
Abstract
Breast cancer is the most commonly diagnosed cancer and the second leading cause of cancer death among women in the United States. Recently, interest has grown in the role of epigenetics in breast cancer development and progression. Epigenetic changes such as DNA methylation, histone modifications, and abnormal expression of non-coding RNAs emerged as novel biomarkers in breast cancer diagnosis, therapy, and prevention. This review focuses on the most recent mechanistic findings underlying epigenetic changes in breast cancer development and their role as predictors of breast cancer risk. The rapid progress in our understanding of epigenetic findings in breast cancer has opened new avenues for potential therapeutic approaches via identification of epigenetic targets. We highlight the development of novel epigenetically targeted drugs, relevant clinical trials in breast cancer patients, and recent approaches combining epigenetic agents with chemotherapy and/or endocrine therapy that may incrementally improve long-term outcomes in appropriately selected breast cancer patients. Biomarkers of response are needed, however, to identify patient subsets that are most likely to benefit from epigenetic treatment strategies.
Key messages
Abnormal epigenetic regulation plays a role in breast cancer development, and epigenetic changes may emerge as predictors of risk.
Epigenetic drugs show promise as therapeutic primers for, and as combination therapies with, chemotherapy and/or endocrine therapy.
Epigenetic marks, particularly DNA methylation and histone modifications, are potential biomarkers of response to epigenetic therapies.
Dysregulation of epigenetic marks in breast cancer
Alterations of breast cancer epigenome
DNA methylation
Breast cancer arises from an accumulation of abnormal genetic and epigenetic events. Epigenetic alterations are modifications to chromatin which do not change the DNA sequence and include both DNA methylation and histone modifications. The dynamic nature of these pathways is now emerging. DNA is methylated by DNA methyltransferases (DNMTs) which include three family members: DNMT1, DNMT3a, and DNMT3b. DNMT3a and 3b are responsible for de novo DNA methylation, while DNMT1 maintains the methylation state during cell replication. DNMT enzymes use methyl groups from S-adenosylmethionines (SAM) to methylate CpG dinucleotides. Clusters of these CpG dinucleotides, which may be differentially methylated, are known as CpG islands and typically exist in gene promoters, but are also found elsewhere in the genome (Citation1,Citation2). It has been recently discovered that DNA methyl marks can be removed through a multistep process by the ten-eleven translocation (TET) proteins (Citation3–5). The TET family consists of three members, TET1, TET2, and TET3, and their mechanistic role in cancer has been minimally studied. TET2 has been shown to be mutated and thereby inactivated in multiple cancer types (Citation6). Additionally, mRNA expression of all three TET family members and DNA demethylase activity have been shown to be decreased in human breast tumors compared to matched normal breast tissue (Citation6,Citation7). Breast cancer frequently presents with hypermethylation of DNA at promoters of specific genes, such as tumor suppressor genes (TSGs), that ultimately leads to silencing of these genes with progression of disease (Citation8). The reversal of the hypermethylated state can lead to silencing of oncogenes and expression of TSGs. Hence, the use of agents that inhibit DNMT activity holds great promise for breast cancer therapy.
The contribution of DNA methylation to breast cancer development has been extensively studied. In breast cancer, abnormal DNA hypermethylation is frequently associated with epigenetic silencing of important genes such as TSGs and growth regulatory genes. These genes include estrogen receptor alpha (ERα), progesterone receptor (PR), retinoic acid receptor-β (RARβ), BRCA1, SFRPs, E-cadherin, MLH1, cyclin D2, p16, GSTP1, and 14.3.3 sigma (Citation9,Citation10). Silencing of RARβ leads to transformation of mammary epithelial cells as evidenced by depolarization and dedifferentiation (Citation11,Citation12). Pruitt et al. demonstrated that inhibition of the class III histone deacetylase (HDAC) reactivated epigenetically silenced tumor suppressive SFRP1, SFRP2, E-cadherin, and CRBP1 genes in human breast cancer cells (Citation13). Silencing of ERα is not only associated with poor prognosis (Citation14,Citation15), but leaves fewer treatment options for patients as their tumors will not respond to targeted hormonal therapies. High ERα DNA methylation correlates with decreased gene expression in human breast tumors (Citation16), and DNMT inhibition may lead to demethylation of DNA and ERα re-expression (Citation17). Better understanding of the mechanisms underlying dysregulation of DNA methylation and their contribution to epigenetic gene silencing in breast cancer is important for the development of novel targeted therapy for this disease.
Histone modifications as emerging targets in breast cancer
Multiple post-translational modifications (PTMs) can be covalently attached to histone tails typically on lysine residues; these include methylation, acetylation, phosphorylation, sumoylation, biotinylation, and ubiquitination. Histone methyltransferases (HMTs), acetylases (HATs), demethylases (KDMs), and deacetylases (HDACs) write and erase the histone code and are responsible for maintaining a balance of histone marks, ultimately regulating gene expression (). Histone methylation/demethylation and acetylation/deacetylation have been extensively studied in cancer. Histone modifications can activate or repress gene transcription. For example, active marks (H3K4me, H3K36me, AcH4K16, or AcH3K9/18) open chromatin conformation and recruit machinery to activate transcription, whereas repressive marks (H3K9me, H4K20me, or H3K27me) recruit repressive complexes and condense chromatin ().
Figure 1. Proposed model of epigenetic regulation of gene expression in breast cancer. DNA methylation is generally associated with transcription repression, while histone modifications can be either active or repressive. DNA is methylated by DNMTs and demethylated by a multistep process by the TETs. There are multiple post-translational modifications that occur on histone tails. A multitude of enzymes function to methylate (HMT: Set8, Set9, SUV39H1, EZH2), demethylate (KDM: JARID1B, LSD1/2, JMJD2C, JMJD3), acetylate (HAT), or deacetylate (HDAC) histones. DNA hypermethylation at a gene promoter in association with abnormal histone modifications in breast cancer cells may lead to the anomalous loss of genes that are important in curbing tumor growth. Established or investigational agents that can block many of the enzymes are shown in green boxes.
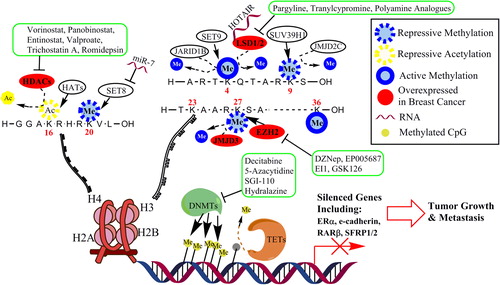
Histone acetylation
The level of histone acetylation is the result of the counterbalancing activity of HATs and HDACs. Abnormally enhanced activity of HDACs has been implicated in breast tumorigenesis in part through the anomalous loss of expression of critical genes that are important in curbing breast tumor growth (Citation10,Citation18). Muller et al. investigated expression of HDAC1, 2, and 3 in a breast cancer patient cohort consisting of 238 tumors. They found that protein expression of HDAC1 is increased in hormone receptor-positive breast tumors, HDAC2 expression is associated with higher-grade tumors, and HDAC3 expression is correlated with both hormone receptor-negative tumors and higher-grade tumors (Citation19). These findings suggest that the use of drugs that inhibit HDACs and re-express silenced genes may hold promise for breast cancer therapy (Citation10,Citation18,Citation20).
Histone methylation
Recent studies have shown that dysregulated histone methylation/demethylation contributes to breast cancer development. One study demonstrated that protein levels of repressive histone marks, H3K9me3 and H4K20me3, in 15 breast cancers were increased compared to breast tissues derived from 28 healthy controls (Citation21). The HMT enhancer of zeste homolog 2 (EZH2) is the key component of the polycomb repressive complex 2. EZH2 has been shown to silence the tumor suppressor RKIP in breast cancer cells, and patients with breast tumors with a high level of EZH2 and a low level of RKIP displayed a significantly reduced 5-year event-free survival (Citation22). EZH2 has also been implicated in breast cancer cell metastasis. Knockdown of EZH2 by RNAi in MDA-MB-231 cells showed significantly decreased cell invasion accompanied by increased expression of the epithelial markers, E-cadherin and CK-18, and decreased expression of the mesenchymal markers, vimentin and snail-1 (Citation23). Further EZH2 knockdown decreased lung metastasis after orthotopic mammary gland xenografts of MDA-MB-231 cells (Citation23). In human breast cancer cells, it has been shown that H3K9me3 is needed to promote tumor cell migration. H3K9 is methylated by the HMT SUV39H1, and blocking SUV39H1 decreased H3K9me. Inhibition of SUV39H1 by chaetocin or siRNA leads to reduced cell motility in a scratch assay, while overexpression of SUV39H1 increases H3K9me and cell migration across a scratch. These results indicate that activities of SUV39H1 and H3K9me3 are essential for cell migration and metastasis, and thus SUV39H1 is a potential epigenetic target for breast cancer therapy (Citation24).
Our recent work showed that the FAD-dependent histone demethylase, LSD1 (lysine-specific demethylase 1), may also have potential as a target in breast cancer therapy (Citation25–27). LSD1, also known as AOF2 or KDM1A, is the first identified histone demethylase capable of specifically demethylating H3K4me1 and H3K4me2 (Citation28,Citation29). LSD1 is typically associated with a transcriptional repressor complex that includes HDAC1/2, coREST, and BHC80 (Citation28). Enhanced expression and activity of LSD1 has been implicated in the tumorigenesis of various cancers, including breast cancer (Citation30–33). We have recently interrogated The Cancer Genome Atlas (TCGA) database and found that LSD1 mRNA expression levels were greatly increased in triple-negative breast cancer (TNBC, tumors that lack expression of ER, PR, and HER2) in comparison to non-TNBC tumors (P < 0.0001), suggesting that its expression may correlate with worse prognosis. A recent study supported this hypothesis by showing that LSD1 is significantly overexpressed in high-grade ductal carcinoma in situ (DCIS) versus low-grade DCIS and in invasive ductal breast carcinoma versus low/intermediate DCIS (Citation34). Lim et al. also reported that LSD1 is highly expressed in ER-negative breast cancers (Citation31). These studies point to a potential role for LSD1 as an effective therapeutic target in a subtype of breast cancer with a more aggressive biology.
A second amine oxidase domain containing histone demethylase was more recently discovered. LSD2, also known as KDM1B or AOF1, is upregulated in human breast tumors compared to normal mammary gland tissue in two microarray studies according to the Oncomine database (oncomine.org). While LSD1 has been shown to interact with HDACs, LSD2 may interact with DNMTs. Loss of LSD2 in oocytes has been shown to reduce de novo DNA methylation of imprinted genes, suggesting that LSD2 is needed for DNMTs to function (Citation35) and further demonstrating the interaction between multiple epigenetic pathways.
In mouse mammary gland cells and human MCF10A cells, inhibition of KDM6B, a member of the Jmj-C domain containing KDMs (JMJD3), abrogated the ability of transforming growth factor (TGF)-β to induce epithelial mesenchymal transition (EMT), while KDM6B overexpression induced EMT in MCF10A cells (Citation36). Blocking KDM6B led to reduced invasion of MDA-MB-231 breast cancer cells, while its overexpression promoted invasion. Additionally, Oncomine gene expression data showed that KDM6B was significantly overexpressed in invasive breast cancer in three studies (Finak & Radvanyi Oncomine) (Citation36).
MicroRNA and long non-coding RNA as novel epigenetic regulators in breast cancer
MicroRNAs (miRNA) are 21–23-nucleotide non-coding RNA molecules that function as negative regulators of gene expression by modulating the stability of target mRNAs. Long non-coding RNAs (lncRNA) are non-protein-coding transcripts longer than 200 nucleotides. Both miR and lncRNA have been shown to play a role in epigenetic regulation in breast cancer. A novel function for miR-7 was recently reported, namely that miR-7 represses Set8, a HMT targeting the repressive H4K20me, thereby reducing H4K20me and inhibiting EMT in invasive breast cancer cells (Citation37). Zhang et al. reported that ERα regulates breast tumor-initiating cells by downregulating miR-140, which targets the transcription factor SOX2 (Citation38); miRs have also been shown to be involved in HDAC function. In a recent study, the HDACi trichostatin A (TSA) was shown to have a great effect on miRNA expression in breast cancer cell lines (Citation39). After treatment with TSA, several miRs known to be tumor suppressive, anti-tumor migration/metastasis, and anti-EMT were upregulated in the drug-resistant breast cancer cell line MCF7TN-R, suggesting that inhibition of HDAC leads to an anti-tumorigenic miRNA profile in drug-resistant breast cancer cells. In another study, the HDACi entinostat induced expression of miR-215a, miR-251b, and miR-205, leading to post-transcriptional downregulation of erbB2 and erbB3 protein expression and enhanced apoptosis in breast cancer cells (Citation40).
A recently identified lncRNA, HOTAIR, has been shown to act as a scaffold binding polycomb repressive complex 2 at its 5’ end and LSD1/coREST at its 3′ end, thereby aiding in assembly of histone-modifying complexes (Citation41). HOTAIR expression is induced by TGFβ, and blocking its induction prevents TGFβ from inducing EMT in breast and colon cancer cells (Citation42). Together, these data imply a role for certain RNAs in regulating epigenetic function and that alterations in expression of key RNAs can aid in tumorigenesis, potentially demonstrating a new target for cancer therapeutics.
Epigenetic predisposition to breast cancer
The fetal environment and breast cancer risk
The in utero environment is known to predispose offspring to diseases later in life, including breast cancer. Specifically, the relationship between maternal diet during fetal development and breast cancer risk in the offspring has been well investigated. There is evidence that the mechanism of disease susceptibility involves epigenetic alterations. Fetal development is a critical period in creating the epigenetic code. During early gestation, the genome is demethylated, followed by de novo methylation of appropriate DNA loci (Citation43–45). Protein restriction during fetal development has been shown to increase susceptibility to mammary carcinogenesis in rodent models, culminating in reduced latency in response to a carcinogen (Citation46). It was also shown that fetal protein restriction led to DNA hypomethylation and reduced DNMT1 expression, which can be rescued by folic acid intake during pregnancy (Citation47,Citation48). Additionally, supplementation of the maternal diet with the methyl donor choline during fetal development rescued the protein restriction effects on mammary tumor susceptibility and led to decreased tumor growth in the offspring (Citation49). In offspring exposed to protein restriction in utero, promoter histone modifications are altered in the mammary gland, including decreases in both H3K4me2 and AcH3, representing a shift toward a pattern consistent with gene repression (Citation50). Additionally, a maternal high-fat diet has also been shown to increase mammary tumor susceptibility in offspring, as manifested by an increased number of mammary cancers in response to the chemical carcinogen DMBA in adulthood (Citation51). This effect of in utero high-fat diet was additionally shown to be transgenerational (Citation51).
Toxin exposure in utero can also affect disease state of offspring later in life, even if the offspring appear perfectly normal at birth (Citation52). Alcohol exposure in utero has been shown to increase susceptibility to mammary tumorigenesis in adult offspring in response to a chemical carcinogen in multiple rodent models (Citation53,Citation54). Animals exposed to alcohol in utero develop increased mammary cancers during adulthood in response to the chemical carcinogen NMU (N-nitrosomethylurea), as well as more ER-negative mammary cancers (Citation54). Alterations induced by fetal alcohol or high-fat diet in rodent models have been shown to be transgenerational and induce epigenetic dysregulation (Citation55,Citation56). Fetal alcohol exposure induces both histone modifications and DNA methylation, which can be rescued by choline supplementation during gestation (Citation55). The level of H3K4me is decreased in the hypothalamus of fetal alcohol-exposed offspring, while H3K9me3 and AcH3K9 are increased, indicating a shift to gene repression. These alterations were accompanied by increased protein expression of DNMT1 and DNMT3a and altered mRNA expression of a number of HMTs. These effects were largely eliminated by maternal choline supplementation during fetal development. Therefore, data suggests that maternal diet, as well as alcohol exposure during fetal development, may affect epigenetic programming in the fetus as well as susceptibility to mammary tumorigenesis during adulthood.
Diet and breast cancer risk
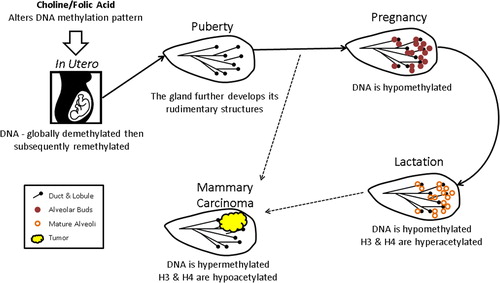
Epigenetic regulation plays a role in breast development and may contribute to cancer development. During pregnancy, DNA becomes hypomethylated, and subsequently, during lactation, H3 and H4 become hyperacetylated, thereby contributing to activation of genes involved in milk production and lactation () (Citation57). It is well known that environmental factors such as pesticides and diet can affect epigenetic regulation (Citation58–60). Dietary methyl donors such as folic acid (folate), a water-soluble vitamin, and choline, a water-soluble essential nutrient, contribute to the activity of SAM, which is ultimately necessary for methylating DNA. Folate deficiency may play a causal role in the development of certain types of cancer (Citation61–65). Folate deficiency not only leads to alterations in DNA methylation, but also disrupts the machinery involved in DNA methylation, increasing protein levels of DNMT1, DNMT3b, MBD2, and MBD4 (Citation66). In the Women's Circle of Health Study cohort, dietary intake was assessed using a Food Frequency Questionnaire (FFQ) in 1582 African American women (749 newly diagnosed breast cancer cases and 833 healthy controls) and 1434 European American women (744 newly diagnosed breast cancer cases and 690 healthy controls). It was found that in both European and African American women the level of natural food folate intake was significantly inversely associated with ER-positive breast cancer risk (Citation67). This relationship was particularly strong in premenopausal African American women. A second recent study used a FFQ to evaluate folate intake in 20,756 women from the Melbourne Collaborative Cohort study and found that women with both high folate and high methionine intake were at a reduced breast cancer risk (Citation68). Additionally, in the 4-Corners Breast Cancer Study of 2325 breast cancer cases (798 Hispanic and 1527 non-Hispanic) and 2535 controls (924 Hispanic and 1601 non-Hispanic), folate intake was associated with decreased development of ER-negative breast cancer, particularly in postmenopausal women. In addition, women who had high folate intake and low alcohol intake were significantly protected against breast cancer of any hormone receptor status (Citation69). Finally, the Long Island Breast Cancer Study Project of 1,508 breast cancer patients and 1,556 controls demonstrated that increased choline consumption in women decreased breast cancer risk by 24%, and women with a SNP in PEMT, an enzyme responsible for endogenous choline biosynthesis, incurred a 30% increase in breast cancer risk (Citation70). Therefore, multiple studies with large cohorts of women have shown associations between reduced breast cancer risk and consumption of dietary folate or choline. These data indicate that lifestyle choices can affect the epigenetic code, ultimately altering breast cancer risk.
Figure 2. Epigenetic predisposition to breast cancer. Epigenetic regulation is instrumental in mammary gland development from the time the mammary streak is created in utero through rounds of pregnancy and lactation, and it is clearly involved in breast cancer development. Early in fetal development the genome is globally demethylated and remethylated in the appropriate patterns, but this process can be disrupted by toxin exposure and poor diet. The effects exerted by poor diet and toxin exposure can be dampened by maternal intake of folic acid or choline. During pregnancy the mammary gland DNA becomes globally hypomethylated to allow active transcription of genes needed to reorganize the tissue structure to develop alveolar buds and prepare for lactation. Once lactation commences and alveoli are mature, H3 and H4 in the chromatin of the gland become additionally hyperacetylated, allowing for activation of milk production genes. During breast cancer development, this process is abrogated and DNA is typically hypermethylated while H3 and H4 are generally hypoacetylated.
Another well studied dietary compound found in Brassica vegetables (broccoli, Brussels sprouts, cabbage) which is known to prevent cancer is sulphoraphane. Sulphoraphane has been shown to inhibit growth and HDAC activity in multiple human breast cancer cell lines (Citation71). Treatment with sulphoraphane also prevented mammary tumor formation in a rat model. After a carcinogenic dose of DMBA, sulphoraphane-treated animals had 35% or 26% risk of developing cancer (75 or 150 μM sulphoraphane, respectively), significantly decreased from 68% in control animals. Tumor multiplicities were also reduced to 0.45 and 0.26 (75 or 150 μM sulphoraphane, respectively) compared to 1.56 in control animals (Citation72). Recently, sulphoraphane was shown to act synergistically with exemestane, an inhibitor of estrogen biosynthesis, to induce antioxidant and anti-inflammatory responses that could lead to cancer prevention (Citation73). These data indicate that sulphoraphane may prevent breast cancer in preclinical models.
Peripheral blood DNA methylation as a biomarker for breast cancer risk
Investigations have been ongoing to determine if epigenetic abnormalities could serve as biomarkers for breast cancer risk, with special attention to the role of DNA methylation in peripheral blood cell DNA. One study of 730 breast cancer patients and 353 healthy controls showed differential DNA methylation patterns in 49 genes in peripheral blood between patients and controls (Citation74). It was unclear whether the observed differences in DNA methylation were potential biomarkers of breast cancer risk versus detection of circulating tumor cell DNA. Similarly, LUMA analysis, a measurement of global promoter methylation, has shown potential association between DNA methylation and breast cancer risk, as breast cancer patients displayed a 57.3% methylation, compared to 52.4% methylation in peripheral blood DNA of healthy controls (Citation75). There have also been two studies in healthy women to determine if DNA methylation could predict breast cancer risk. One study examined breast tissue collected from healthy women with a mean age of 35 and found that women with a family history of breast cancer demonstrated increased DNA hypermethylation of both p16INK4 and BRCA1 in the breast (Citation76). A second study utilized DNA isolated from peripheral blood from the Sister Study. In 910 healthy women, each with a sister with breast cancer, evidence for differential DNA methylation was found between women who went on to develop breast cancer in comparison to women who remained cancer-free (Citation77). Taken together, evidence supports that epigenetic dysregulation in response to environmental factors plays an important role in breast cancer development.
Novel agents that target the dysregulated epigenetic pathways
DNMT inhibitors
Azacitidine (Aza) is a pyrimide nucleoside analogue of cytidine, and decitabine (DAC) is a deoxycytidine analogue activated by deoxycytidine kinase. In breast cancer, both compounds have been examined for their ability to inhibit DNA methylation, inhibit tumor cell growth, and re-express aberrantly silenced genes such as ERα, E-cadherin, and SFRPs (Citation15,Citation17,Citation78,Citation79). Despite the advantages of these drugs in treatment of leukemia, significant challenges remain in the use of these compounds for treatment of solid tumors, including breast cancer. One of their disadvantages is the instability of both compounds in aqueous solutions, as well as their rapid inactivation through deamination by cytidine deaminase. Therefore, novel targeted DNMT inhibitors with improved therapeutic efficacy are continually being pursued. Two novel DNMT inhibitors, SGI-110 and NPEOC-DAC, have been developed; they display enhanced resistance to cytidine deaminase and an extended half-life (Citation80,Citation81). After metabolic activation and incorporation into DNA, SGI-110 inhibits DNMT activity, causes DNA hypomethylation, and inhibits tumor cell growth. Another putative agent that inhibits both DNMT and cytidine deaminase is zebularine. Zebularine forms complexes between DNMT protein and zebularine-substituted DNA that may act primarily as a trap for DNMT protein (Citation82). One study suggested that zebularine is a potent DNMT inhibitor and DNA demethylating agent in human breast cancer cells and potentiates the inhibitory effect of other epigenetic therapeutics on cell proliferation and colony formation (Citation83). Unfortunately, development of zebularine for human clinical trials has been halted because of concerns about an unfavorable toxicity profile.
HDAC inhibitors
HDACs regulate the expression of numerous genes involved in breast cancer initiation and progression. One likely mechanism of the contribution of HDACs to breast tumorigenesis is that abnormally enhanced activity of HDACs decrease chromatin-activating marks and subsequently silence important genes, such as TSGs, that curb breast tumor growth. Hence, the use of drugs that inhibit HDACs holds promise for breast cancer therapy. Two synthetic HDACis, vorinostat (SAHA) and romidepsin (FK-228), have been approved by the US FDA for the clinical treatment of cutaneous T-cell lymphoma (CTCL). Some HDACis have been investigated for their effects on chromatin structure and re-expression of aberrantly silenced genes in breast cancer cells (Citation13,Citation84). For example, in ER-negative breast cancer cells, inhibition of HDAC activity by specific HDACi reactivates silenced ERα and PR genes (Citation84–87). Inhibition of the class III HDAC, SIRT1, has been reported to restore epigenetically silenced tumor suppressive SFRP1, SFRP2, E-cadherin, and CRBP1 genes in ER-negative breast cancer cells (Citation13). These preclinical data indicate that the use of HDACi holds promise for breast cancer treatment, presumably in part through the re-expression of epigenetically silenced genes. However, it is clear that HDACi will not be useful as monotherapy, suggesting that it is critically important to investigate the role of combination strategies to enhance cell killing with the goal of improving response rates and long-term outcomes of HDACi treatment in breast cancer.
KDM and KMT inhibitors
A role for the enhanced expression or activity of key histone-modifying enzymes such as recently emerging KDMs has been implicated in breast tumor tumorigenesis. Our recent studies have shown that treatment with the LSD1-inhibiting polyamine analogues altered global H3K4me2 and induced expression of silenced genes in breast cancer cells (Citation25). Monoamine oxidase inhibitors such as tranylcypromine and pargyline were also shown to potently inhibit LSD1 activity and inhibit growth of breast cancer cells (Citation26,Citation31). A recent screen of a large library of inhibitory scaffolds identified two compounds which inhibit lysine-specific demethylase 4C (KDM4C or JMJD2C) (Citation88). Wang et al. reported that a novel inhibitor of α-ketoglutarate, JIB-04, specifically inhibits the activity of the Jumonji family of KDMs in cancer cells, including breast cancer (Citation89). It is anticipated that these types of novel agents that target the histone demethylation pathway may lead to the development of a new generation of epigenetically active drugs with considerable clinical potential in breast cancer therapy.
HMTs may also be effective targets for cancer therapy. 3-Deazaneplanocin A (DZNep) has been shown to deplete EZH2 levels, inducing apoptosis in human breast cancer cells without affecting normal cells (Citation90). In addition, BRCA1-deficient tumors depend on EZH2, and BRCA1-deficiency sensitized tumors to the EZH2 inhibitor DZNep (Citation91). A novel SAM competitive inhibitor of EZH2 was recently developed and shown to be effective in killing large B-cell lymphoma cells. EI1 is a specific EZH2 inhibitor which inhibits cellular H3K27me and induces gene expression of EZH2-targeted genes (Citation92). A second EZH2 inhibitor, EPZ005687, is also a SAM competitive inhibitor and effectively eliminates H3K27me, ultimately inhibiting lymphoma cell growth (Citation93). GSK126 is a third selective EZH2 inhibitor which significantly decreased tumor growth and increased survival in a mouse xenograft model of diffuse large B-cell lymphoma (Citation94). While all of these EZH2 inhibitors have been developed in lymphoma models, these drugs may also have promise for breast cancer studies. A summary of these novel agents as well as beneficial dietary compounds are depicted in .
Table I. Select epigenetic agents with potential clinical applications in breast cancer. A list of epigenetic agents, their epigenetic targets, and characteristics and clinical implications is presented.
Epigenetic combination therapy in breast cancer
Targeting multiple epigenetic pathways
It is now clear that epigenetic pathways work in tandem, and thus targeting multiple epigenetic pathways simultaneously could enhance efficacy. Our recent study shows that stable knockdown of the histone demethylase LSD1 by shRNA (short hairpin RNA) led to a decrease in breast cancer cell growth which is further reduced by combining LSD1 knockdown with HDAC inhibition by vorinostat (Citation27). LSD1 knockdown reduced HDAC levels and activity, and its effect was potentiated by use of HDACi, especially in TNBC cells. Another study found that the combination of the HDAC inhibitor romidepsin with DAC synergistically killed TNBC cells (Citation95). The combination was shown to induce expression of the tumor suppressor, SFRP1, and it was determined that SFRP1 was involved in the mechanism leading to the synergistic cell death (Citation95).
Combining epigenetic drugs with current breast cancer therapies
Further evidence implies that epigenetic pathways working in concert also interface with non- epigenetic pathways. Thus, targeting an epigenetic pathway in combination with a cancer driving pathway could significantly inhibit cancer cell growth and result in therapeutic benefit. Combining either decitabine (DAC), a DNMT inhibitor, or vorinostat, an HDAC inhibitor, with doxorubicin, one of the most common breast cancer chemotherapeutic agents, led to synergistic cell death which was more pronounced with DAC (Citation96). This effect was observed in MCF7, BT474, and MDA-MB-231 breast cancer cells, but the effect was most dramatic in drug-resistant MCF7 cells (MCF7/ADR). This indicates that pretreatment with the epigenetic drug sensitized the drug-resistant cells, resulting in a therapeutic response to doxorubicin (Citation96).
Combining active antitumor dietary compounds with current therapies
Several dietary constituents are felt to have epigenetic properties. In a recent study, the soybean phytoestrogen, genistein, given at safe levels, led to epigenetic re-expression of the ER and response to tamoxifen in vitro and in vivo in MDA-MB-231 TNBC cells (Citation97). Animals fed genistein developed smaller tumors and had a reduced latency in a spontaneous breast cancer mouse model, and orthotropic MDA-MB-231 xenografted tumors completely regressed when treated with both genistein and tamoxifen. Spontaneous mammary tumor formation and tumor staining for proliferating cell nuclear antigen (PCNA) were also attenuated with combined genistein and tamoxifen treatment (Citation97). While ERα expression was increased in all genistein-exposed tumors, HDAC1 and DNMT1 expression and activity were reduced in the combination therapy tumors from both mammary tumor mouse models (Citation97).
Phenethyl isothiocyanate (PEITC) is an anticancer agent found in cruciferous vegetables which has been found to inhibit HDAC activity and induce cell death. In both ER/PR-positive MCF7 and triple-negative MDA-MB-231 cells, PEITC reduced cell growth, and this effect was accentuated in a synergistic manner with paclitaxel treatment in both cell lines (Citation98). These studies suggest that combining dietary constituents with documented epigenetic effects with commonly used cytotoxics can augment the antitumor effects in preclinical models.
Inducing tumor suppressor expression in combination with epigenetic treatments
ING1 is a tumor suppressor family known to be downregulated in many breast cancers. A group recently investigated the effect of treating cancer with a combination of this tumor suppressor with epigenetic drugs. They administered ING1 in an adenoviral construct (ad-ING1) that infected the cells and induced a high expression of ING1. In breast cancer cells, combination of 5-azacytidine (AZA) and panobinostat has shown synergy in inhibiting growth of tumor cells, but combined use of ad-ING1 and panobinostat displayed more significant growth inhibitory effect than AZA/panobinostat combination. This finding was replicated in a tumor xenograft mouse model where ad-ING1 and AZA greatly reduced tumor volume compared to any controls or either alone in MDA-MB-468 tumor xenografts (Citation99).
Clinical development of epigenetic strategies in breast cancer
The potentially reversible nature of epigenetic changes makes them attractive therapeutic targets. Specifically, in breast cancer preclinical studies, DNMT inhibitors and HDAC inhibitors act synergistically to reactivate ERα transcription (Citation100), resulting in sensitization of TNBC cells to tamoxifen induced cell death (Citation101). A number of trials in breast cancer have been completed (), and others are ongoing. They have explored epigenetic agents alone, in combination with each other, or in combination with chemotherapy or endocrine therapy. Several pivotal trials are highlighted below.
Table II. Trials of epigenetic therapies in breast cancer. Selected clinical trials using epigenetic agents for the treatment of breast cancer, trial details, and available results are presented.
HDAC inhibitors
A phase II study of the HDAC inhibitor vorinostat (suberoylanilide hydroxamic acid) in 14 patients with advanced breast cancer with a median of 1.5 chemotherapeutic regimens for metastatic disease showed no partial responses, resulting in closure after the first stage. However, stable disease was observed in four patients over a range of 4–14 months, indicating potential for clinical benefit despite the lack of RECIST-defined response (Citation102). Next, a proof of principle preoperative window study examined the ability of vorinostat as a single agent to modulate biomarkers in breast tumor tissue from patients with newly diagnosed breast cancer who received preoperative vorinostat 300 mg orally (PO) BID × 6 doses versus untreated controls (Citation103). Gene methylation and expression (RT-PCR using the 21-gene recurrence score, and IHC for Ki67 and cleaved caspase 3) were analyzed in pre- and post-vorinostat breast tumors in comparison to untreated control tumor samples. Vorinostat led to significantly decreased expression by RT-PCR of proliferation-associated genes such as Ki67, cyclin B1, and STK15. However, it did not significantly affect Ki67 by IHC, expression of other genes in the Oncotype DX 21-gene assay, or methylation of candidate genes. This important trial demonstrated that vorinostat significantly affected proliferation-associated gene expression, despite the fact that only a limited amount of pre- and post-treatment paired biopsy samples were available for analysis, indicating potential for its activity in breast cancer. Moreover, as the authors note, the observed vorinostat concentrations in this trial were lower than those needed preclinically to result in gene re-expression, making further studies warranted for determination of optimal dosing strategies and combination approaches with other agents.
HDAC inhibitors in combination with DNMT inhibitors
We participated in a recently completed phase II study of AZA and entinostat (MS-275) in patients with advanced breast cancer that was either hormone-resistant and HER2-negative, or triple-negative (Citation104). Enrolled patients received 40 mg/m2 subcutaneously of AZA on days 1–5 and 8–10, and entinostat 7 mg orally on day 2 and 10 of a 28-day cycle. Based on preclinical findings showing the ability of these agents to re-sensitize breast cancer cells to tamoxifen and aromatase inhibitors, at the time of progression on study, patients were able to continue study therapy in an optional continuation phase with the addition of hormonal therapy at the discretion of the treating physician. The most common grade 3 and 4 toxicities observed were a 23% rate of leukopenia and neutropenia. In the triple-negative cohort, no clinical response was observed at the first-stage interim analysis, and the cohort was closed. One partial response was observed in the first stage in the hormone-resistant cohort, thus allowing this cohort to proceed to full enrollment. Final analysis of clinical findings, as well as results from embedded correlative studies obtained from mandatory tumor biopsies and circulating DNA that will examine candidate gene re-expression and methylation, should be available soon.
Combination of HDAC inhibitors and endocrine therapy
Based on preclinical models, a phase II study of vorinostat in combination with tamoxifen (400 mg daily vorinostat for 3 of 4 weeks with 20 mg continuous daily tamoxifen) was performed in 43 ER-positive metastatic breast cancer (MBC) patients who had progressed on endocrine therapy (58% had prior adjuvant tamoxifen; 98% had progressed on an aromatase inhibitor [AI] therapy). The objective response rate (ORR) was 19%, and the clinical benefit rate (CBR, defined as response or stable disease > 24 weeks) was 40% (Citation105). Interestingly, patients with responsive or stable disease had significantly increased histone H4 acetylation in peripheral blood mononuclear cells (PBMCs) (430% increase [CI 219–633]) versus non-responders (6% increase [confidence interval (CI) –7–21]; P = 0.042). Responders also had higher baseline HDAC2 levels in PBMCs than non-responders (P = 0.04). The durable responses observed in endocrine-resistant patients, as well as the identification of potential biomarkers predictive of response, were highly encouraging in this trial. The investigators postulated that patients with either low baseline HDAC2 expression or no increased histone acetylation after treatment might either need higher dose therapy or not benefit from the therapy.
A second study of the combination of an HDAC inhibitor with an endocrine agent enrolled women with ER-positive MBC who had progressive disease on AI therapy to continued treatment on the AI on which they had progressed with addition of 5 mg of the HDAC inhibitor entinostat (SNDX-275), weekly (Citation106). Of the 27 patients, there was one patient with a partial response, and one patient with stable disease > 6 months, and 41% received study treatment for > 4 months, indicating potential for clinical benefit. Grade 3 or higher toxicities included nausea, fatigue, and diarrhea. Biomarker analysis from this study documented increased lysine acetylation in PBMCs with entinostat. In addition, a recently reported randomized, double-blind, phase II study of exemestane with entinostat (5 mg orally once weekly) versus exemestane plus placebo targeted 130 patients with advanced ER-positive disease who had previously progressed on an AI. Combination therapy with exemestane and entinostat yielded only a modest improvement in progression-free survival (PFS) (4.3 months versus 2.3 months; HR 0.73; CI 0.50–1.07; P = 0.055) (Citation107). However, median overall survival (an exploratory end-point) was 28.1 months with exemestane and entinostat versus 19.8 months with exemestane plus placebo (0.59; CI 0.36–0.97; P = 0.036), indicating that epigenetic therapy may improve long-term outcomes for patients, even in the absence of short-term response by conventional RECIST assessment. Again, increased histone lysine hyperacetylation was correlated with improved PFS, further indicating the potential for use of histone acetylation as a biomarker of response. A phase I/II study of the non-selective HDAC inhibitor panobinostat in combination with letrozole (NCT01105312) in patients with triple-negative MBC has completed accrual but not yet reported phase II results. The phase I portion of this trial reported a main dose-limiting toxicity (DLT) of thrombocytopenia in three of six patients at the 30 mg dose level, and the recommended dose for the phase II portion was 20 mg orally three times per week in combination with standard dose letrozole (Citation108). Finally, an ongoing study of the HDAC inhibitor vorinostat (NCT0172062) in patients with MBC who have progressed on AI therapy has reported enrollment of 4 of 20 planned patients. The trial is examining serial fluoroestradiol (FES) PET imaging as a potential biomarker of vorinostat effect on ER expression and tumor metabolism. Patients receive vorinostat 400 mg orally BID for 2 weeks, followed by re-treatment with their previous AI for 6 weeks, with FES, FDG, and clinical assessment at baseline, and after treatment. Patients who demonstrate clinical benefit will continue to be treated with 2 weeks of vorinostat followed by 6 weeks of AI in 8-week cycles until progression (Citation109).
Combination of HDAC inhibitors and chemotherapeutic agents
A number of trials are exploring combination therapy with epigenetic and chemotherapeutic agents. In one proof-of-principle study in 16 patients with locally advanced breast cancer (77% ER-positive, 30% HER2-positive), patients were typed for an acetylator phenotype and then treated with hydralazine (182 mg if they were rapid –acetylators; 83 mg if they were slow – acetylators) plus magnesium valproate at 30 mg/kg, starting 1 week before chemotherapy until chemotherapy concluded (Citation110). Chemotherapy consisted of four cycles of standard-dose of doxorubicin and cyclophosphamide. Core biopsies of the breast tumors were obtained at diagnosis and on day 8. The study reported a clinical ORR of 81% [31% complete clinical response (CR), and 50% partial response]. Only one patient (6.6%) had a pathologic complete response (pCR), but 70% of patients had < 3 cm of residual disease. It is difficult to know whether the addition of the epigenetic agents in this trial affected the clinical response to chemotherapy, but there was a significant decrease in global 5mC content and HDAC activity in tumors. Microarray analysis on pre- and post-treatment clinical samples showed that TP53 transcript was increased 7.7-fold after treatment, indicating that the combined epigenetic therapy could induce the re-expression of the important tumor suppressor TP53.
In a phase I–II study of first-line therapy for 54 patients with MBC, patients received vorinostat (200 or 300 mg orally BID) on days 1–3, 8–10, and 15–17, plus paclitaxel (90 mg/m2) on days 2, 9, 16, and bevacizumab (10 mg/kg) on days 2 and 16 every 28 days (Citation111). In phase I, no DLTs were observed, and the recommended dose of vorinostat was 300 mg BID. In phase II, 24 objective responses were observed in 44 patients (55%; CI 39%–70%). Paired tumor biopsies were assessed for seven patients before and after the third vorinostat dose, and they showed increased acetylation of Hsp90 and α-tubulin, and functional inhibition of Hsp90, a chaperone protein known to be involved in activity of numerous oncogenes (Citation112).
In another phase I study of vorinostat with capecitabine (Citation113) in 23 patients with advanced breast cancer, investigators postulated that the two agents would act synergistically based on preclinical observations showing that the HDACi TSA downregulates thymidylate synthase. Of the 14 patients who were evaluable for response, no objective responses were seen, but 3 patients had stable disease longer than 6 months, and 1 patient completed 42 cycles of therapy. The DLT of the combination was fatigue, and the maximum tolerable dose was 1500 mg of capecitabine combined with 200 mg of vorinostat twice a day (Citation113). Pre- and post-treatment biopsies were analyzed, and preliminary results showed changes in pathways involving extracellular matrix and TGFβ, both of which are involved in tissue remodeling to enable progression to metastasis.
Other trials combining vorinostat with chemotherapeutic agents including capecitabine, ixabepilone, nab-paclitaxel, and carboplatin are ongoing. A phase II double-blind study of carboplatin and nab-paclitaxel with or without vorinostat as preoperative systemic therapy in HER2-negative operable breast cancer examined response and surrogate biomarkers. In the phase II portion of this study (Citation114), women with clinical stage II–III HER2-negative breast cancer were randomly assigned to chemotherapy plus placebo or vorinostat. Final results are awaited, but an initial report showed similar pCR rates in the two arms (27.6% in the vorinostat arm, 26.7% in the placebo arm). Secondary end-points from the trial examined correlation of baseline and change in standardized uptake values on positron emission tomography with pCR and clinical CR, and the trial will also analyze changes in candidate gene methylation, gene expression, and histone acetylation in tumor tissue as potential biomarkers of response.
Other studies have examined epigenetic agents in combination with anti-HER2 therapy. In a phase I/II trial of vorinostat 200 mg orally BID in combination with trastuzumab 6 g/kg every 21 days in women with heavily pre-treated HER2-positive MBC, no DLTs were observed in 16 enrolled patients. The combination was well tolerated, with grade 3/4 toxicities including dyspnea (n = 2) and thrombocytopenia (n = 2). No objective responses were observed in 11 patients with centrally confirmed HER2-positive disease, but one objective response was observed in a patient with centrally confirmed HER2-negative disease. Correlative studies, including hyperacetylation status of PBMCs, CTC enumeration, and methylation studies are ongoing (Citation115). Panobinostat is also being studied in combination with chemotherapy and trastuzumab in ongoing trials.
Conclusion
In summary, unprecedented advances have been made in the understanding of epigenetic regulatory mechanisms in breast cancer. A number of novel agents targeting epigenetic changes have recently been developed. However, no epigenetic agents have moved into clinical practice for breast cancer, suggesting an urgent need to explore further the implications of the basic science findings for translation into the clinic. It is clear that monotherapy with an epigenetic modifying agent does not yield high response rates in patients with advanced breast cancer, but early information suggests that combination therapy with chemotherapy and/or endocrine therapy may improve long-term outcomes in appropriately selected patients. Several trials have shown prolonged stable disease in individual cases, indicative of potential for clinical benefit. Further studies are needed to identify biomarkers of response to therapy in individual patients. For example, assessment of PBMC histone acetylation seems to be a promising predictor of response to HDAC inhibitor therapy. Patient selection will be critical for future trials, as will identification of potent epigenetic therapies, the determination of optimal timing and sequence of such therapies, and their integration with other classes of drugs for breast cancer management.
Declaration of interest: The authors report no conflicts of interest.
References
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–13.
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21.
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7.
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5.
- Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–9.
- Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–37.
- van Hoesel AQ, Sato Y, Elashoff DA, Turner RR, Giuliano AE, Shamonki JM, et al. Assessment of DNA methylation status in early stages of breast cancer development. Br J Cancer. 2013;108:2033–8.
- Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–82.
- Stearns V, Zhou Q, Davidson NE. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 2007;25:659–65.
- Ren M, Pozzi S, Bistulfi G, Somenzi G, Rossetti S, Sacchi N. Impaired retinoic acid (RA) signal leads to RARbeta2 epigenetic silencing and RA resistance. Mol Cell Biol. 2005;25:10591–603.
- Bistulfi G, Pozzi S, Ren M, Rossetti S, Sacchi N. A repressive epigenetic domino effect confers susceptibility to breast epithelial cell transformation: implications for predicting breast cancer risk. Cancer Res. 2006;66:10308–14.
- Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40.
- Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–5.
- Nass SJ, Herman JG, Gabrielson E, Iversen PW, Parl FF, Davidson NE, et al. Aberrant methylation of the estrogen receptor and E-cadherin 5’ CpG islands increases with malignant progression in human breast cancer. Cancer Res. 2000;60:4346–8.
- Lapidus RG, Ferguson AT, Ottaviano YL, Parl FF, Smith HS, Weitzman SA, et al. Methylation of estrogen and progesterone receptor gene 5’ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res. 1996;2: 805–10.
- Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995;55:2279–83.
- Huang Y, Shaw PG, Davidson NE. Inhibition of histone deacetylases. Methods Mol Biol. 2011;791:297–311.
- Muller BM, Jana L, Kasajima A, Lehmann A, Prinzler J, Budczies J, et al. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer. 2013;13:215.
- Huang Y, Nayak S, Jankowitz R, Davidson NE, Oesterreich S. Epigenetics in breast cancer: what’s new? Breast Cancer Res. 2011;13:225.
- Leszinski G, Gezer U, Siegele B, Stoetzer O, Holdenrieder S. Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res. 2012;32:2199–205.
- Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012;72:3091–104.
- Moore HM, Gonzalez ME, Toy KA, Cimino-Mathews A, Argani P, Kleer CG. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res Treat. 2013;138:741–52.
- Yokoyama Y, Hieda M, Nishioka Y, Matsumoto A, Higashi S, Kimura H, et al. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013;104:889–95.
- Zhu Q, Huang Y, Marton LJ, Woster PM, Davidson NE, Casero RA Jr. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids. 2012;42:887–98.
- Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat. 2012;131:777–89.
- Vasilatos SN, Katz TA, Oesterreich S, Wan Y, Davidson NE, Huang Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis. 2013;34:1196–207.
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53.
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–5.
- Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–18.
- Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–20.
- Metzger E, Wissmann M, Yin N, Muller J, Schneider R, Peters A, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–9.
- Huang Y, Marton LJ, Woster PM, Casero RA. Polyamine analogues targeting epigenetic gene regulation. Essays Biochem. 2009;46: 95–110.
- Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, Buettner R. Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin Pathol. 2012;12:13.
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, et al. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–18.
- Ramadoss S, Chen X, Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem. 2012; 287:44508–17.
- Yu N, Huangyang P, Yang X, Han X, Yan R, Jia H, et al. microRNA-7 suppresses the invasive potential of breast cancer cells and sensitizes cells to DNA damages by targeting histone methyltransferase SET8. J Biol Chem. 2013;288:19633–42.
- Zhang Y, Eades G, Yao Y, Li Q, Zhou Q. Estrogen receptor alpha signaling regulates breast tumor-initiating cells by down-regulating miR-140 which targets the transcription factor SOX2. J Biol Chem. 2012;287:41514–22.
- Rhodes LV, Nitschke AM, Segar HC, Martin EC, Driver JL, Elliott S, et al. The histone deacetylase inhibitor trichostatin A alters microRNA expression profiles in apoptosis-resistant breast cancer cells. Oncol Rep. 2012;27:10–16.
- Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, et al. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat- induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013;4:e556.
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93.
- Alves CP, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, et al. The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cells lines. Stem Cells. 2013;31:2827–32.
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93.
- Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, et al. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992;6:705–14.
- Santos F, Dean W. Epigenetic reprogramming during early development in mammals. Reproduction. 2004;127:643–51.
- Fernandez-Twinn DS, Ekizoglou S, Gusterson BA, Luan J, Ozanne SE. Compensatory mammary growth following protein restriction during pregnancy and lactation increases early-onset mammary tumor incidence in rats. Carcinogenesis. 2007;28:545–52.
- Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–73.
- Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr. 2008;100:278–82.
- Kovacheva VP, Davison JM, Mellott TJ, Rogers AE, Yang S, O’Brien MJ, et al. Raising gestational choline intake alters gene expression in DMBA-evoked mammary tumors and prolongs survival. FASEB J. 2009;23:1054–63.
- Zheng S, Rollet M, Yang K, Pan YX. A gestational low-protein diet represses p21(WAF1/Cip1) expression in the mammary gland of offspring rats through promoter histone modifications. Br J Nutr. 2012;108:998–1007.
- de Assis S, Warri A, Cruz MI, Laja O, Tian Y, Zhang B, et al. High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Nat Commun. 2012;3:1053.
- Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–17.
- Hilakivi-Clarke L, Cabanes A, de Assis S, Wang M, Khan G, Shoemaker WJ, et al. In utero alcohol exposure increases mammary tumorigenesis in rats. Br J Cancer. 2004;90:2225–31.
- Polanco TA, Crismale-Gann C, Reuhl KR, Sarkar DK, Cohick WS. Fetal alcohol exposure increases mammary tumor susceptibility and alters tumor phenotype in rats. Alcohol Clin Exp Res. 2010;34:1879–87.
- Bekdash RA, Zhang C, Sarkar DK. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res. 2013;37:1133–42.
- Hilakivi-Clarke L, de Assis S. Fetal origins of breast cancer. Trends Endocrinol Metab. 2006;17:340–8.
- Rijnkels M, Kabotyanski E, Montazer-Torbati MB, Hue Beauvais C, Vassetzky Y, Rosen JM, et al. The epigenetic landscape of mammary gland development and functional differentiation. J Mammary Gland Biol Neoplasia. 2010;15:85–100.
- Guerrero-Bosagna C, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol Cell Endocrinol. 2012;354:3–8.
- Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–9.
- Skinner MK. Endocrine disruptors and epigenetic transgenerational disease etiology. Pediatr Res. 2007;61(5 Pt 2):48R–50R.
- Duthie SJ, Narayanan S, Sharp L, Little J, Basten G, Powers H. Folate, DNA stability and colo-rectal neoplasia. Proc Nutr Soc. 2004;63:571–8.
- Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004;13:511–19.
- Duthie SJ. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011;34:101–9.
- Bistulfi G, Vandette E, Matsui S, Smiraglia DJ. Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells. BMC Biol. 2010;8:6.
- Stefanska B, Karlic H, Varga F, Fabianowska-Majewska K, Haslberger A. Epigenetic mechanisms in anti-cancer actions of bioactive food components—the implications in cancer prevention. Br J Pharmacol. 2012;167:279–97.
- Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, et al. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. 2006;136:1522–7.
- Gong Z, Ambrosone CB, McCann SE, Zirpoli G, Chandran U, Hong CC, et al. Associations of dietary folate, vitamins B6 and B12 and methionine intake with risk of breast cancer among African American and European American women. Int J Cancer. 2014;134: 1422–35.
- Bassett JK, Baglietto L, Hodge AM, Severi G, Hopper JL, English DR, et al. Dietary intake of B vitamins and methionine and breast cancer risk. Cancer Causes Control. 2013;24:1555–63.
- Yang D, Baumgartner RN, Slattery ML, Wang C, Giuliano AR, Murtaugh MA, et al. Dietary intake of folate, B-vitamins and methionine and breast cancer risk among Hispanic and non-Hispanic white women. PLoS One. 2013;8:e54495.
- Xu X, Gammon MD, Zeisel SH, Lee YL, Wetmur JG, Teitelbaum SL, et al. Choline metabolism and risk of breast cancer in a population-based study. FASEB J. 2008;22:2045–52.
- Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–21.
- Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci U S A. 1994;91:3147–50.
- Liu H, Talalay P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc Natl Acad Sci U S A. 2013;110:19065–70.
- Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, et al. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008;3:e2656.
- Xu X, Gammon MD, Hernandez-Vargas H, Herceg Z, Wetmur JG, Teitelbaum SL, et al. DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012;26:2657–66.
- Dumitrescu RG, Marian C, Krishnan SS, Spear SL, Kallakury BV, Perry DJ, et al. Familial and racial determinants of tumour suppressor genes promoter hypermethylation in breast tissues from healthy women. J Cell Mol Med. 2010;14:1468–75.
- Xu Z, Bolick SC, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013; 105:694–700.
- Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–9.
- Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98:1147–56.
- Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, et al. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–8.
- Byun HM, Choi SH, Laird PW, Trinh B, Siddiqui MA, Marquez VE, et al. 2’-Deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5-azacytidine: a novel inhibitor of DNA methyltransferase that requires activation by human carboxylesterase 1. Cancer Lett. 2008; 266:238–48.
- Hurd PJ, Whitmarsh AJ, Baldwin GS, Kelly SM, Waltho JP, Price NC, et al. Mechanism-based inhibition of C5-cytosine DNA methyltransferases by 2-H pyrimidinone. J Mol Biol. 1999;286:389–401.
- Billam M, Sobolewski MD, Davidson NE. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res Treat. 2010;120:581–92.
- Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66:6370–8.
- Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2’-deoxycytidine. Breast Cancer Res Treat. 2003;81:177–86.
- Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther. 2007;6:64–9.
- Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000; 60:6890–4.
- Leurs U, Clausen RP, Kristensen JL, Lohse B. Inhibitor scaffold for the histone lysine demethylase KDM4C (JMJD2C). Bioorg Med Chem Lett. 2012;22:5811–13.
- Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035.
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63.
- Puppe J, Drost R, Liu X, Joosse SA, Evers B, Cornelissen-Steijger P, et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11:R63.
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–5.
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12.
- Cooper SJ, von Roemeling CA, Kang KH, Marlow LA, Grebe SK, Menefee ME, et al. Reexpression of tumor suppressor, sFRP1, leads to antitumor synergy of combined HDAC and methyltransferase inhibitors in chemoresistant cancers. Mol Cancer Ther. 2012;11:2105–15.
- Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm. 2013;10:337–52.
- Li Y, Meeran SM, Patel SN, Chen H, Hardy TM, Tollefsbol TO. Epigenetic reactivation of estrogen receptor-alpha (ERalpha) by genistein enhances hormonal therapy sensitivity in ERalpha-negative breast cancer. Mol Cancer. 2013;12:9.
- Liu K, Cang S, Ma Y, Chiao JW. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer Cell Int. 2013;13:10.
- Thakur S, Feng X, Qiao Shi Z, Ganapathy A, Kumar Mishra M, Atadja P, et al. ING1 and 5-azacytidine act synergistically to block breast cancer cell growth. PLoS One. 2012;7:e43671.
- Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–9.
- Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66:6370–8.
- Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14:7138–42.
- Stearns V, Jacobs LK, Fackler M, Tsangaris TN, Rudek MA, Higgins M, et al. Biomarker modulation following short-term vorinostat in women with newly diagnosed primary breast cancer. Clin Cancer Res. 2013;19:4008–16.
- Connolly RM, Jankowitz RC, Zahnow CA, Zhang Z, Rudek MA, Jeter SC, et al. A phase 2 study investigating the safety, efficacy and surrogate biomarkers of response of 5-azacitidine (5-AZA) andentinostat (MS-275) in patients with triple-negative advanced breast cancer. Cancer Res. 2013;73(8 Suppl 1).
- Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–35.
- Wardley AM, Stein R, McCaffrey J, Crown J, Malik Z, Rea D, Barrett-Lee PJ, et al. Phase II data for entinostat, a class 1 selective histone deacetylase inhibitor, in patients whose breast cancer is progressing on aromatase inhibitor therapy. 2010 ASCO Annual Meeting Abstract. J Clin Oncol. 2010;28(15 suppl):1052.
- Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–35.
- Tan W, Allred JB, Moreno-Aspitia A, Northfelt DW, Ingle JN, Perez EA. Phase I study of panobinostat (LBH589) and letrozole in post-menopausal women with metastatic breast cancer. J Clin Oncol. 2012;30(suppl); abstr e13501.
- Linden HM, Kurland BF, Link J, Gadi VK, Specht JM, Gralow J, et al. Vorinostat to restore sensitivity to aromatase inhibitor therapy in metastatic breast cancer: a phase II clinical trial with ER imaging correlates. J Clin Oncol. 2012;30(suppl): abstr TPS3109.
- Arce C, Perez-Plasencia C, Gonzalez-Fierro A, de la Cruz-Hernandez E, Revilla-Vazquez A, Chavez-Blanco A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:e98.
- Ramaswamy B, Fiskus W, Cohen B, Pellegrino C, Hershman DL, Chuang E, et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: evidence for vorinostat- induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res Treat. 2012;132:1063–72.
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49.
- James ES, Chung GG, DiGiovanna M, Sanft TB, Hofstatter EW, Sowers N, et al. Phase I study of the HDAC inhibitor vorinostat in combination with capecitabine in a biweekly schedule in advanced breast cancer. J Clin Oncol 2013;31(suppl); abstr 2587.
- Connolly RM, Jeter S, Zorzi J, Zhang Z, Armstrong DK, Fetting JH, et al. A multi-institutional double-blind phase II study evaluating response and surrogate biomarkers to carboplatin and nab-paclitaxel (CP) with or without vorinostat as preoperative systemic therapy (PST) in HER2- negative primary operable breast cancer (TBCRC008). 2010 ASCO Annual Meeting Abstracts. J Clin Oncol. 2010;28(15 suppl):TPS111.
- Swaby R, Sparano J, Bhalla K, Meropol N, Falkson C, Pellegrino C, et al. A phase II study of the histone deacetylase inhibitor, vorinostat, in combination with trastuzumab in patients with advanced metastatic and/or local chest wall recurrent HER-2 amplified breast cancer resistant to transtuzumab-containing therapy: (E1104) a trial of the Eastern Cooperative Oncology Group. Cancer Res. 2009;69(24 Suppl):5084.
- Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284:17775–82.