
Abstract
The primary unseptated heart tube undergoes extensive remodeling including septation at the atrial, atrioventricular, ventricular, and ventriculo-arterial level. Alignment and fusion of the septal components is required to ensure full septation of the heart. Deficiencies lead to septal defects at various levels. Addition of myocardium and mesenchymal tissues from the second heart field (SHF) to the primary heart tube, as well as a population of neural crest cells, provides the necessary cellular players. Surprisingly, the study of the molecular background of these defects does not show a great diversity of responsible transcription factors and downstream gene pathways. Epigenetic modulation and mutations high up in several transcription factor pathways (e.g. NODAL and GATA4) may lead to defects at all levels. Disturbance of modulating pathways, involving primarily the SHF-derived cell populations and the genes expressed therein, results at the arterial pole (e.g. TBX1) in a spectrum of ventricular septal defects located at the level of the outflow tract. At the venous pole (e.g. TBX5), it can explain a variety of atrial septal defects. The various defects can occur as isolated anomalies or within families. In this review developmental, morphological, genetic, as well as epigenetic aspects of septal defects are discussed.
Key messages
The atrial and ventricular septum develops, by way of various mechanisms, from several components. Defects can be due to 1) deficient looping of the heart tube and subsequent malalignment of septal components; 2) abnormal or absent fusion of the endocardial cushions; 3) deficiency of second heart field (SHF) and neural crest cell contributions to outflow and inflow tract; and 4) hypoplasia of myocardial septal components.
Morphogenetically important genes for septal defects are predominantly expressed in the derivatives of the SHF population and to a lesser extent in the cardiac neural crest cells.
Gain or loss of function of a relatively small number of morphogenetic transcription and growth factors affects the various embryonic cell populations leading, based on their temporo-spatial expression, to atrial, atrioventricular, and/or ventricular septal defects.
The majority of the septal defects seen in the human population are the result of genetic modulation by epigenetic factors including histone methylation, miRNA regulation, environmental factors, and dosage effects that all interact in the very complicated gene regulatory pathways.
The in-depth understanding of the mechanisms leading to a specific septal defect or a spectrum of defects within families needs a systems biology approach.
Introduction
Atrial septal defects (ASDs), ventricular septal defects (VSDs), and atrioventricular septal defects (AVSDs) are major contributors to the spectrum of congenital heart disease (CHD). The incidence of congenital heart disease is an estimated 10 per 1000 live births. Of these, 11.6% are ASDs, 30.7% are VSDs, and 2% AVSDs (Citation1). Novel insights into the development of the heart and the contributing embryonic cell types allow for a better understanding of the observed variety in septal defects. This progress is mainly due to the use of animal models of which the avian and the (transgenic) mouse models are leading in the field. It should be taken into account that most mouse models present with a complete knock-out phenotype which might be embryolethal in the human population. However, in humans the increased possibilities for exome and whole-genome screening link relevant morphogenetic genes gathered from mouse transgenic lines (Citation2–4) to our understanding of genetic, epigenetic (Citation5,Citation6), and environmental (Citation7–11) causes of human CHD. There is a continuous increase in our knowledge, with new data emerging from the various novel approaches in genetics including the relevance of modifying genes (Citation3,Citation12).
In this paper we will allude to the current knowledge and incorporate concepts on the morphogenetic and molecular causes of ASDs, VSDs, and AVSDs in human CHD. Development of terminology particularly in human septal defects has expanded over the years from pure morphologically based (Citation13–15) to additional developmentally influenced terminology (Citation16). With regard to development-based concepts, new findings lead to use of new terms and concepts which may be confusing, also because the nomenclature in the avian and mouse model literature is not always identical (Citation17). In general the very informative segmental analysis for clinical use and attempting to bridge the gap between basic scientists and clinicians (Citation18) is not designed for understanding the developmental background of cardiac septation and the involved genes. In a short introduction we will provide the developmental concepts followed by morphology, pathology, and homology of terms in this field before venturing into underlying molecular considerations.
Developmental concepts of cardiac septation
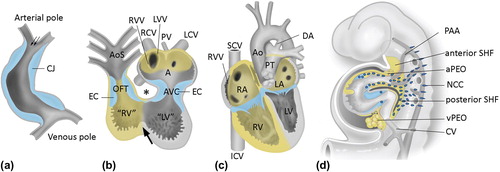
A real step forward in our understanding of cardiac development including cardiac septation was made by the recognition of the embryonic second heart field (SHF) population () adding new myocardium and endocardium to the cardiac outflow (anterior SHF-derived) and inflow tract (posterior SHF-derived) of the heart (Citation19). The so-called first heart field (FHF) forms the primary heart tube which connects the cardiac inflow tract, the sinus venosus, via the atrioventricular (AV) canal to the primitive ventricle and the arterial pole (). The primary heart tube loops to the right with an inner curvature in between the inflow and outflow tract. At this inner curvature is situated part of the bulboventricular or primary fold that encircles the primary heart tube (, arrow).
Figure 1. The developing heart; contributions from the second heart field. a: Primary heart tube, consisting mainly of the putative left ventricle (LV) and only very limited parts of the inflow tract (atria, sinus venosus) and outflow tract of the heart. The myocardium of the primary heart tube is derived from the splanchnic mesoderm of the primitive plate, the so-called ‘first heart field’ (FHF). Fetal blood enters the blood at the venous pole, and via peristaltic movements is propelled to the arterial pole of the heart tube. The tube consists at this stage of endocardial and myocardial cells (depicted in grey), with cardiac jelly (CJ, depicted in blue) in between. No epicardial covering is present at this stage. b: The heart tube after looping has been initiated. The tube is now U-shaped. The inner curvature is indicated by an asterisk and is continuous with the bulboventricular or primary fold (arrow) that encircles the heart tube between the primitive left ventricle (“LV”) and primitive right ventricle (“RV”). FHF-derived parts of the myocardium are depicted in grey. In addition, myocardium has been recruited to the arterial and venous heart tube from the mesenchyme situated behind the heart during development, the so-called ‘second heart field’ (SHF, also see panel d). SHF-derived structures are depicted in yellow. The CJ has concentrated at the AV canal (AVC) and outflow tract (OFT) to the endocardial cushion (EC) and will contribute to, for example, the cardiac valves during further development. The venous pole consists of the right cardinal vein (RCV, putative superior caval vein), left caval vein (LCV, that will form part of the putative coronary sinus), and vitelline veins entering the tube. Atrial and ventricular septation have not occurred, yet. The blood from the common atrium (A) passes through the AVC into “LV”, and via the interventricular foramen to the primitive right ventricle “RV” and OFT. The latter is initially positioned entirely above the “RV”. c: Situation after atrial and ventricular septation has been completed. A right atrium (RA) to RV connection has now been formed by an expansion of the AV canal to the right side, thus forming the tricuspid ostium. The OFT has been separated into an aorta (Ao) connecting to the LV and a pulmonary trunk (PT) still connecting to the RV. SHF-derived myocardium is depicted in yellow. d:Scheme of the embryo (lateral view) demonstrating the cellular contributions to the heart. The SHF mesenchyme is depicted in yellow, as are structures derived from this mesenchyme. SHF contributing to the arterial pole of the heart is referred to as ‘anterior SHF’, whereas cells are recruited to the venous pole from the ‘posterior SHF’. The pro-epicardial organ (PEO), that is present at both the venous (vPEO) and arterial (aPEO) pole of the heart (Citation33), is also derived from the SHF. Neural crest cells (NCC, indicated by dark blue dots) reflect an extracardiac contribution to the heart. For additional explanation see text. (AoS = aortic sac; CV = cardinal vein; DA = ductus arteriosus; ICV = inferior caval vein; LA = left atrium; LVV = left venous valve; PAA = pharyngeal arch arteries; PV = primitive pulmonary vein; SCV = superior caval vein; RVV = right venous valve).
Development of the atrial septum
The human atrial septum consists of several components including the septum primum that grows in from cranio-posterior as a thin muscular structure with an endocardial cushion-like mesenchymal cap lining the free under-rim () (Citation17,Citation20). The primordium of the pulmonary veins arises directly on the left side of the septum primum. It is commonly accepted that the SHF-derived myocardium surrounding the sinus venosus is incorporated into the posterior right atrial wall, flanked by two valve flaps, the right and left venous valves. Whether or not this sinus venosus contribution expands to the posterior wall of the left atrium (LA), the wall of the coronary sinus, the common pulmonary vein area, and the septum primum () has been a matter of contention for a long time (Citation21). Recently, however, this problem has been solved by cardiac progenitor tracing studies (Citation22) supporting the separate origin of the right myocardial covering of the cardinal veins including the dorsal right atrial wall, versus the left cardinal vein, left atrium, and pulmonary vein area from the posterior SHF, which has implications for understanding the molecular background of ASDs.
Figure 2. Atrial septation. a: Early developmental stage. Atrial septation starts with formation of a muscular septum primum (SP), that grows out from the roof of the atrium towards the AV canal (AVC). The lower rim of the SP consists of mesenchymal tissue, the mesenchymal cap (MC, indicated in dark blue) that is continuous with the endocardial cushions in the AV canal (light blue). Initially, the lower part of the SP will not connect to the tissues in the AV canal, leaving an opening, the ostium primum (OP). b: During further development, the OP will be closed by fusion of the endocardial cushion with the mesenchymal cap, as well as with second heart field (SHF) mesenchyme protruding into the heart at the base of the atrial septum. This protrusion is referred to as dorsal mesenchymal protrusion (DMP). Meanwhile, the ostium secundum (OS) in the muscular SP has developed by a process of apoptosis. The septum secundum (SS) is formed at the right side of the SP by an infolding of the roof of the atrium, but will never entirely reach the AVC. The free rim of the septum secundum forms the limbus of the foramen ovale. During the entire fetal phase the foramen ovale (consisting of the opening between the lower rim (limbus) of the SS and the OS secundum) will allow shunting of blood from the right atrium (RA) to the left atrium (LA) (arrow in panel b). c: Schematic overview of the three structures responsible for proper closure of the OP during development: the atrioventricular endocardial cushions (EC, light blue), the mesenchymal cap (MC, dark blue), and the dorsal mesenchymal protrusion (DMP, yellow). d–f: Transverse sections stained with the myocardial marker myosin light chain 2a (MLC2a) of murine embryonic wild-type hearts, stage E12.5. d. is the most superior and f. the most inferior section. Elements contributing to normal atrial septation are indicated. (AS = atrial septum; LV = left ventricle; LCV = left cardinal vein; LVV = left venous valve; MO = mitral ostium; PV = pulmonary vein; TO = tricuspid ostium; RV = right ventricle; RVC = right cardial vein; RVV = right venous valve; VS = ventricular septum). a and b: modified after (Citation116).
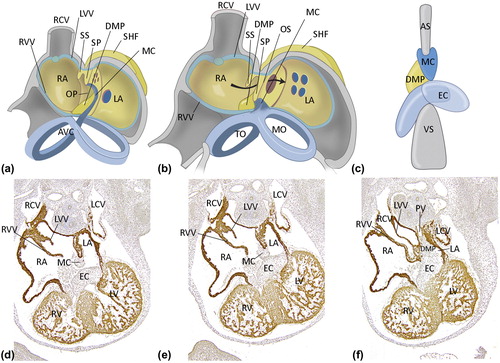
The mesenchymal cap of the septum primum fuses with the AV cushions and the dorsal mesenchymal protrusion (DMP), also derived from the posterior SHF, eventually forming a firm muscular basis of the atrial septum (). The septum secundum is an infolding of the cranial and anterior wall of the right atrium (RA) which is present in mouse embryos, but lacking in avian embryos. In conclusion the atrial septum is almost in its entirety derived from the posterior SHF.
Development of the ventricular septum
In an evolutionary concept we are confronted with species that lack both atrial and ventricular septation like fishes. For ventricular septation different possibilities are observed between species varying from incomplete septation, harboring a horizontal and a vertical septum between three cava in most reptiles like turtles and snakes (Citation23), to complete septation in birds and mammals including humans. It is tempting to learn from an evo-devo approach although we still do not know whether the reptilian horizontal and vertical septa are homologous to, for instance, the anterior and inlet septum components in birds and mammals.
Three main concepts on development of the ventricular septum have been advocated between the late 1970s up till recent time. The first one distinguishes three septal components during development (, ): 1) the anterior or trabecular septum having the septal band (trabecula septomarginalis) as posterior rim; 2) the posteriorly located inlet septum that has a separate origin below the inferior AV cushion; and 3) the outflow tract (OFT) septum (synonymous to septum aorto-pulmonale or conotruncal septum) which develops as the last part (Citation24,Citation25).
Figure 3. Ventricular septation. a: Schematic overview of ventricular septation. Remodeling of the primary fold (PF) is essential for the formation of the right ventricular (RV) inlet compartment and positioning of the right side of the AV canal (AVC) above the RV, which will result in formation of the tricuspid orifice (TO). The mitral orifice (MO) will remain above the left ventricle (LV). Formation of the TO and MO is achieved by fusion of the atrioventricular endocardial cushions (indicated in pale blue). The anterior trabeculated part of the ventricular septum (TS) derives from infolding of the anterior part of the primitive ventricle, whereas the inlet septum (IS) and inlet cavity of the RV are formed by expansion of the posterior part of the PF. The borderline between the RV inlet compartment and folding septum is visible as the septal band (SB). b and c: In the outflow tract (OFT), the endocardial cushions (indicated in pale blue) fuse to separate the left and right ventricular OFT in an aorta (Ao) and pulmonary trunk (PT). During normal septation, neural crest cells migrate into the cushions and orchestrate the formation of an OFT septum before they go into apoptosis (not shown). At the ventricular level, the primary interventricular foramen (double arrow) is still patent but will be closed with completion of OFT septation and connection of the Ao to the LV. (ICV = inferior caval vein; LA = left atrium; LV = left ventricle; MO = mitral orifice; SCV = superior caval vein; RA = right atrium; TO = tricuspid orifice). Modified after: Crawford Cardiology (Citation117).
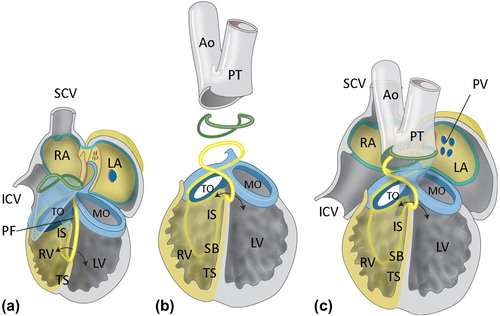
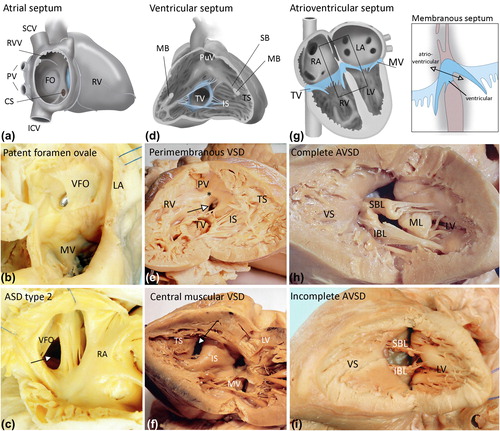
Figure 4. The cardiac septa in the adult and morphology of septal defects. a: Normal atrial septum after birth, right lateral view. A fenestration has been drawn to allow visualization of the atrial septum. The foramen ovale is closed and can be recognized as the fossa ovalis (FO). b: Patent foramen ovale, as seen from the left atrium (LA). The valvula foraminis ovalis (VFO, derived from the embryological septum primum) is competent but has not fused with the septum secundum, thus allowing the probe to be inserted from the right side. c: Atrial septal defect type 2, as viewed from the right atrium (RA). The valvula foraminus ovalis (VFO, septum primum) is deficient, thus leaving a septum defect (arrow). d: Normal ventricular septum, as viewed from the right side. The different septal components can be recognized and include the ventricular inlet septum (IS), below the tricuspid orifice, and the anterior and apical trabeculated parts (TS). The septal band (SB) and moderator band (MB, which has been cut in order to open the RV) are indicated. e: Perimembranous ventricular septal defect (arrow), as seen from the right ventricle (RV). f: Central muscular ventricular septal defect (arrow), as seen from the left ventricle (LV). The defect is classically situated at the border between two septal components, i.e. the inlet septum (IS) and the trabeculated septum (TS). g: Normal atrioventricular septum. Note the differential insertion of the tricuspid valve (TV) and mitral valve (MV), resulting in the length of the AV septum between the right atrium and left ventricle. The membranous septum consists of an atrioventricular (double arrow) and interventricular part. h: Complete AVSD. The anterior bridging leaflet (ABL) and inferior bridging leaflet (IBL) are not attached to the atrial septum (not shown) and ventricular septum (VS), allowing shunting at both the atrial and ventricular level. i: Incomplete AVSD. Both the superior bridging leaflet (SBL) and inferior bridging leaflet (IBL) are attached to the ventricular septum (VS), therefore shunting can only occur at the atrial level (not shown). (CS = coronary sinus; ICV = inferior caval vein; ML = mural leaflet; MV = mitral valve; PV = pulmonary veins; PuV = pulmonary valve; RVV = right venous valve; SCV = superior caval vein; VS = ventricular septum).
The second concept of ventricular septal development does not appreciate the origin of a separate inlet septum but holds the primary (bulboventricular) fold (, ) responsible for both the anterior trabecular and the inlet septum. In this latter situation the tricuspid orifice has to move over this primary fold to the right side in order to reach the inlet of the right ventricle (RV) (Citation26,Citation27). In a third concept, the tricuspid orifice forms a small slit-like RV inlet component within the posterior part of the primary fold () (Citation17,Citation28), that will expand during further development. Up till now no definitive answers have been found to decide upon the most realistic concept. Recent investigations from our group incorporating molecular and immunohistochemical analysis of ventricular septation, using an evo-devo approach, support concepts one and three, which advocate a separate origin of the inlet septum (Poelmann, 2014). These concepts allow for the best explanation of the inlet septal defects including the defects seen in straddling tricuspid valve, as well as the central muscular defect, and will therefore serve as basis for our developmental considerations on VSD.
The so-called atrioventricular septum, situated partly between the left ventricle (LV) and RV and partly between LV and RA, is composed of both muscular and membranous (fibrous) components. It is not a separate entity but can be distinguished because of the remodeling of the area between the LV outflow tract and the attachment of the septal leaflet of the tricuspid valve to the membranous septum ().
The RV myocardium is derived from the anterior SHF. Details on the origin of the anterior trabecular and inlet septum are confusing as some claim the right side of the ventricular septum to be anterior SHF-derived while others indicate that the complete ventricular septum is SHF-derived (Citation29).
Development of the OFT septum has been adequately described using avian and mouse models without much discussion on possible concepts (Citation30–32). This structure is derived from the OFT endocardial cushions and underlying myocardium, which are from anterior SHF origin. The distal (truncal) part of the cushions is mainly recruited for semilunar valve development, while the proximal (conal) septal and parietal OFT endocardial ridges extend into the myocardial OFT. Neural crest cells that migrate into the arterial pole region of the heart are important for the septation at the level of the aortic and pulmonary orifice. They also contribute to the differentiation of the semilunar valves, and the two prongs that extend into the proximal part of the OFT ridges are important for the induction of the myocardialization of the subpulmonary OFT endocardial cushion tissue. This myocardium in fact realizes the separation of a long subpulmonary infundibulum and a short subaortic OFT. In a normal heart the actual OFT septum is very small and in fact only recognizable as a separate structure in CHD with VSDs at the level of the OFT (Citation17).
Data from our group show a major role for the epicardium in the process of ventricular septation (Poelmann, 2014). The anterior septal component develops as a folding structure between the RV and LV incorporating the overlying epicardium and subepicardial tissue. The inlet septum is a more solid myocardial structure harboring many epicardium-derived cells (EPDCs) similar to the lateral myocardial walls of the RV and LV () (Citation33). The apical trabecular septum expands during ventricular cavity formation, and an extensive network of epicardial and endocardial lined vascular structures are harbored within this region.
In conclusion the anterior trabecular and inlet septal components originate from both the FHF and the SHF. The OFT septum is derived from the anterior SHF and also incorporates the neural crest cells that induce myocardialization of the OFT endocardial cushions.
Morphology and morphogenesis of septal defects
The terminology used below is for the major part consistent between development and (patho)morphology so that an optimal relation between both disciplines is achieved. In the completely septated heart the components distinguished during development are best viewed from the right side both in the atrium and the ventricle ().Variations in human septal defects considered from a developmental point of view and supported by data from human, avian, and mouse models will be presented in the following paragraphs.
Atrial septum and ASDs
In the mature atrial septum, the various components contributing during development can still be recognized (). The atrial septum shows anteriorly a marked muscular ridge, the limbus fossa ovalis (the lower rim of the embryonic septum secundum) bordering the fossa ovalis and the valve of the foramen ovale (derived from the embryonic septum primum). More posteriorly these structures merge fluently with the dorsal atrial wall. The base of the atrial septum is a thick muscular ridge that runs from the dorsal to the post-aortic area. In this region the ostium of the coronary sinus connects to the RA (). Normally, the foramen ovale closes after birth due to increased pressure in the LA after the initiation of the pulmonary circulation. The morphologically distinguished types of ASDs have been extensively described for surgical purposes (Citation13). We will focus on the most important ones in relation to development and known molecular background.
Atrial septum defects at the level of the ostium secundum/foramen ovale (ASD type 2)
ASD type 2 defects are present in the center of the atrial septum and are the result of inadequate connection of the embryonic septum primum and the infolded septum secundum (Citation34). These defects can be the result of a deficiency of the embryonic septum primum, the septum secundum, or both. Cases where there is a non-fusion of the (septum primum-derived) valve of the foramen ovale with the free rim of the septum secundum are referred to as delayed closure of the foramen ovale or as a persistent foramen ovale (PFO) (). In these cases the valvula foraminis ovalis is properly developed to close the foramen ovale, although a fusion with the septum secundum has not occurred, as is reported in up to 27% of adults with structurally normal hearts (Citation35). Since there is no shunting of blood during normal physiology, PFO is not considered an ASD. In some cases, a PFO is difficult to distinguish from an ASD type 2. In ASD type 2, the valvula foraminis ovalis is deficient and therefore incompetent to close the foramen ovale (). Likewise, fenestrations in the valve of the foramen ovale can lead to an ASD type 2 situation which can even be combined with either a competent (PFO) or incompetent valve of the foramen ovale.
In the genetic background, both in mouse models (Citation36,Citation37) as well as in the human population (Holt–Oram syndrome) (Citation38), the TBX5 gene plays a crucial role. Other mutations linked to ASD type 2 in both mouse models and humans are mutations in the NKX2.5 gene () (Citation39).
Table I. Genes encoding for transcription factors and proteins related to human congenital heart disease. Special attention is given to the NODAL (Δ) and the VEGF pathways (*).
Atrial septum defects at the level of the ostium primum (ASD type 1)
Closure of the ostium primum requires fusion of three embryonic components () (Citation40). It is rarely encountered as a solitary malformation and is mostly seen in the setting of AVSD () described below.
Atrial sinus venosus defects
These ASDs occur classically outside the region of the foramen ovale. The ASD is closely related to the ostium of the superior or inferior caval vein. Both superior and inferior sinus venosus ASDs are often associated with partial or total anomalous drainage of the pulmonary veins (Citation21). In human and animal studies abnormal drainage patterns at the venous pole of the heart concurrent with ASDs have been linked to mutations in the PDGFRα gene (Citation41).
Common atrium
This is a rare condition in which septation at the atrial level is lacking entirely. Usually, situs determination is still possible due to the presence of morphological right and left atrial appendages (Citation42).
Genes involved in human ASD formation are summarized in and depicted in . Many genetic mutations encountered in the posterior SHF have only been reported in mouse models, e.g. Podoplanin (Citation43) and Shox2 (Citation44). In human these mutations are most probably embryolethal.
Figure 5. Overview of genes involved in cardiac septation. Modified after (Citation2).
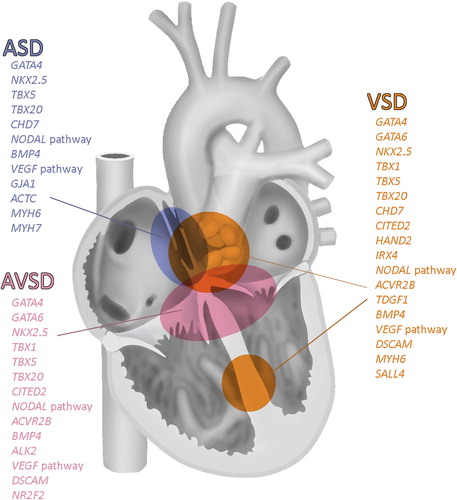
Ventricular septum and VSDs
Viewed from the LV the septum is a smooth structure without clear indication of its constituting muscular septal components with the exception of the membranous septum where the interventricular and atrioventricular component can be appreciated (, right panel). When VSDs are present the LV view may reveal some aspects of these muscular components. The ventricular septum, as viewed from the right side (), provides the best indication of the three proposed ventricular septal components. At the border of the anterior trabecular and inlet septum a muscular structure (variably pronounced) is found, referred to as the septal band or trabecula septomarginalis (). This band has an extension that crosses the cavity of the RV abutting on the right lateral wall, referred to as the moderator band ().
VSDs are mainly positioned on the border of the septal components (), and they may extend into various directions but can also be present within the myocardium of the components (some muscular VSDs). A detailed classification and terminology has been provided for clinicians (Citation15). We will describe a simplified version with information relevant for the developmental and molecular background.
Outflow tract ventricular septal defects
After the formation of the primary heart tube from the FHF there is a genetically determined primary dextral looping followed by secondary looping in which the aorta becomes wedged between the tricuspid and mitral orifices. Disturbances of this secondary looping process lead to a more dextral and anterior position of the aorta and complete or partial persistence of the AV canal above the LV. Some variations in morphological OFT defects can be appreciated dependent on the extension and positioning of the OFT septum, separating the aortic sac into an ascending aorta and pulmonary trunk. The most extreme situation is the so-called common arterial trunk (CAT) also called persistent truncus arteriosus, missing at least the intracardiac part of this OFT septum. This malformation is therefore always accompanied by a subarterial VSD. Initially based on avian studies with ablation of the cardiac neural crest population (Citation45), this abnormality was attributed to genes expressed in the neural crest cell population. Specific mouse models with neural crest cell mutations like Pax3 (Citation46) and Semaphorin3 (Citation47) lead to CAT, but also many variations with a hypoplastic or malaligned OFT septum were found (Citation48). A breakthrough was made based on deletion of the q11 region on chromosome 22 (22q11), a human genetic defect initially referred to by a number of syndromic names including the DiGeorge syndrome (Citation49). Research on the gene(s) responsible for the observed CAT and the variation in OFT VSDs resulted in recognition of the importance of the transcription factor TBX1 (Citation50,Citation51). However, this gene was not expressed in neural crest cells but in cells of the anterior SHF (Citation52). Thus, it became clear that a proper interaction of neural crest cells and SHF was essential for normal OFT septation. Other important morphogenetic genes expressed in these populations (, ) could lead to similar OFT VSDs.
In tetralogy of Fallot (TOF) a separation of the aorta and pulmonary trunk has taken place, but the myocardium of the OFT septum is deviated or malaligned to the remainder of the ventricular septum. The amount of deviation of the OFT septum contributes to the severity of the septal defect, aortic overriding, and subpulmonary stenosis. The VSD in TOF is often perimembranous, although muscular VSDs can also be observed. The VSDs are subaortic in case of a more or less prominent muscular OFT septum, and doubly committed (Citation53) in case of a small fibrotic OFT septum. TOF has been attributed to many genetic mutations (). Also copy number variations (CNV), mostly leading to gain of protein function are linked to TOF. However, a clear responsible gene or gene pathway covering most cases is missing, as yet. Sometimes it is possible to predict the cause of TOF by observing associated anomalies, e.g. as in the 22q11 deletion setting where there is a clear preponderance for accompanying left pulmonary artery obstruction as well as a right aortic arch.
In addition to the expected genetic mutations, also environmental factors can influence ventricular OFT septation which will be discussed in the paragraph on genetic and epigenetic considerations.
Outflow tract ventricular septal defects with transposed great arteries
During development of the OFT it is a prerequisite that the aorta becomes connected to the LV, while the RV connects to the pulmonary trunk and orifice, that during this remodeling process achieves an anteriorly more elevated position resulting in crossing of the great arteries. Many papers have alluded to the necessary OFT and endocardial cushion rotation (Citation55) to achieve this situation. Using novel data from the expression patterns of the SHF marker NKX2.5, we recently reported an asymmetric addition of myocardium to the future pulmonary trunk and right ventricular OFT. This ‘pulmonary push’ principle is held responsible for the observed rotation (Citation56). A deficient or abnormal asymmetric contribution to the OFT may underlie OFT anomalies, where the normal left anterior position of pulmonary trunk is not achieved, as is observed in e.g. double outlet right ventricle (DORV), or transposition of the great arteries. In transposition of the great arteries (TGA) combined with a VSD, the position of the VSD can vary. When the VSD is situated below the pulmonary orifice, it thus constitutes a subpulmonary VSD with a spectrum of mild to severe malalignment of the OFT septum. This is also referred to as Taussig–Bing malformation. Genetic information on the background of TGA is scarce (). There are a few mouse models like the Perlecan (Citation57), the Pitx2 (Citation58), and the Zic3 (Citation59) mutants, but these genes are also involved in left–right asymmetry (Citation60) and are not specific for the OFT VSDs seen in this malformation.
Outflow tract ventricular septal defects and double outlet right ventricle
Deficient looping and tightening of the inner curvature as well as deficient asymmetry in addition of second heart field myocardium can lead to DORV.
Double outlet right ventricle (DORV) always presents with a malalignment OFT VSD. Nevertheless it is neither a morphologic nor a morphogenetic entity. DORV only means that both great arteries are connected completely or for their major part to the RV.
Two main groups can be distinguished: 1) with crossed great arteries presenting with the aortic orifice more or less right posterior and with a subaortic VSD, that includes the TOF with DORV; and 2) with transposed great arteries with the aortic orifice more or less right anterior and with a subpulmonary VSD, also known as Taussig–Bing malformation. Thus, TOF and DORV can coexist in cases with extreme dextroposition of the aorta. Likewise, extreme dextroposition of the pulmonary orifice in Taussig–Bing malformation deserves the designation DORV. Specific genetic information on the two types of DORV is linked to the data on VSDs in TOF and TGA. It is remarkable that in both avian and mouse models for CHD the DORV with crossed (or ‘normally related’) great arteries is a common phenomenon.
The membranous and perimembranous VSDs
Closure of the interventricular foramen is achieved by fusion of the atrioventricular endocardial cushions with the septal and parietal OFT endocardial ridges together forming the substrate of the already described muscular OFT septum and part of the muscular subpulmonary infundibulum. At the cross-roads of the fusing endocardial cushions a membranous septum is formed, which usually has an interventricular and an atrioventricular (between the left ventricular OFT and the RA) part () (Citation61). A membranous VSD mostly implies a small defect in the interventricular part with still a complete encircling of the defect by the fibrous tissue.
A perimembranous VSD refers to a deficient membranous septum, although the posterior wall of the defect will always contain the fibrous tissue of the mitral to tricuspid (endocardial cushion-derived) connection () (Citation62).
Most OFT VSDs underneath the OFT septum can also be referred to as perimembranous VSDs. In case of additional deviation of the OFT septum either in the RV or LV they can be referred to as malalignment VSDs as well.
In case the interventricular and the atrioventricular part of the membranous septum are deficient we are dealing with an AVSD (see below).
Muscular ventricular septal defects
This type of defect, with a completely muscular border, can be found 1) along the anterior rim of the anterior trabecular septum, 2) isolated within the inlet septum and along the posterior ventricular wall, and 3) on the border between the anterior and the inlet septum, the so-called central muscular VSDs (). Our recent data on the role of the epicardium in fusion of the RV and LV side of the anterior trabecular septum can explain multiple deficiencies in the muscular anterior septum. Epicardial deficiency in animal models results in a spongy septum, usually also showing a thin compact myocardium of the lateral ventricular walls (Citation43,Citation63). The central muscular VSD is special in that it is present on the border between the anterior and inlet septum (). In the RV it is found between the trabeculations next to or even behind the septal band. From the LV it can be seen in the smooth LV septum as if there is overlapping of muscular components () (Citation64). In mouse models muscular VSDs are encountered in cases with a spongy septum indicative of deficient epicardial to myocardial interaction (Citation43).
The atrioventricular septal defect (AVSD)
The AV septum contains both muscular and fibrous tissues. The fibrous part is made up of the membranous septum, flanked by atrial and ventricular myocardium (). If this septum is intact, there is a connection and alignment of the atrial and ventricular septa in the vertical plane.
AVSDs form a special category of CHD in which the atrial septum does not connect with the ventricular inlet septum. In the most severe form, the so-called complete AVSD, an open communication exists between the four chambers of the heart with a common valve between the atria and the ventricles. The common valve consists of an anterior and a posterior bridging AV leaflet () and on the lateral sides of the annulus, the anterosuperior and inferior leaflets (right side), and a mural valve leaflet (left side). These leaflets represent the parts of the abnormally developed endocardial AV cushions. The chordae tendinea are connected to papillary muscles in the RV and LV and on the rim of the ventricular septum. Sometimes the anterior and posterior bridging leaflets themselves adhere to the rim of the septum. Variations in these connections have led to nomenclature subdivisions (Citation14). In the intermediate type of AVSDs the AV leaflets adhere in part to the free rim of the muscular ventricular inlet septum. The atrial component of the defect, an ASD type 1 as an isolated malformation, is the result of non-fusion of the lower rim of the septum primum with AV cushions and/or with the DMP (, ). A less frequent form of partial AVSD presents with attachment of the common valve only to the lower rim of the atrial septum; in these cases shunting occurs only at the ventricular level.
The search for the developmental background has engendered extensive research focusing on a primary fusion defect of the AV endocardial cushions, explaining the often-used term endocardial cushion defects (Citation65). Other options besides an endocardial cushion fusion problem have been investigated. First, a deficient muscular inlet septum may render the distance between the AV septum and the atrial septum too large, thus physically inhibiting fusion. Secondly, an abnormal formation of the base of the atrial septum is seen in human embryos with AVSD (Citation66) and in mouse embryos (Citation20) with a deficient formation of the DMP (formerly called vestibular spine or spina vestibuli). In live-born children with Down syndrome the incidence of AVSD is as high as 15%. The most frequently described associated gene in both patients with and without Down syndrome is CRELD1 on chromosome 3. Interestingly, CRELD+/− mice do not have septal defects. After crossing into the Down syndrome mice Ts65Dn, they do develop septal defects (Citation67). Very often AVSDs are part of syndromes () or of the spectrum of anomalies observed in heterotaxia syndromes as reported in 6% of all AVSD patients (Citation68), with a higher incidence in right isomerism (90%) (Citation69) than in left isomerism (60%–70%) (Citation70). This indicates that the genesis of at least some cases of AVSD may be related to the loss of normal right–left asymmetry in the body, which is addressed in the next paragraph. Interestingly, the copy number variations (CNV) observed in isolated VSDs do not seem applicable as cause for syndromic AVSD (Citation71).
Table II. Syndromes associated with congenital heart disease with mutation in genes that are linked to the SHH pathway.
Molecular considerations
Heart development is very sensitive to the temporo-spatial regulation of many genes in concert. As a consequence the frequent occurrence of congenital cardiac malformations does not come as a surprise. To some extent the above-provided data on normal and abnormal cardiac development that are meant to add to our understanding of the mechanisms leading to septal defects might seem disappointing with regard to a clear understanding of relatively simple genetic pathways underlying molecular mechanisms. As it turns out a relatively limited number of important cardiac transcription factors seem to govern the development of a variety of septal defects (). This is evident from both the current knowledge on genetic mutations in humans () as well as from data on cardiac morphogenetic gene modifications in (transgenic) mouse models.
Genetic considerations
Morphogenetic and molecular pathways that lead to cardiac septal defects reveal highly complex backgrounds where state-of-the-art genetic approaches provide new insights (Citation3,Citation39). De novo mutations based on exome sequencing indicate that in 10% of CHD a significant enrichment of mutations in the genes is involved in histone methylation relevant for chromatin biology (Citation4). Furthermore CNV mostly related to functional gains showed a clear relation to CHD, specifically to a subset of TOF. These CNV could be linked to important cardiac transcription factor pathways like WNT and GATA4 signaling (Citation3). A specific study on the role of TBX5 and the cis-regulatory elements revealed a single nucleotide variant related to VSD development (Citation72). Of interest, TGA relates to laterality genes (). For AVSD many responsible mutated genes have been proposed () which in part do not overlap with the more common genetic pathways for ASDs and VSDs, although the latter are also found in the syndromic families that present with an AVSD. Interestingly the sonic hedgehog (SHH)-GLI pathway plays an important role in many syndromes associated with septal defects (). There is an indication that the primary cilium (Citation73,Citation74), present on many cells including the endocardium and the epicardium, is involved. The SHH pathway acts on the OFT via contribution of endocardial cushion cells from the anterior SHF, as well as on the inflow tract via DMP contribution from the posterior SHF (Citation75).
These genetic considerations as such do not give credit to the complex interactions of modulating genes and factors which most probably play a very important role.
Epigenetic regulations
Chromatin remodeling
At the epigenetic level the regulation of transcription factors depends on the assembly of DNA in chromatin structures which is a complex structure efficiently wrapping the long strands of DNA around a core of histones. Euchromatin refers to an open state allowing for active gene expression, whereas heterochromatin presents a tightly packed form of DNA concurrent with repression of transcription. Currently, we lack detailed knowledge of how chromatin remodeling and DNA methylation co-ordinately govern expression patterns of groups of genes, including Tbx5, Gata4, Baf60c, during differentiation of cardiomyocytes (Citation76). Studies, mainly in cultured cells, have shown that cell differentiation states are correlated with histone modifications and that disrupted epigenetic regulation corresponds to CHD including abnormal cardiac septation. Genes important for heart development such as Tbx5 and Nkx2.5 interact with histone-modifying genes (Citation77,Citation78). Established chromatin patterns, involving methylation states of H3K4 and H3K27 are associated with expression patterns of genes involved in metabolism, signaling, and muscle contraction. It is postulated that early deposition of H3K4me1 at specific cardiac genes is a regulated step facilitating the ensuing activation of these genes (Citation6). It is necessary to identify the chromatin regulators catalyzing this modification to get more insight into their role in development of CHD. The multiple interactions including the important cardiac transcription factors Tbx5, Nkx2.5, and the GATA-family have been addressed to some extent as these orchestrate many processes in early differentiation and morphogenesis. Mutations in these genes have been reported to lead to septal defects (). Novel signaling networks include GATA-binding partnering with MEIS transcription factors (Citation6). The plethora of interactions at multiple levels calls for a systems biology approach providing insights into the combinatorial regulation by cardiac transcription factors and shows that they can partially compensate each other's function (Citation5,Citation12).
Environmental factors
The gene-modulating effects as described for epigenetic regulation also account for the effect of a number of environmental factors. These can also lead to histone modifications and can lead to gene modulation. Examples are found in the interaction of Tbx5, Gata4, and the environmentally linked variations in eNos (Citation37) where the posterior SHF-derived myocardium and the endocardium play a role.
Important environmental factors interacting with eNos and the already mentioned cilium-related genes are the hemodynamic influences on cardiac remodeling.
Simple changes in the inflow tract blood flow in the avian embryo model will lead to a variety of VSDs both in the subarterial and in the muscular infundibular position. The disturbed genes have been tracked down to the flow-dependent endothelin pathway (Citation79). Besides their being influenced by hemodynamics, the SHH-GLI pathway and cilium development themselves are involved in the extracardiac cholesterol metabolism which has been suggested to be linked to CHD (Citation80) and even adult cardiovascular disease (Citation81). The observed relevance of the epigenetic modulation of the histone methylation pathways is also of importance during normal differentiation of cells and can be influenced through the environmental homocysteine concentration and by diabetes (Citation3,Citation82,Citation83). In rat (Citation8), mouse (Citation84), and avian (Citation7) models with maternal hyperglycemia the incidence of CHD can be up to 80% of the embryos born. Both in avian (Citation7) and mouse (Citation84) embryos it has been shown that reducing the reactive oxygen species by N-acetylcysteine can lead to a reduction to 20% of CHD in maternal hyperglycemic offspring. These findings open up new options to explain the remaining 80%–85% of cases with CHD (including septal defects) that are considered to be multifactorial, implying a combination of genetic susceptibility and environmental factors.
The field of regulatory genetic and epigenetic factors becomes even more complex when also the role of microRNAs is taken into account.
Regulation by microRNAs
Recent investigations have attributed critical functions in post-transcriptional regulation of gene expression by microRNAs (miR) in many aspects of cardiac development. MiRs, consisting of a large group of > 1000 molecules that have been described so far, are a new class of non-protein-coding RNA that affect mRNA stability and translation. They are considered ‘molecular switches’ of gene expression, also intertwined in complex cardiac signaling and transcription (Citation85). MiRs, typically 20–26 nucleotides, are generated from longer precursors by enzymes, including Drosha and Dicer (Citation86). In mice, global deletion of these enzymes is lethal in early gestation, but conditional knock-out in specific cell lineages revealed essential roles in cardiac development. Specific deletion of Dicer influencing global miR processing in cardiac neural crest resulted in anomalies resembling human CHD including VSD (Citation87). More selective deletion of the miR-133a-1/2 complex causes VSDs with lethality in approximately 50% of double mutants. The others that survive to adulthood succumb to cardiomyopathy and sudden cardiac death (Citation86). Other compound mutant mouse embryos lacking miR-17∼92 and miR-106b∼25 clusters die at mid-gestation and present with VSDs and ASDs (Citation88).
Converging molecular denominators in septal defects
There seems to be support for convergence of transcription factors and downstream pathways (, and ) involved in atrial and ventricular septation, in which genomic variation in combination with epigenetic and environmental factors is responsible for the complex interacting pathways (Citation89). A dose-dependent modulation by regulatory genes for TBX1, TBX5, NKX2.5, GATA4, and ZIC3 has been shown (Citation90). TBX5, NKX2.5, and GATA4 interact in specified time windows and can work in combination to enhance effects (Citation91). Overall, a number of important transcription factors form a critical panel in development () in which all types of defect can occur when a mutation is found. There seems to be a differentiation in influence on OFT and inflow tract defects (). These can be linked to the expression of genes in the anterior (e.g. Tbx1) or posterior (e.g. Tbx5) SHF-derived populations being endocardium, myocardium, and epicardium. For example the severe outflow tract defect seen in CAT is not related to the inflow tract-expressed gene TBX5, whereas TBX1 does not seem to be linked to ASDs. An early perturbation of genes expressed in cardiac progenitor cells of both (anterior and posterior) heart fields, however, may explain the co-occurrence of inflow and outflow cardiac anomalies, as was recently suggested for the commonly observed co-occurrence of AVSD and tetralogy of Fallot in patients with Down syndrome (Citation92). A number of morphogenetic transcription factors (e.g. NODAL) are involved in left–right asymmetry and interact with e.g. PITX2 (Citation93), ZIC3 (Citation59), and the GLI-SHH pathways () (Citation94). The development of the cardiac septal structures and their common background with regard to the involved cell populations and their temporo-spatial specific gene expression mutations the defects can be observed at the atrial, ventricular, as well as at the AV level. The variation in the observed defects is not so much a specific link of a certain gene to a specific septal defect but relates more to the origin of the embryonic heart field population with a central role for the SHF.
In conclusion
It is becoming evident that regulation of cardiac development is a multi-faceted landscape at cellular and molecular levels. This includes cell interactions involving pharyngeal endoderm, FHF and SHF, cardiac neural crest, and others to provide for the precursors of the various components of the heart such as endocardial, myocardial, epicardial, smooth muscle, fibroblasts, endothelial, and neuronal cells. Many of these must interact harmoniously to build the components of the cardiac septa. At the level of morphogenesis much has been learned about the various cell populations that contribute to the septum components. Here cell tracing in (conditional) transgenic mouse models as well as the use of quail-chicken chimera models have been crucial. These data can be used to support understanding of the pathomorphology of septal defects in the fully grown human heart. Also many genetic defects have been detected both in the human population and in mouse models in which important cardiac morphogenetic genes have been modulated. The real understanding of the in-depth mechanisms is, however, still to be achieved. The molecular level encompasses the combination of genetic (transcription factors, post-translational processing) and epigenetic (chromatin remodeling, DNA methylation, miRNAs) regulation, including influences of the uterine environment related to life-style and genetic make-up of the mother. In view of the multiple interactions it is not surprising that focusing on a single factor or gene, although having unveiled many of the components, does not reveal the complexity or dynamics of the developing cardiovascular system. Integration and combination of data from the different levels will be necessary to understand further the etiology of cardiovascular malformations and septal defects in particular.
Funding
E. E. Calkoen was funded by the Willem-Alexander Children's Foundation (kinderfonds) and M. R. M. Jongbloed by a personal research grant from the ZonMw/NWO (project number 90700368).
Declaration of interest: The authors report no conflicts of interest.
References
- Morris C. Epidemiology of congenital heart disease. In: Crawford MH, DiMarco JP, Paulus WJ, editors. Cardiology. 3rd ed. Philadelphia: Mosby; 2010. p. 1381–9.
- Clark KL, Yutzey KE, Benson DW. Transcription factors and congenital heart defects. Annu Rev Physiol. 2006;68:97–121.
- Gelb BD. Recent advances in understanding the genetics of congenital heart defects. Curr Opin Pediatr. 2013;25:561–6.
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–3.
- Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, Krueger T, et al. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011;7:e1001313.
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20.
- Roest PAM, van Iperen L, Vis L, Wisse LJ, Poelmann RE, Steegers-Theunissen RGM, et al. Exposure of neural crest cells to elevated glucose leads to congenital heart defects, an effect that can be prevented by N-acetylcysteine. Birth Defects Res A Clin Mol Teratol. 2007;79:231–5.
- Molin DGM, Roest PA, Nordstrand H, Wisse LJ, Poelmann RE, Eriksson UJ, et al. Disturbed morphogenesis of cardiac outflow tract and increased rate of aortic arch anomalies in the offspring of diabetic rats. Birth Defects Res A Clin Mol Teratol. 2004;70:927–38.
- Verkleij-Hagoort AC, Verlinde M, Ursem NTC, Lindemans J, Helbing WA, Ottenkamp J, et al. Maternal hyperhomocysteinaemia is a risk factor for congenital heart disease. BJOG. 2006;113:1412–18.
- Beauchesne LM, Warnes CA, Connolly HM, Ammash NM, Grogan M, Jalal SM, et al. Prevalence and clinical manifestations of 22q11.2 microdeletion in adults with selected conotruncal anomalies. J Am Coll Cardiol. 2005;45:595–8.
- Liu Y, Feng Q. NOing the heart: role of nitric oxide synthase-3 in heart development. Differentiation. 2012;84:54–61.
- Sperling SR. Systems biology approaches to heart development and congenital heart disease. Cardiovasc Res. 2011;91:269–78.
- Jacobs JP, Quintessenza JA, Burke RP, Mavroudis C. Congenital Heart Surgery Nomenclature and Database Project: atrial septal defect. Ann Thorac Surg. 2000;69(4 Suppl):S18–24.
- Jacobs JP, Burke RP, Quintessenza JA, Mavroudis C. Congenital Heart Surgery Nomenclature and Database Project: atrioventricular canal defect. Ann Thorac Surg. 2000;69(4 Suppl):S36–43.
- Jacobs JP, Burke RP, Quintessenza JA, Mavroudis C. Congenital Heart Surgery Nomenclature and Database Project: ventricular septal defect. Ann Thorac Surg. 2000;69(4 Suppl):S25–35.
- Van Praagh R. The segmental approach to diagnosis in congenital heart disease. Birth Defects. 1972;8:4–23.
- Gittenberger-de Groot AC, Bartelings MM, DeRuiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169–76.
- Anderson RH, Becker AE, Freedom RM, Macartney FJ, Quero-Jiménez M, Shinebourne EA, et al. Sequential segmental analysis of congenital heart disease. Pediatr Cardiol. 1984;5:281–8.
- Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–35.
- Snarr BS, Wirrig EE, Phelps AL, Trusk TC, Wessels A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev Dyn. 2007;236:1287–94.
- Douglas YL, Jongbloed MR, DeRuiter MC, Gittenberger-de Groot AC. Normal and abnormal development of pulmonary veins: State of the art and correlation with clinical entities. Int J Cardiol. 2010;147: 13–24.
- Lescroart F, Mohun T, Meilhac SM, Bennett M, Buckingham M. Lineage tree for the venous pole of the heart: clonal analysis clarifies controversial genealogy based on genetic tracing. Circ Res. 2012; 111:1313–22.
- Jensen B, van den Berg G, van den Doel R, Oostra RJ, Wang T, Moorman AF. Development of the hearts of lizards and snakes and perspectives to cardiac evolution. PLoS One. 2013;8:e63651.
- Wenink ACG. Embryology of the ventricular septum. Separate origin of its components. Virchows Arch. 1981;390:71–9.
- van Mierop LHS, Kutsche LM. Comparative anatomy of the ventricular septum. In: Wenink ACG, editor. The ventricular septum of the heart. Hague: Martinus Nijhoff; 1981. p. 35–46.
- Lamers WH, Viragh S, Wessels A, Moorman AF, Anderson RH. Formation of the tricuspid valve in the human heart. Circulation. 1995;91:111–21.
- Wenink ACG, Wisse BJ, Groenendijk PM. Development of the inlet portion of the right ventricle in the embryonic rat heart: the basis for tricuspid valve development. Anat Rec. 1994;239:216–23.
- Jongbloed MR, Wijffels MC, Schalij MJ, Blom NA, Poelmann RE, van der Laarse A, et al. Development of the right ventricular inflow tract and moderator band: a possible morphological and functional explanation for Mahaim tachycardia. Circ Res. 2005;96:776–83.
- Vincent SD, Buckingham ME. How to make a heart: the origin and regulation of cardiac progenitor cells. Curr Top Dev Biol. 2010;90:1–41.
- Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: differentiation and apoptosis. Dev Dyn. 1998;212:373–84.
- Poelmann RE, Groot AC, Vicente-Steijn R, Wisse LJ, Bartelings MM, Everts S, Hoppenbrouwers T, et al. Evolution and development of ventricular septation in the amniote heart. Plos One. 2014 Epub.
- van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, et al. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–90.
- Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, et al. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281:78–90.
- Gittenberger-de-Groot AC, Winter EM, Bartelings MM, Goumans MJ, DeRuiter MC, Poelmann RE. The arterial and cardiac epicardium in development, disease and repair. Differentiation. 2012;84:41–53.
- Blom NA, Ottenkamp J, Jongeneel TH, DeRuiter MC, Gittenberger-de Groot AC. Morphogenetic differences of secundum atrial septal defects. Pediatr Cardiol. 2005;26:338–43.
- Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59:17–20.
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–21.
- Nadeau M, Georges RO, Laforest B, Yamak A, Lefebvre C, Beauregard J, et al. An endocardial pathway involving Tbx5, Gata4, and Nos3 required for atrial septum formation. Proc Natl Acad Sci U S A. 2010;107:19356–61.
- Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, et al. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol. 1999;211:100–8.
- Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707–20.
- Briggs LE, Kakarla J, Wessels A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation. 2012;84:117–30.
- Bleyl SB, Saijoh Y, Bax NA, Gittenberger-de Groot AC, Wisse LJ, Chapman SC, et al. Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: integrating evidence from human genetics and model organisms. Hum Mol Genet. 2010;19:1286–301.
- Anderson RH, Ho SY. Sequential segmental analysis – description and categorisation for the millenium. Cardiol Young. 1997;7:98–116.
- Mahtab EAF, Wijffels MCEF, van den Akker NMS, Hahurij ND, Lie-Venema H, Wisse LJ, et al. Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: correlation with abnormal epicardial development. Dev Dyn. 2008;237:847–57.
- Blaschke RJ, Hahurij ND, Kuijper S, Just S, Wisse LJ, Deissler K, et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007;115:1830–8.
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–61.
- Conway SJ, Henderson DJ, Kirby ML, Anderson RH, Copp AJ. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc Res. 1997;36:163–73.
- Feiner L, Webber AL, Brown CB, Min Lu M, Jia L, Feinstein P, et al. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–70.
- Waldo K, Miyagawa-Tomita S, Kumiski D, Kirby ML. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: aortic sac to ventricular septal closure. Dev Biol. 1998;196:129–44.
- Epstein JA. Developing models of DiGeorge syndrome. Trends Genet. 2001;17:S13–17.
- Baldini A. DiGeorge’s syndrome: a gene at last. Lancet. 2003;362: 1342–3.
- Wang H, Chen D, Ma L, Meng H, Liu Y, Xie W, et al. Genetic analysis of the TBX1 gene promoter in ventricular septal defects. Mol Cell Biochem. 2012;370:53–8.
- Huynh T, Chen L, Terrell P, Baldini A. A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis. 2007;45:470–5.
- Lev M, Eckner FAO. The pathologic anatomy of tetralogy of Fallot and its variations. Dis Chest. 1964;45:251–61.
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–5.
- Pexieder T. Conotruncus and its septation at the advent of the molecular biology era. In: Clark EB, Markwald RR, Takao A, editors. Developmental mechanisms of heart disease. Futura, Armonk, New York; 1995. p. 227–47.
- Scherptong RW, Jongbloed MR, Wisse LJ, Vicente-Steijn R, Bartelings MM, Poelmann RE, et al. Morphogenesis of outflow tract rotation during cardiac development: the pulmonary push concept. Dev Dyn. 2012;241:1413–22.
- Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–64.
- Franco D, Campione M. The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc Med. 2003;13:157–63.
- Purandare SM, Ware SM, Kwan KM, Gebbia M, Bassi MT, Deng JM, et al. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development. 2002;129:2293–302.
- Unolt M, Putotto C, Silvestri LM, Marino D, Scarabotti A, Valerio M, et al. Transposition of great arteries: new insights into the pathogenesis. Front Pediatr. 2013;1:11.
- Allwork AP, Anderson RH. Developmental anatomy of the membranous part of the ventricular septum in the human heart. Brit Heart J. 1979;41:275–80.
- Soto B, Becker AE, Moulaert AJ, Lie JT, Anderson RH. Classification of ventricular septal defects. Brit Heart J. 1980;43:332–43.
- Bax NA, Bleyl SB, Gallini R, Wisse LJ, Hunter J, Van Oorschot AA, et al. Cardiac malformations in Pdgfralpha mutant embryos are associated with increased expression of WT1 and Nkx2.5 in the second heart field. Dev Dyn. 2010;239:2307–17.
- Wenink ACG. Muscular ventricular septal defect in a human embryo of 11 mm crown-rump length (six weeks)-no case of developmental arrest. Cardiol Young. 1996;6:181–3.
- Moskowitz IP, Wang J, Peterson MA, Pu WT, Mackinnon AC, Oxburgh L, et al. Cardiac-specific transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. Proc Natl Acad Sci U S A. 2011;108:5921.
- Blom NA, Ottenkamp J, Wenink AG, Gittenberger-de Groot AC. Deficiency of the vestibular spine in atrioventricular septal defects in human fetuses with Down syndrome. Am J Cardiol. 2003;91:180–4.
- Li H, Cherry S, Klinedinst D, DeLeon V, Redig J, Reshey B, et al. Genetic modifiers predisposing to congenital heart disease in the sensitized Down syndrome population. Circ Cardiovasc Genet. 2012;5:301–8.
- Christensen N, Andersen H, Garne E, Wellesley D, Addor MC, Haeusler M, et al. Atrioventricular septal defects among infants in Europe: a population-based study of prevalence, associated anomalies, and survival. Cardiol Young. 2012;1–8.
- Phoon CK, Neill CA. Asplenia syndrome: insight into embryology through an analysis of cardiac and extracardiac anomalies. Am J Cardiol. 1994;73:581–7.
- Peoples WM, Moller JH, Edwards JE. Polysplenia: a review of 146 cases. Pediatr Cardiol. 1983;4:129–37.
- Priest JH, Phillips CN, Wang Y, Richmond A. Chromosome and growth factor abnormalities in melanoma. Cancer Genet Cytogenet. 1988; 35:253–62.
- Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet. 2012;21:3255–63.
- Hierck BP, Van der Heiden K, Alkemade FE, van de Pas S, van Thienen JV, Groenendijk BCW, et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008;237:725–35.
- Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, et al. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ Res. 2011;108:1093–101.
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. sonic hedgehog is required in pulmonary endoderm for atrial septation. Development. 2009;136:1761–70.
- van Weerd JH, Koshiba-Takeuchi K, Kwon C, Takeuchi JK. Epigenetic factors and cardiac development. Cardiovasc Res. 2011; 91:203–11.
- Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008;22:2980–93.
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–91.
- Groenendijk BCW, Van der Heiden K, Hierck BP, Poelmann RE. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology (Bethesda). 2007;22:380–9.
- Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–9.
- Alkemade FE, Gittenberger-de Groot AC, Schiel AE, VanMunsteren JC, Hogers B, van Vliet LS, et al. Intrauterine exposure to maternal atherosclerotic risk factors increases the susceptibility to atherosclerosis in adult life. Arterioscler Thromb Vasc Biol. 2007;27:2228–35.
- Boot MJ, Steegers-Theunissen RP, Poelmann RE, van Iperen L, Gittenberger-de Groot AC. Cardiac outflow tract malformations in chick embryos exposed to homocysteine. Cardiovasc Res. 2004;64:365–73.
- Liu S, Joseph KS, Lisonkova S, Rouleau J, Van den Hof M, Sauve R, et al. Association between maternal chronic conditions and congenital heart defects: a population-based cohort study. Circulation. 2013;128: 583–9.
- Moazzen H, Lu X, Ma NL, Velenosi TJ, Urquhart BL, Wisse LJ, et al. N-Acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014;13:46.
- Porrello ER. MicroRNAs in cardiac development and regeneration. Clin Sci (Lond). 2013;125:151–66.
- Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–25.
- Huang ZP, Chen JF, Regan JN, Maguire CT, Tang RH, Dong XR, et al. Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler Thromb Vasc Biol. 2010;30:2575–86.
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86.
- Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segre AV, et al. Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc Natl Acad Sci U S A. 2012;109:14035–40.
- Bruneau BG, Srivastava D. Congenital heart disease: entering a new era of human genetics. Circ Res. 2014;114:598–9.
- Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–8.
- Nguyen HH, Jay PY. A single misstep in cardiac development explains the co-occurrence of tetralogy of Fallot and complete atrioventricular septal defect in Down syndrome. J Pediatr. 2014;165:194–6.
- Piedra ME, Icardo JM, Albajar M, Rodriguez-Rey JC, Ros MA. Pitx2 participates in the late phase of the pathway controlling left-right asymmetry. Cell. 1998;94:319–24.
- Hildreth V, Webb S, Chaudhry B, Peat JD, Phillips HM, Brown N, et al. Left cardiac isomerism in the Sonic hedgehog null mouse. J Anat. 2009;214:894–904.
- DeLuca A, Sarkozy A, Ferese R, Consoli F, Lepri F, Dentici ML, et al. New mutations in ZFPM2/FOG2 gene in tetralogy of Fallot and double outlet right ventricle. Clin Genet. 2011;80:184–90.
- Posch MG, Perrot A, Berger F, Ozcelik C. Molecular genetics of congenital atrial septal defects. Clin Res Cardiol. 2010;99:137–47.
- Ackerman C, Locke AE, Feingold E, Reshey B, Espana K, Thusberg J, et al. An excess of deleterious variants in VEGF-A pathway genes in Down-syndrome-associated atrioventricular septal defects. Am J Hum Genet. 2012;91:646–59.
- Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, et al. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci U S A. 2009;106:13933–8.
- Sheng W, Qian Y, Zhang P, Wu Y, Wang H, Ma X, et al. Association of promoter methylation statuses of congenital heart defect candidate genes with tetralogy of Fallot. J Transl Med. 2014;12:31.
- Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z. T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51:580–7.
- van de Laar I, Dooijes D, Hoefsloot L, Simon M, Hoogeboom J, Devriendt K. Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A. 2007;143A:2712–15.
- Ma L, Selamet Tierney ES, Lee T, Lanzano P, Chung WK. Mutations in ZIC3 and ACVR2B are a common cause of heterotaxy and associated cardiovascular anomalies. Cardiol Young. 2012;22:194–201.
- Lambrechts D, Devriendt K, Driscoll DA, Goldmuntz E, Gewillig M, Vlietinck R, et al. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: a family based association study. J Med Genet. 2005;42:519–22.
- Barlow GM, Chen X-N, Lyons GE, Kurnit DM, Celle L, Spinner NB, et al. Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genet Med. 2001;3:91–101.
- Marion V, Stoetzel C, Schlicht D, Messaddeq N, Koch M, Flori E, et al. Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci U S A. 2009;106:1820–5.
- Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, et al. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet. 2013;22:1358–72.
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565–75.
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72.
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43:601–6.
- Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–53.
- Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. 2012;23: 823–35.
- Sund KL, Roelker S, Ramachandran V, Durbin L, Benson DW. Analysis of Ellis van Creveld syndrome gene products: implications for cardiovascular development and disease. Hum Mol Genet. 2009;18:1813–24.
- Petrova R, Garcia AD, Joyner AL. Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J Neurosci. 2013;33: 17490–505.
- Currier DG, Polk RC, Reeves RH. A Sonic hedgehog (Shh) response deficit in trisomic cells may be a common denominator for multiple features of Down syndrome. Prog Brain Res. 2012;197: 223–36.
- Ahlgren SC, Thakur V, Bronner-Fraser M. Sonic hedgehog rescues cranial neural crest from cell death induced by ethanol exposure. Proc Natl Acad Sci U S A. 2002;99:10476–81.
- Gittenberger-de Groot AC, Jongbloed MRM, Poelmann RE. Normal and abnormal cardiac development. In: Moller JH, Hoffman JIE, editors. Pediatric cardiovascular medicine. 2nd ed. West Sussex: Wiley-Blackwell, Hoboken, New Yersey; 2011. p. 1–22.
- Gittenberger-de Groot AC, Jongbloed MRM, DeRuiter MC, Bartelings MM, Poelmann RE. Embryology of congenital heart disease. In: Crawford MH, DiMarco JP, Paulus WJ, editors. Cardiology. 3rd ed. Philadelphia: Mosby; 2010. p. 1391–402.