, while a neutrally charged citrulline residue would be accepted. Indeed, it has been shown for several peptides that the conversion of an arginine to a citrulline enhances the affinity of these peptides for the HLA-SE molecules (Citation71). Thus, citrullination of proteins could potentially allow for the presentation of citrullinated peptides that cannot be presented in their native form. These peptides could then be a target for citrulline-directed CD4 + T-cells that escaped negative selection.
The cross-reactive nature of ACPA hampers the identification of relevant citrullinated T-cell epitopes involved in helping B-cells. Currently, most work is performed on vimentin, aggrecan, and enolase. Using epitope prediction software, Vim59-71 and Vim66-78 were identified as potential T-cell epitopes with an arginine at p4. Upon immunization of HLA-DR4 humanized mice with cit-Vim59-78 (a peptide that combines the two epitopes) cit-Vim59-78-directed T-cells could be identified (Citation71,Citation72). T-cells interacting with vimentin-peptide-containing tetramers can also be detected in small numbers in HLA-DR4 + healthy controls and RA patients with similar frequencies. These T-cells have an activated phenotype (Citation72,Citation73). Similar observations were made for a mutated citrullinated aggrecan peptide cit-aggrecan84-103G92Y (Citation73–75).
Future work should be focused on identifying the importance of these tetramer-reactive cell populations in RA since important questions are still unanswered. It is unclear if the studied T-cell epitopes can be generated upon processing of the citrullinated antigen, or if the sites of citrullination of the studied epitopes are truly citrullinated in vivo. Furthermore, the presence of citrulline-directed T-cells was, thus far, only shown using single-colour tetramer stainings. These T-cells have not been isolated or clonally expanded to study their peptide reactivity and their ability to help ACPA-producing B-cells. Thus, future research should aim at further investigating the relevance of these epitopes and citrulline-directed CD4 + T-cells in ACPA+ RA.
Concluding remarks
In recent years, much work has been done on elucidating the long-known HLA class II association as an understanding would likely offer critical insights in the pathogenesis of ACPA + RA. Recent insights in the HLA class II association are now reshaping our understanding of the contribution of this locus to the pathogenesis of ACPA+ RA. Excitingly, CD4 + T-cells have been proposed to contribute to the effect of HLA class II on RA, and data in support of these hypotheses are accumulating. Many questions remain unanswered, including the exact role and relative importance of newly identified autoreactive T-cell populations, a problem also faced in other autoimmune diseases including multiple sclerosis. Nevertheless, future studies in this area will likely offer novel insights in the pathogenesis of ACPA+ RA and the influence of the HLA class II locus in shaping the autoimmune response.
Declaration of interest: The authors report no conflicts of interest.
References
- Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–108.
- Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GM, van Veelen PA, et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci U S A. 2011;108:17372–7.
- van der Linden MP, van der Woude D, Ioan-Facsinay A, Levarht EW, Stoeken-Rijsbergen G, Huizinga TW, et al. Value of anti-modified citrullinated vimentin and third-generation anti-cyclic citrullinated peptide compared with second-generation anti-cyclic citrullinated peptide and rheumatoid factor in predicting disease outcome in undifferentiated arthritis and rheumatoid arthritis. Arthritis Rheum. 2009;60:2232–41.
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81.
- van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Toes RE, Huizinga TW. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res Ther. 2005;7:R949–58.
- Huizinga TW, Amos CI, van der Helm-van Mil AH, Chen W, van Gaalen FA, Jawaheer D, et al. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005;52:3433–8.
- van der Helm-van Mil AH, Huizinga TW. Advances in the genetics of rheumatoid arthritis point to subclassification into distinct disease subsets. Arthritis Res Ther. 2008;10:205.
- Snir O, Widhe M, Hermansson M, von Spee C, Lindberg J, Hensen S, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62: 44–52.
- Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint- derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. 2013;210:445–55.
- van der Woude D, Syversen SW, van der Voort EI, Verpoort KN, Goll GL, van der Linden MP, et al. The ACPA isotype profile reflects long-term radiographic progression in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1110–16.
- Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. 2012;122:1791–802.
- Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, et al. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis. 2014;73:854–60.
- Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–73.
- Laurent L, Clavel C, Lemaire O, Anquetil F, Cornillet M, Zabraniecki L, et al. Fcgamma receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann Rheum Dis. 2011;70:1052–9.
- van Steendam K, Tilleman K, de Ceuleneer M, de Keyser F, Elewaut D, Deforce D. Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res Ther. 2010;12:R132.
- Suwannalai P, van de Stadt LA, Radner H, Steiner G, El-Gabalawy HS, Zijde CM, et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012;64: 1323–8.
- Suwannalai P, Britsemmer K, Knevel R, Scherer HU, Levarht EW, van der Helm-van Mil AH, et al. Low-avidity anticitrullinated protein antibodies (ACPA) are associated with a higher rate of joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2014;73:270–6.
- Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009;60:1923–31.
- Young BJ, Mallya RK, Leslie RD, Clark CJ, Hamblin TJ. Anti- keratin antibodies in rheumatoid arthritis. Br Med J. 1979;2:97–9.
- Nienhuis RL, Mandema E. A new serum factor in patients with rheumatoid arthritis; the antiperinuclear factor. Ann Rheum Dis. 1964;23:302–5.
- Sebbag M, Simon M, Vincent C, Masson-Bessiere C, Girbal E, Durieux JJ, et al. The antiperinuclear factor and the so-called antikeratin antibodies are the same rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1995;95:2672–9.
- Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol. 2001;166:4177–84.
- Despres N, Boire G, Lopez-Longo FJ, Menard HA. The Sa system: a novel antigen-antibody system specific for rheumatoid arthritis. J Rheumatol. 1994;21:1027–33.
- Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81.
- Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. 1999;162:585–94.
- Vossenaar ER, Despres N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther. 2004;6:R142–50.
- Anzilotti C, Pratesi F, Tommasi C, Migliorini P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev. 2010;9:158–60.
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–58.
- Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402.
- Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. 2008;67:1488–92.
- Darrah E, Giles JT, Ols ML, Bull HG, Andrade F, Rosen A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci Transl Med. 2013; 5:186ra65.
- Verpoort KN, Cheung K, Ioan-Facsinay A, van der Helm-van Mil AH, de Vries-Bouwstra JK, Allaart CF, et al. Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheum. 2007;56:3949–52.
- Ioan-Facsinay A, el-Bannoudi H, Scherer HU, van der Woude D, Menard HA, Lora M, et al. Anti-cyclic citrullinated peptide antibodies are a collection of anti-citrullinated protein antibodies and contain overlapping and non-overlapping reactivities. Ann Rheum Dis. 2011;70:188–93.
- van de Stadt LA, van Schouwenburg PA, Bryde S, Kruithof S, van Schaardenburg D, Hamann D, et al. Monoclonal anti-citrullinated protein antibodies selected on citrullinated fibrinogen have distinct targets with different cross-reactivity patterns. Rheumatology (Oxford). 2013;52:631–5.
- van Beers JJ, Schwarte CM, Stammen-Vogelzangs J, Oosterink E, Bozic B, Pruijn GJ. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum. 2013;65:69–80.
- Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1421–9.
- Tabushi Y, Nakanishi T, Takeuchi T, Nakajima M, Ueda K, Kotani T, et al. Detection of citrullinated proteins in synovial fluids derived from patients with rheumatoid arthritis by proteomics-based analysis. Ann Clin Biochem. 2008;45(Pt 4):413–17.
- Haag S, Schneider N, Mason DE, Tuncel J, Andersson IE, Peters EC, et al. Mass spectrometric analysis of citrullinated type II collagen reveals new citrulline-specific autoantibodies, which bind to human arthritic cartilage. Arthritis Rheumatol. 2014 Jan 27. [Epub ahead of print]
- Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–6.
- Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–9.
- Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol. 2012;8:144–52.
- Shiina T, Inoko H, Kulski JK. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens. 2004;64:631–49.
- Stastny P. Mixed lymphocyte cultures in rheumatoid arthritis. J Clin Invest. 1976;57:1148–57.
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–13.
- Gregersen PK, Shen M, Song QL, Merryman P, Degar S, Seki T, et al. Molecular diversity of HLA-DR4 haplotypes. Proc Natl Acad Sci U S A. 1986;83:2642–6.
- Winchester RJ, Gregersen PK. The molecular basis of susceptibility to rheumatoid arthritis: the conformational equivalence hypothesis. Springer Semin Immunopathol. 1988;10:119–39.
- Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–6.
- Singal DP, D’Souza M, Reid B, Bensen WG, Kassam YB, Adachi JD. HLA-DQ beta-chain polymorphism in HLA-DR4 haplotypes associated with rheumatoid arthritis. Lancet. 1987;2:1118–20.
- Bondinas GP, Moustakas AK, Papadopoulos GK. The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics.2007;59:539–53.
- Bax M, van Heemst J, Huizinga TW, Toes RE. Genetics of rheumatoid arthritis: what have we learned?Immunogenetics. 2011;63:459–66.
- Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med. 1994;180:157–63.
- Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS is essential for effective T-helper-cell responses. Nature. 2001;409:105–9.
- Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–63.
- Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30: 324–35.
- Bos WH, Ursum J, de Vries N, Bartelds GM, Wolbink GJ, Nurmohamed MT, et al. The role of the shared epitope in arthralgia with anti-cyclic citrullinated peptide antibodies (anti-CCP), and its effect on anti-CCP levels. Ann Rheum Dis. 2008;67:1347–50.
- Hensvold HA, Magnusson PK, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. 2013 Nov 25. [Epub ahead of print]
- Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol. 2009;9:833–44.
- Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–19.
- Cunin R, Glansdorff N, Pierard A, Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986;50:314–52.
- Zuniga M, Perez G, Gonzalez-Candelas F. Evolution of arginine deiminase (ADI) pathway genes. Mol Phylogenet Evol. 2002;25: 429–44.
- Wegner N, Lundberg K, Kinloch A, Fisher B, Malmstrom V, Feldmann M, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233:34–54.
- Koning F. Celiac disease: caught between a rock and a hard place. Gastroenterology. 2005;129:1294–301.
- Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58:3009–19.
- Pratesi F, Tommasi C, Anzilotti C, Chimenti D, Migliorini P. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54:733–41.
- Munz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG, et al. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J Exp Med. 2000;191:1649–60.
- Lunemann JD, Frey O, Eidner T, Baier M, Roberts S, Sashihara J, et al. Increased frequency of EBV-specific effector memory CD8 + T cells correlates with higher viral load in rheumatoid arthritis. J Immunol. 2008;181:991–1000.
- Kimachi K, Croft M, Grey HM. The minimal number of antigen-major histocompatibility complex class II complexes required for activation of naive and primed T cells. Eur J Immunol. 1997;27:3310–17.
- Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–5.
- Enouz S, Carrie L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med. 2012;209:1769–79.
- Hitchon CA, El-Gabalawy HS. Infection and rheumatoid arthritis: still an open question. Curr Opin Rheumatol. 2011;23:352–7.
- Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003;171: 538–41.
- Snir O, Rieck M, Gebe JA, Yue BB, Rawlings CA, Nepom G, et al. Identification and functional characterization of T cells reactive to citrullinated vimentin in HLA-DRB1*0401-positive humanized mice and rheumatoid arthritis patients. Arthritis Rheum. 2011;63:2873–83.
- Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210: 2569–82.
- von Delwig A, Locke J, Robinson JH, Ng WF. Response of Th17 cells to a citrullinated arthritogenic aggrecan peptide in patients with rheumatoid arthritis. Arthritis Rheum. 2010;62:143–9.
- Law SC, Street S, Yu CH, Capini C, Ramnoruth S, Nel HJ, et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Res Ther. 2012;14:R118.