
Abstract
Background. Biomarkers of T-cell receptor excision circles (TRECs) and immunoglobulin κ-deleting recombination excision circles (KRECs) reflect naïve T and B cell emigrants. This study assessed the biomarkers in patients with primary immunodeficiency diseases (PIDs) to determine the lymphocyte output disturbance and the correlation to lymphocytes.
Methods. A standard plasmid was constructed to calculate TRECs and KRECs in 250 ng genomic DNA from whole blood of PIDs patients. These were correlated to naïve and memory lymphocytes for further classification and adequate treatment.
Results. In 69 studied patients, the low TRECs mainly included those with severe combined T and B immunodeficiency (SCID, 7/8), combined immunodeficiency (CID, 4/4), and common variable immunodeficiency (CVID, 6/7). The diminished KRECs was in SCID (4/8), CID (4/4), CVID (7/7), Bruton's tyrosine kinase mutation (Btk, 3/4), anti-B cell deletion (by anti-CD20 antibody in 1), and Behçet syndrome under steroid treatment (1). The TRECs and KRECs positively correlated to absolute naïve T (CD4 + CD45RA+) and naïve B (CD19 + CD27−), and to memory B (CD19 + CD27+) numbers, respectively.
Conclusion. This study validates that low TRECs and KRECs values reflect low naïve T and B lymphocytes in ‘combined immunodeficiencies’ and in some CVID patients with the potential to develop the CID phenotype.
Key messages
Biomarkers of T-cell receptor excision circles (TRECs) and immunoglobulin κ-deleting recombination excision circles (KRECs) can be used to classify patients with primary immunodeficiency diseases (PIDs) for elucidating the naïve lymphocyte output disturbance for effective treatment.
The TRECs and KRECs values in 250 ng genomic DNA isolated from peripheral whole blood positively correlated to absolute counts of naïve T and B cell lymphocytes, respectively.
Low TRECs and KRECs values reflect low counts of naïve T and B lymphocytes in ‘combined T and B immunodeficiencies’, as well as in some CVID patients with the potential to develop the CID phenotype.
Introduction
Normal T cell differentiation in the thymus is characterized by the rearrangement of T cell receptor (TCR) genes, leaving the unused V, D, and J gene segments to join together as T-cell receptor excision circles (TRECs). The one specific δRec-ΨJα signal joint TRECs are produced from approximately 70% of all T cells that express the α/β T cell receptor (Citation1,Citation2). Reasonably, in the process of B cell maturation, immunoglobulin κ-deleting recombination excision circles (KRECs) are also created during the κ-deleting recombination allelic exclusion and isotype exclusion of the λ chain (Citation3). The TRECs and KRECs are not replicated during cell division (Citation3,Citation4) so that their measurements from isolated peripheral blood cells in patients with primary immunodeficiency diseases (PIDs) reflect naïve T and B cell emigrant defects. Low TRECs and low KRECs often herald critical infections in patients with T and B cell deficiency, and therefore, in time, prompt for effective interventions of suitable hematopoietic stem cell transplantation (HSCT) in those with low TRECs and/or regular intravenous immunoglobulin (IVIG) supplementation in those with low KRECs (Citation1,Citation5–8).
The updated PIDs categories have been revised every two years since 1970, with increasing diseases every year. The recent 2014 revision substituted ‘combined immunodeficiencies with associated or syndromic features’ for ‘well-defined syndromes with immunodeficiency’ in 2011 or ‘other well-defined immunodeficiency syndromes’ in 2009 and referred to the OMIM system for a friendly approach to the clinical spectrum and early diagnosis (Citation9,Citation10). Although much more is known now than before, it is still challenging to comprehensively recognize 266 complex orphan PIDs caused by 244 genetic defects (Citation11) and classify some diseases with two or more defective immune cells into more than one kind of category, such as the Omenn-like phenotype, hyper IgM syndromes (HIGM), and hyper IgE recurrent infection syndromes (HIES) (Citation9,Citation10). The success of newborn screening for naïve T cell emigrant defects and arrests in B cell maturation by measuring TRECs (Citation5–8) and KRECs (Citation12) from neonatal Guthrie cards prompt early diagnosis and optimal treatment despite controversies in the revision of the update category.
For adequate treatment and prevention of irreversible complications, this study detected TRECs and KRECs from peripheral blood in referred patients with PIDs upon diagnosis to investigate the disturbance of naïve T and B cell development and the correlation of TRECs and KRECs values to naïve lymphocytes.
Methods
Patients
Since 2003, 727 individuals (573 suspected cases and 154 related persons) were referred to the Primary Immunodeficiency Care And Research (PICAR) Institute for functional, molecular, and genetic diagnosis of PIDs. A total of 246 patients were identified as PIDs. Based on the 2014 updated PIDS categories, ‘combined immunodeficiencies with associated or syndromic features’ (previously termed as ‘well-defined syndromes with immunodeficiency’) (n = 89) had become the most frequent PIDs in Taiwan, followed by ‘predominantly antibody deficiencies’ (n = 57), ‘combined T and B cell immunodeficiencies’ (n = 40), congenital defects of phagocytes (n = 35), complement deficiencies (n = 15), disease of immune dysregulation (n = 6), defects in innate immunity (n = 2), and auto-inflammatory disorders (n = 2).
Basic immunologic functions, including serum immunoglobulins, lymphocyte counts, and subsets of naïve and memory cells (CD4 + CD45RA+, CD8 + CD45RA+, and CD19 + CD27- representative for naïve cells; CD4 + CD45RO+, CD8 + CD45RO+ and CD19 + CD27+ for memory cells), phagocytic superoxide production, and lymphocyte proliferation tests were evaluated. For each patient, an EBV-transformed B lymphoblastoid cell line and an IL-2-dependent cell line were established (Citation13). Candidate genes were investigated and sequenced from isolated genomic DNA and complement DNA as previously described (Citation14–16).
The Chang Gung Human Investigation Committee approved this study, and the patients’ parents or guardians provided written and verbal informed consent.
Evaluation of TRECs and KRECs
Preparation of plasmid-containing inserts of TRECs and KRECs as reference gene
Fragments of TRECs, KRECs, and T-cell receptor alpha constant gene (TRACs, as internal controls) were obtained by amplifying nucleic acid with primers specific for TRECs, KRECs, and TRACs as previously described () (Citation17,Citation18). The TRAC- and KREC-specific primers contained Spe I and Hind III restriction sites at the 5’-end that were not present in the canonical TRECs, KRECs, and TRACs sequences. Briefly, the PCR product of TRECs was inserted in the T-A acceptor site of pCR®2.1-TOPO Vector (Invitrogen, Carlsbad, CA, USA). The TRACs fragment was then ligated into the Spe I restriction site and the KRECs fragment into the Hind III restriction site.
Table I. Forward and reverse primers for the evaluation of TRECs, KRECs, and TRACs.
The DNA plasmid was transformed in DH5 alpha cells, and colonies containing the three fragments were identified by PCR. Direct sequencing demonstrated that the inserted DNA had no mutations in terms of canonical sequences and that each insert was present in single copy. One of the identified colonies was expanded, and plasmid DNA was rapidly purified using the Bioman Plasmid DNA Midiprep Kit (Bioman, Taipei, Taiwan).
Real-time PCR to quantify the TRECs and KRECs values
The numbers of TRECs and KRECs were simultaneously detected by duplex real-time PCR assay performed on the 7500 Fast Real-Time PCR (Applied Biosystems, Bio-Rad, Hercules, CA, USA). The PCR reactions were developed in a final volume of a 25-μL mixture, consisting of 250 ng genomic DNA solution from whole blood, 12.5 μL of 2 × TaqMan Universal PCR master mix containing AmpErase UNG (Applied Biosystems), and primers and probes for TRECs and KRECs () at a final concentration of 900 nM and 200 nM, respectively. Assay for the three genes was run with the same protocol consisting in a first step at 50°C for 2 min, initial heating at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 15 s and a combined primer/probe annealing and elongation at 60°C for 1 min.
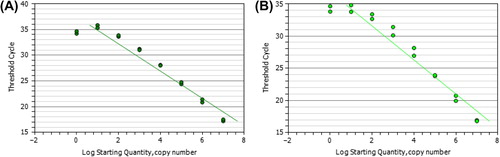
The TRECs and KRECs numbers were obtained by extrapolating the respective sample quantities from the standard curve obtained by serial dilutions (107,106, 105, 104,103, 102, 101, and 100) of the linear triple-insert plasmid DNAs amplified in each PCR plate (). The absolute numbers of TRECs or KRECs per 250 ng genomic DNA of whole peripheral blood were calculated.
Figure 1. The reference standards of KRECs, TRECs, and TRACs (as internal controls) fragments were inserted into DH5 alpha cells, and each pCR®2.1-TOPO Vector clone contained the three fragments (each single copy) identified and confirmed by whole-vector PCR. The KRECs and TRECs numbers were shown by extrapolating the respective sample quantities from the standard curve obtained by serial dilutions (107,106, 105, 104,103, 102, 101, and 100) and the linear triple-insert plasmid DNA (A: KRECs, and B: TRECs) that were amplified in each PCR plate. The absolute number of TRECs or KRECs per 250 ng whole blood was calculated from the linear slope by y = –3.327x + 37.373 for TRECs (R2 = 0.9947) and y = –3.0727x + 38.526 (R2 = 0.9907) for KRECs.
Statistical analysis
The TRECs and KRECs were calculated from 40 healthy controls to set up the normal ranges (). Those < 5% of the normal range were defined as the ‘low’ level. The correlation between TRECs or KRECs and lymphocyte subsets (naïve and memory subpopulations) was performed by SPSS Advanced Statistical™ 17 (2009, Chicago, IL). Statistical significance was set at P<0.05.
Table II. Normal TRECs and KRECs values from healthy controls (n = 40).
Results
Characteristics of enrolled patients
Among 246 PIDs patients, 69 were admitted to the institute for further treatment. They received TRECs and KRECs evaluation to investigate naïve emigrant T and B cell development from the thymus and/or bone marrow (). Overall, 15 patients had ‘predominantly antibody deficiencies’ (Bruton's tyrosine kinase mutation (Btk) mutation in four, common variable immunodeficiency (CVID) in seven, and one each with activation-induced cytidine deaminase (AICDA), chromosome 1q42 deletion with CVID-HIGM phenotype, hypo-gamma-globulinemia after anti-CD20 antibody (rituximab) deletion, and transient hypo-gamma-globulinemia in infancy), 14 had combined T and B immunodeficiencies (severe combined T and B immunodeficiency (SCID) in eight, combined T and B immunodeficiency in four, and CD40L mutation in two), 10 had congenital defects of phagocyte disorders (CYBB mutation in five, NCF1 mutation in one, and ELANE mutation in four), eight had disorders of immune dysregulation (HLH phenotype with unknown genetic defects in six, XIAP mutation in one, and FOXP3 mutation in one), and 13 had combined immunodeficiencies with associated or syndromic features (WASP mutation in nine, and one each with DOCK mutation, STAT3, HIGE without identified molecular defects, and DiGeorge syndrome (DGS)). One had complement (C3) deficiency, one had defects in innate immunity (NEMO mutation), and another had auto-inflammatory disorders (cold-induced auto-inflammatory syndrome (CIAS)). Two patients with chronic diarrhea and another four with ectodermal dysplasia, Behçet syndrome, Kabuki syndrome, and recurrent cutaneous infections were enrolled.
Table III. Copy numbers of TRECs, KRECs, and naïve and memory lymphocytes in patients with primary immunodeficiency diseases.
Establishment of standard reference values of TRECs and KRECs
From the 40 healthy controls (), the ‘low’ value was defined as < 5% of normal ranges, which corresponded to 124.0 for TRECs and 157.7 for KRECs.
TRECs assessment
Like in the neonatal Guthrie card blood dot screening test in previous reports (Citation6,Citation7,Citation11), patients with profound T cell deficiency mainly encompassed SCID, combined T and B immunodeficiency (CID), and CD40L-hyper IgM syndrome had low TRECs (< 124.0 copy number) except for the SCID5-SCT patient with unknown genetic defects. Notably, low TRECs was also detected in CVID patients (6/7), two HLH-phenotype patients, and one WAS patient with a larger deletion in the WASP gene (including the promoter and exons 1 and 2).
The DGS patient with typical face dysmorphology and hypocalcemia-induced convulsion but without lymphopenia had relatively appropriate TRECs (168 copy number in 250 ng genomic DNA isolated from whole blood) and normal lymphocyte proliferation. One patient with Behçet syndrome taking prednisolone inhibited naïve T and B cell emigrant output.
KRECs assessment
With hypo-gamma-globulinemia and increased susceptibility to recurrent sino-pulmonary infections, patients classified as ‘predominantly antibody deficiencies’ and CID had disturbed B cell development with decreased KRECs (< 157.7 copy number). However, some patients with Btk mutation (Btk-1) and RAG1 mutations (SCID6) had leaky phenotype with higher KRECs. There were no low KRECs in the study cohort among the ‘combined immunodeficiencies with associated or syndromic features’ category.
Relationship of TRECs and KRECs to naïve and memory cells
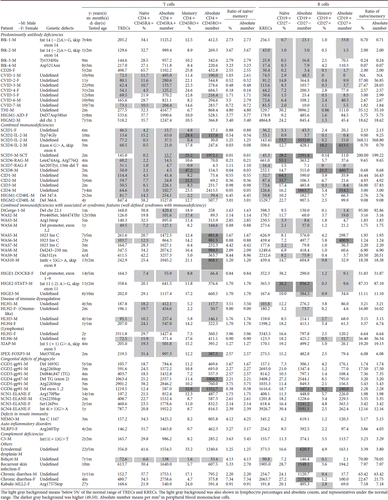
The copy numbers of TRECs and KRECs representatives for naïve emigrants of T and B cells were positively proportional to the absolute numbers and percentages of naïve CD4 + CD45RA+ T cells and the absolute number of naïve CD19 + CD27- B cells, respectively (; ). In the KRECs correlation, the absolute number of CD19 + CD27+ memory B cells was positively proportional to CD19 + CD27- naïve B cells (P = 0.027) () and indirectly positively correlated to KRECs (). However, although not statistically significant, the TRECs value tended to be opposite of memory CD4 + CD45RO+ and memory CD8 + CD45RO+ T cell percentages because the total T cell pool remained relatively constant and was mainly made up of both naïve CD4 + CD45RA+ T cells and memory CD4 + CD45RO+ T cells, while waxing memory T cells paralleled the weaning of naïve T cells by age.
Figure 2. The representative significant linear correlation between A: TRECs versus absolute naïve CD4 + CD45RA+ T cells percentage; B: TRECs versus absolute naïve CD4 + CD45RA+ T cells; C: KRECs versus absolute naïve CD19 + CD27- B cells; D: absolute naïve CD19 + CD27- B cells versus absolute memory CD19 + CD27+ B cells; E: KRECs versus absolute memory CD19 + CD27+ B cells; and F: TRECs versus the ratio of CD4 + plus CD8 + naïve/memory percentages.
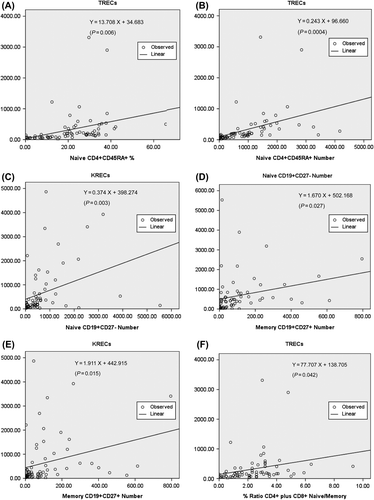
Table IV. The copy number of TRECs and naïve and memory CD8 + lymphocytes in patients with primary immunodeficiency diseases.
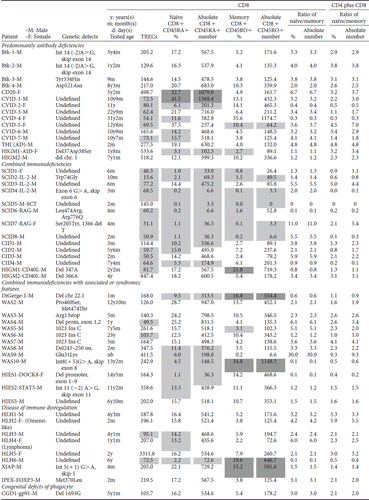
Table IV. (Continued)
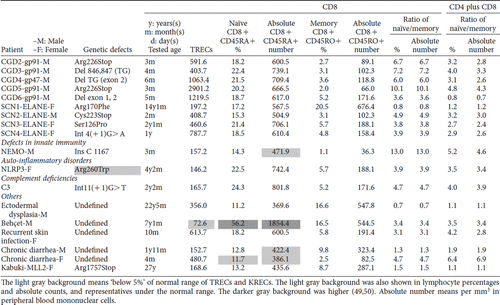
In contrast, relatively fewer but gradually expanding total B cell pool (including both naïve CD19 + CD27- cells and memory CD19 + CD27+ cells) by age made the naïve/memory B cell ratio inversely proportional to the KRECs value, although not statistically significant (). This implied the possibility that in peripheral blood, memory CD19 + CD27+ B cells increased more than that of naïve CD19 + CD27- B cells by age, although both positively correlated to KRECs.
Table V. The relationship of TRECs and KRECs with the respective naïve and memory cells.

Naïve CD8 + CD45RA+ and naïve CD8 + CD45RO+ were also evaluated () but did not significantly correlate with the TRECs value (). Comparing all naïve CD3 + T cells by taking naïve CD4 + and CD8 + T cells together, the TRECs values were positively proportional to the ratio of CD4 plus CD8 naïve/memory percentages (). But unlike naïve CD4 + cells, the TRECs values in naïve CD8 + dynamics are more disturbed by co-morbidity of autoimmunity, allergic characteristics, and/or co-infection in PIDs patients and warrant further investigations.
Discussion
Utilizing TRECs and KRECs to evaluate naïve T and B cell development has the potential of classifying heterozygous PIDs into four components by the low and not-so-low values of TRECs and KRECs to guide physicians in reconstructing immunity (Citation19–21). If patients with low TRECs compatible to low absolute naïve (CD4 + CD45RA+) T cell count and profoundly impaired lymphocyte proliferation (Citation6) present with opportunistic infections, failure to thrive, and refractory course, then HSCT is still a practical solution if suitable donors are available (Citation20,Citation22,Citation23).
For those with low KRECs as well as decreased naïve B lymphocyte subsets (CD19 + CD27-) (Citation24) and/or below the physiologic level of immunoglobulins, regular IVIG supplementation decreases the frequency of recurrent sino-pulmonary infections and prevents the sequelae of recurrent otitis media causing hearing impairment, severe sinus complication, bronchiectasis, and even cor pulmonale (Citation25,Citation26). Such assessment for naïve emigrant lymphocytes has benefits for patients who lack a family history but later deteriorate into life-threatening catastrophes (Citation27).
It is easy to understand how low TRECs and KRECs are detected in the ‘combined T and B immunodeficiencies’ category (Citation9,Citation10) and low KRECs in the ‘predominant antibody deficiencies’. In the ‘combined immunodeficiencies with associated and syndromic features’, approximately 5% of DGS patients with deletion chromosome 22q11.2 have thymic hypoplasia leading to decreased naïve T cell output, low TRECs, and SCID phenotype (Citation28–30). Moreover, DOCK8-HIES patients with extremely low TRECs from neonatal Guthrie cards may be classified into the ‘combined immunodeficiency’ category (Citation31). In contrast, the DGS patient here (DiGeorge-1-M) has no lymphopenia but has normal lymphocyte proliferation with normal TRECs value. The DOCK8-patient here (HIES1-DOCK8-F) also has normal TRECs and KRECs levels despite severe opportunistic infections (CMV and molluscum contagiosum), bronchiectasis, intra-cranial vasculitis, and cutaneous malignant transformation (Citation32). Although patients with the DOCK8 mutation are thought to be candidates for HSCT because of defective CD8 development and dysfunction against viral infection (Citation33,Citation34), TRECs from whole blood in adolescence may not be consistent with that of the neonatal Guthrie card, suggesting that the compensation for TRECs value is by other naïve T cells rather than by naïve CD8 + T cells.
Phenotypic spectrums may exist in the same mutant location even within a family. The severity in different mutations of the same gene depends on whether or not critical domains are involved. Thus, some with IL-2RG (Citation7,Citation35), RAG (Citation36), and adenosine deaminase deficiency (ADA) mutations outside such critical domains (Citation7,Citation35–37) have normal TRECs in the neonatal Guthrie card. Those with NEMO mutations have lower borderline TREC values from the neonatal Guthrie card (Citation38,Citation39) but normal values from whole blood beyond the newborn stage in the NEMO-M patient here whose mutation may be outside critical domains responsible for naïve lymphocyte development. In the ‘disease of immune dysregulation’ category, including six infection-associated HLH phenotypes without identified molecular defects, one with XIAP mutation, and one with FOXP3 mutation, only two without molecular defects had low TRECs, while another had low KRECs.
Of the ‘predominantly antibody deficiencies’ category, CVID is the most popular, with half as adult-onset (Citation40,Citation41). Approximately 5%–10% of CVID patients are identified as overlap syndromes (with Btk, CD40L, and SAP/SHD1A mutations) or have molecular defects (ICOS, TACI, BAFF-R, and CD19) (Citation42). In most CVID treatment, regular IVIG infusions treat hypo-gamma-globulinemia due to B cell defects to prevent irreversible sequelae of bronchiectasis and cor pulmonale (Citation41). However, overlapping with leaky SCID and CID phenotypes with T cell dysfunctions, the subgroup of CVID patients develop nodular granulomatous pulmonary diseases (Citation43–45) and increased susceptibility to opportunistic infections (Citation46), compatible with low TRECs and/or decreased lymphocyte proliferation.
In the six CVID patients with low TRECs in the present study, the phenotype of T cell defects occurred in four patients, including Takayasu arteritis (CVID-1), splenomegaly (CVID-3), and failure to thrive (CVID-6 and CVID-7). Although HSCT is not successful in patients with predominant antibody deficiencies caused by Btk mutations (Citation47), this subgroup of CVID patients with T cell defects and low TRECs may have leaky SCID or CID phenotypes and warrant optimal intervention of HSCT (Citation46). Thus, tracing longitudinal TRECs values in such CVID patients should remind physicians of timely HSCT to attenuate susceptibility to opportunistic infection and reconstruct their immunity.
The incidence of PIDs is around 1–5 per 100,000. They are considered orphan disorders (Citation40). Underestimation and delayed diagnosis often makes patients suffer from irreversible complications and mortality. Neonatal Guthrie card screening by TRECs from the blood dots rescues unfortunate infants with profound T cell deficiency by successful HSCT as early as possible (Citation38,Citation48). Furthermore, neonatal screening by KRECs will be larger-scaled applied after overcoming the technique difficulty of having lower B cell numbers than T cell numbers. A standard reference of plasmid construct consisting of TRECs and KRECs in the present study has been designed to measure the naïve lymphocyte emigrants and echoes the advantage and importance of neonatal Guthrie card screening. This assay demonstrates at least that SCID, CID, and some CVID patients have decreased naïve T cell and naïve B cell developments. Similar to newborn screening, the plasmid construct provides a simple way of investigating dynamic naïve lymphocyte development in complex syndromes with increased susceptibility to infections for the early recommendation of HSCT and IVIG based on four components divided by low and not-low TRECs and KRECs, especially for PIDs patients with early onset age beyond the neonatal period.
Acknowledgements
The authors wish to thank all of the patients and their families for their co-operation, as well as their physicians for the referrals.
Declaration of interest: All of the authors declare no conflicts of interest.
This study was supported by Chang-Gung Medical Research Progress (Grant CMRPG 4B0051) and the National Science Council (Grants NSC 99-2314-B-182-003-MY3 and NSC 102-2314-B-182A-039-MY3).
References
- Douek DC, Vescio RA, Betts MR, Brenchley JM, Hill BJ, Zhang L, et al. Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet. 2000;355:1875–81.
- Verschuren MC, Wolvers-Tettero IL, Breit TM, Noordzij J, van Wering ER, van Dongen JJ. Preferential rearrangements of the T cell receptor-delta-deleting elements in human T cells. J Immunol. 1997;158:1208–16.
- Siminovitch KA, Bakhshi A, Goldman P, Korsmeyer SJ. A uniform deleting element mediates the loss of kappa genes in human B cells. Nature. 1985;316:260–2.
- van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007;204:645–55.
- Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–8.
- Routes JM, Grossman WJ, Verbsky J, Laessig RH, Hoffman GL, Brokopp CD, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465–70.
- Morinishi Y, Imai K, Nakagawa N, Sato H, Horiuchi K, Ohtsuka Y, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal Guthrie cards. J Pediatr. 2009;155:829–33.
- Baker MW, Grossman WJ, Laessig RH, Hoffman GL, Brokopp CD, Kurtycz DF, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:522–7.
- Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2:54.
- Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;22:162.
- Griffith LM, Cowan MJ, Notarangelo LD, Kohn DB, Puck JM, Pai SY, et al.; workshop participants. Primary Immune Deficiency Treatment Consortium (PIDTC) report. J Allergy Clin Immunol. 2014;133:35–47.
- Borte S, von Döbeln U, Fasth A, Wang N, Janzi M, Winiarski J, et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 2012; 119:2552–5.
- Lee WI, Kuo ML, Huang JL, Lin SJ, Wu CJ. Distribution and clinical aspects of primary immunodeficiencies in a Taiwan pediatric tertiary hospital during a 20-year period. J Clin Immunol. 2005;25:162–73.
- Lee WI, Jaing TH, Hsieh MY, Kuo ML, Lin SJ, Huang JL. Distribution, infections, treatments and molecular analysis in a large cohort of patients with primary immunodeficiency diseases (PIDs) in Taiwan. J Clin Immunol. 2006;26:274–83.
- Lee WI, Huang JL, Jaing TH, Shyur SD, Yang KD, Chien YH, et al. Distribution, clinical features and treatment in Taiwanese patients with symptomatic primary immunodeficiency diseases (PIDs) in a nationwide population-based study during 1985-2010. Immunobiology.2011;216:1286–94.
- Lee WI, Huang JL, Yeh KW, Yang MJ, Lai MC, Chen LC, et al. Clinical features and genetic analysis of Taiwanese patients with the hyper IgM syndrome phenotype. Pediatr Infect Dis J. 2013;32:1010–16.
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5.
- Hazenberg MD, Otto SA, de Pauw ES, Roelofs H, Fibbe WE, Hamann D, et al. T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. Blood. 2002;99:3449–53.
- Lipstein EA, Vorono S, Browning MF, Green NS, Kemper AR, Knapp AA, et al. Systematic evidence review of newborn screening and treatment of severe combined immunodeficiency. Pediatrics. 2010;125: e1226–35.
- Kildebeck E, Checketts J, Porteus M. Gene therapy for primary immunodeficiencies. Curr Opin Pediatr. 2012;24:731–8.
- de la Morena MT, Nelson RP Jr. Recent advances in transplantation for primary immune deficiency diseases: a comprehensive review. Clin Rev Allergy Immunol. 2014;46:131–44.
- Horn B, Cowan MJ. Unresolved issues in hematopoietic stem cell transplantation for severe combined immunodeficiency: need for safer conditioning and reduced late effects. J Allergy Clin Immunol. 2013;131:1306–11.
- Cavazzana-Calvo M, André-Schmutz I, Fischer A. Haematopoietic stem cell transplantation for SCID patients: where do we stand? Br J Haematol. 2013;160:146–52.
- Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131:959–71.
- Bonagura VR. Using intravenous immunoglobulin (IVIG) to treat patients with primary immune deficiency disease. J Clin Immunol. 2013;33:S90–4.
- Schroeder HW Jr, Dougherty CJ. Review of intravenous immunoglobulin replacement therapy trials for primary humoral immunodeficiency patients. Infection. 2012;40:601–11.
- Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–19.
- McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). 2011;90:1–18.
- Fernández L, Nevado J, Santos F, Heine-Suñer D, Martinez-Glez V, García-Miñaur S, et al. A deletion and duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Med Genet. 2009;10:48.
- Markert ML, Hummell DS, Rosenblatt HM, Schiff SE, Harville TO, Williams LW, et al. Complete DiGeorge syndrome: persistence of profound immunodeficiency. J Pediatr. 1998;132:15–21.
- Dasouki M, Okonkwo KC, Ray A, Folmsbeel CK, Gozales D, Keles S, et al. Deficient T cell receptor excision circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening. Clin Immunol. 2011;141:128–32.
- Lee WI, Huang JL, Lin SJ, Yeh KW, Chen LC, Ou LS, et al. Clinical, immunological and genetic features in Taiwanese patients with the phenotype of hyper-immunoglobulin E recurrent infection syndromes (HIES). Immunobiology. 2011;216:909–17.
- Al-Mousa H, Hawwari A, Alsum Z. In DOCK8 deficiency donor cell engraftment post-geno-identical hematopoietic stem cell transplantation is possible without conditioning. J Allergy Clin Immunol. 2013;131:1244–5.
- Ghosh S, Schuster FR, Fuchs I, Laws HJ, Borkhardt A, Meisel R. Treosulfan-based conditioning in DOCK8 deficiency: complete lympho-hematopoietic reconstitution with minimal toxicity. Clin Immunol. 2012;145:259–61.
- Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130:177–83.
- Ijspeert H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immuno-biological effects but in different clinical phenotypes. J Allergy Clin Immunol. 2014;13: 1848–4.
- la Marca G, Canessa C, Giocaliere E, Romano F, Duse M, Malvagia S, et al. Tandem mass spectrometry, but not T-cell receptor excision circle analysis, identifies newborns with late-onset adenosine deaminase deficiency. J Allergy Clin Immunol. 2013;131:1604–10.
- Verbsky J, Thakar M, Routes J. The Wisconsin approach to newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:622–7.
- Verbsky JW, Baker MW, Grossman WJ, Hintermeyer M, Dasu T, Bonacci B, et al. Newborn screening for severe combined immunodeficiency: the Wisconsin experience (2008–2011). J Clin Immunol. 2012; 32:82–8.
- Gathmann B, Binder N, Ehl S, Kindle G; ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: update2011. Clin Exp Immunol.2012; 167:479–91.
- Chapel H. Classification of primary immunodeficiency diseases by the International Union of Immunological Societies (IUIS) Expert Committee on Primary Immunodeficiency2011. Clin Exp Immunol.2012;168:58–9.
- Goldacker S, Warnatz K. Tackling the heterogeneity of CVID. Curr Opin Allergy Clin Immunol. 2005;5:504–9.
- Baldovino S, Montin D, Martino S, Sciascia S, Menegatti E, Roccatello D. Common variable immunodeficiency: crossroads between infections, inflammation and autoimmunity. Autoimmun Rev. 2013; 12:796–801.
- Salzer U, Warnatz K, Peter HH. Common variable immunodeficiency - an update. Arthritis Res Ther. 2012;14:223.
- Boursiquot JN, Gérard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al.; DEFI study group. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol. 2013;33:84–95.
- Kamae C, Nakagawa N, Sato H, Honma K, Mitsuiki N, Ohara O, et al. Common variable immunodeficiency classification by quantifying T-cell receptor and immunoglobulin κ-deleting recombination excision circles. J Allergy Clin Immunol. 2013;131:1437–40.e5.
- Howard V, Myers LA, Williams DA, Wheeler G, Turner EV, Cunningham JM, et al. Stem cell transplants for patients with X-linked agammaglobulinemia. Clin Immunol. 2003;107:98–102.
- Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132:140–50.
- Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al.; Pediatric AIDS Clinical Trials Group. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003;112:973–80.
- Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, van der Burg M, et al. Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. 2011;74:502–10.