
Abstract
Human pluripotent stem cells (hPSCs) have practically unlimited proliferation potential and a capability to differentiate into any cell type in the human body. Since the first derivation in 1998, they have been an attractive source of cells for regenerative medicine. Numerous ethical, technological, and regulatory complications have been hampering hPSC use in clinical applications. Human embryonic stem cells (ESCs), parthenogenetic human ESCs, human nuclear transfer ESCs, and induced pluripotent stem cells are four types of hPSCs that are different in many clinically relevant features such as propensity to epigenetic abnormalities, generation methods, and ability for development of autologous cell lines. Propensity to genetic mutations and tumorigenicity are common features of all pluripotent cells that complicate hPSC-based therapies. Several recent advances in methods of derivation, culturing, and monitoring of hPSCs have addressed many ethical concerns and technological challenges in development of clinical-grade hPSC lines. Generation of banks of such lines may be useful to minimize immune rejection of hPSC-derived allografts. In this review, we discuss different sources of hPSCs available at the moment, various safety risks associated with them, and possible solutions for successful use of hPSCs in the clinic. We also discuss ongoing clinical trials of hPSC-based treatments.
Key words::
- Epigenetic aberrations
- good manufacturing practices
- human embryonic stem cells (hESCs)
- human induced pluripotent stem cells (hiPSCs)
- human nuclear transfer embryonic stem cells (NT-ESCs)
- human pluripotent stem cells (hPSCs)
- immune rejection
- parthenogenetic human embryonic stem cells (phESCs)
- stem cell bank
- tumorigenicity
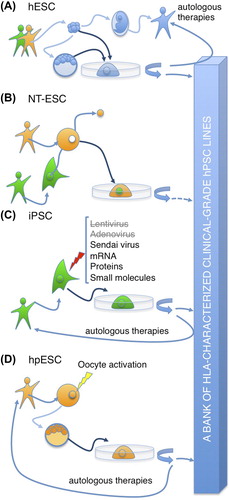
There are several types of human pluripotent stem cells (PSCs), such as human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs), parthenogenetic human embryonic stem cells (phESCs), and human nuclear transfer ESCs (NT-ESCs). As of today, there is no ideal PSC type to use in clinical treatments; each type has advantages and disadvantages ().
The most realistic method for routine PSC therapies, including iPSC therapies, is based on generation of clinical-grade banks of hPSC lines that human leukocyte antigen (HLA)-match a target population. In this context, generation of HLA-homozygous hPSCs is of particular interest.
Human ESCs are the ‘gold standard’ human PSCs and, along with human iPSCs, already used in ongoing clinical trials (). However, those trials are mostly designed to test for safety of therapies involving hPSCs. Recently, methods of hESCs derivation under animal component-free and chemically defined conditions from whole inner cell masses and single blastomeres without a need to destroy human embryos have been developed.
Introduction
Human embryonic stem cells (hESCs) (Citation1) and human induced pluripotent stem cells (hiPSCs) (Citation2) are the most studied types of human pluripotent stem cells (hPSCs), which are already useful for the development of cell lineages for therapeutic purposes. Parthenogenetic human embryonic stem cells (phESCs) (Citation3) and recently reported human nuclear transfer embryonic stem cells (NT-ESCs) (Citation4) are considerably less studied types of hPSCs, but they may be useful for therapeutic purposes in the future. Although those cells share main properties of hPSCs, they are different in many features, which are relevant to the clinical use (). Human ESCs are usually derived from the inner cell mass of human supernumerary in vitro fertilization (IVF) blastocysts (Citation1). Human ESCs are considered as ‘gold standard’ of hPSCs, but they are allogeneic to patients, and derivation of new lines often demands destruction of the embryos. The latter is an important ethical concern, which has been addressed by derivation of hESC lines from single blastomere cells without a need to destroy the parental embryo () (Citation5–7). The human genomes may contain sequences that are associated with predisposition to diseases. Such predispositions are not known for hESC lines derived with destruction of the parental embryos, because usually there is no living individual with the exact genome.
Table I. Properties of hPSC types developed using presently existing technologies.
Parthenogenetic human embryonic stem cells are derived from parthenogenetic embryos. Depending on the oocyte activation protocol, the resulting phESCs may be heterozygous (Citation3) or homozygous (Citation8) for human leukocyte antigens (HLAs). Heterozygous phESCs are autologous, and homozygous phESCs are valuable for banking of cell lines that HLA-match a target population, but abnormal imprinting and loss of heterozygosity (LOH) may complicate their use in regenerative medicine (recently reviewed in 9). Unfertilized human oocytes for generation of new phESC lines should be acquired through an egg donation program involving administration of an ovarian stimulation protocol to the donors. This may raise certain ethical concerns (Citation10).
Induced PSCs (iPSCs) are established by ectopic expression or direct delivery of certain mRNAs or proteins in somatic cells (Citation2,Citation11–13). Since the initial report, the methods of hiPSC generation (reprogramming methods) have demonstrated enormous progress from primitive reprogramming procedures involving random mutagenesis in the cells (Citation2) to methods that may be useful for the development of cells for therapeutic purposes (Citation12,Citation13). In research laboratories, iPSCs are the most commonly used type of hPSCs. Human iPSCs are autologous and free from major ethical concerns, but prone to transcriptional and epigenetic aberrations (Citation14–18). This or other properties may sometimes lead to incomplete differentiation of human iPSCs and generation of only partially functional somatic cells, e.g. cardiomyocytes unresponsive to hypertrophic signals (Citation19). Using currently existing technologies, it is too expensive and time-consuming to generate iPSCs for only one certain patient (Citation20).
Human NT-ESCs are derived by a transfer of a somatic cell nucleus into an enucleated human oocyte (Citation4). The procedure is called somatic cell nuclear transfer (SCNT). NT-ESCs cells are HLA-matched with the donor of the somatic cell, but allogeneic mitochondria, which mainly originate from the oocyte, may trigger an adaptive alloimmune response (Citation21). Similar to generation of phESCs, procurement of unfertilized human oocytes for SCNT may raise certain ethical concerns.
Apart from the special features, there are several general risk factors that are associated with treatments involving all types of PSCs. The cells should be developed under controlled conditions that minimize risks of viral/bacterial contaminations and immunogenic complications. To ensure that, derivation, culturing, and storage of clinical-grade hPSCs should comply with current good manufacturing practices (cGMPs) and chemistry and manufacturing controls (CMCs) (Citation22). Optimally, hPSCs should be developed under xeno-free (animal substance-free) and chemically defined conditions. Prolonged culturing of hPSCs in vitro sometimes leads to adaptive genetic changes that may show similarity to those found in tumors (Citation23,Citation24). Also, efficacy of differentiation protocols is not always absolute, leading to generation of heterogeneous populations of cells containing differentiated somatic cells and residual pluripotent stem cells. The residual PSCs may grow into tumors after transplantation into patients (Citation25). Another issue is that many cells in the human body are organized in three-dimensional structures of interconnected and cooperatively acting cells. Transplantation of only one type of somatic cells may lead to low efficacy of integration into endogenous existing structures and, therefore, lack of significant long-term regenerative effects. Apart from technical issues, regulatory and legal differences between countries are a major hurdle to the development of hPSC-based clinical treatments (Citation22). In this review, we describe properties of various hPSCs as they exist at the present time, safety risks associated with hPSC clinical use, possible solutions that may minimize the risks, and existing examples of hPSC use in clinical trials.
Culturing and derivation of hPSCs
It is necessary that the hPSC culturing environment strongly promotes self-renewal of pluripotent cells and prevents their spontaneous differentiation. This is defined by characterization of cultured pluripotent cells (recently reviewed in 26). The first hESC lines were derived and propagated on a layer of murine fibroblasts in a medium containing bovine serum (Citation1). Such methods are not optimal for development of cells for clinical use. Contact of cultured hESCs with substances that originate from animals may induce immunogenicity of the cells (Citation27). Fibroblasts are cellular factories that produce a variety of growth factors and cytokines in a batch- and time-dependent manner. This may affect cultured hPSCs in unpredictable ways, jeopardizing reproducibility of the methods. Moreover, fibroblasts can be a source of viral and/or bacterial contamination. Since the first derivation, many improvements to the initial hPSC culture method were proposed. Murine fibroblasts were replaced by human feeder cells both for culturing and derivation of hESCs (Citation28,Citation29). This reduced the risk of induced immunogenicity, but the variability of feeder cells was still a significant problem. To avoid feeder cells, murine extracellular extract Matrigel was used as a cell culture substratum for feeder-free hPSC growth (Citation30). Matrigel is a protein extract from Engelbreth–Holm–Swarm (EHS) mouse sarcoma. It is a complex batch-to-batch different composition of proteins containing collagen-IV, laminin-111, other extracellular matrix proteins, and growth factors. Therefore, Matrigel is not an optimal substratum for hPSC growth.
Optimally, development of cells for therapeutic purposes requires methods that enable derivation and culturing of hPSCs under xeno-free and chemically defined condition. Such methods are reproducible, and they minimize the probability of contamination and immune complications. Several xeno-free chemically defined media for long-term culturing of hPSCs are now available on the market (Citation30). Human recombinant extracellular matrix proteins, such as vitronectin (Citation31) and laminin-511 (Citation32), a hydrogel (Citation33), a synthetic polymer (Citation34), and a synthetic peptide-acrylate surface (Citation35) have recently been reported as xeno-free chemically defined substrata for proliferation of hPSCs. E-8 laminin-511 fragment (Citation36) and laminin-521, which has been produced as human recombinant protein in our laboratory (Citation7,Citation37), also enable easy passaging of hPSCs in single cell suspensions as opposed to standard passaging in small cellular clumps. Passaging of cells in single cell suspension may be useful for automation of hPSC culturing. Therefore, several methods for culturing of hPSCs that can comply with cGMPs have been developed to date.
Although similar techniques are used for culturing of established hPSC lines of all types, methods for derivation of new hESC, phESC, human iPSC, and human NT-ESC lines are very different (). Early hESCs lines were derived on feeder cells, e.g. fibroblasts, with destruction of the parental embryo (Citation1). Although the cells derived on human feeders can comply with cGMPs (Citation38), which is necessary for clinical-grade cell lines, it is not an optimal method. It is even possible to apply cGMPs to animal-derived substances if no better options are available (Citation39). Until very recently, there has been no robust method of new hESC line derivation under xeno-free, feeder-free, and chemically defined conditions. Destruction of ex utero embryos during the derivation procedure has been the major ethical concern in many countries in the world. Klimanskaya et al. (Citation5,Citation6) suggested an alternative method of new hESC line derivation using a single blastomere without a need to destruct the parental embryo. The method addressed the ethical concerns of many, although the cell lines were derived on feeder cells in environments containing products of animal origin. Recently, our group has shown that laminin-521/E-cadherin matrix allows new hESC line derivation both from blastocyst inner cell masses and single blastomere cells under completely xeno-free and chemically defined conditions (Citation7). Such cell lines are free from major ethical concerns and can be easily developed in compliance with cGMPs.
The first human iPSC lines were generated with use of viruses, which integrate into the host genome (Citation2). The multiple integration sites created mutations in genomes of iPSCs, and thus such cells could not be used in clinical treatments. Recently, several methods of integration-free human iPSCs generation have been reported (Citation11–13). Such methods may facilitate development of clinical-grade quality cells. Derivation of human iPSCs under xeno-free and chemically defined conditions has also been reported () (Citation40,Citation41). No such method has been reported for generation of human NT-ESCs and phESCs yet, but derivation of the latter cells is similar to methods used for hESCs.
Genetic and epigenetic abnormalities in hPSCs
Human PSCs commonly undergo adaptive genetic changes during prolonged culturing in vitro, such as a gain of chromosomes 17 and 12 (Citation42). If the genetic mutations generate a selective advantage for the cells, such as greater propensity for self-renewal, they become fixed over time. Environmental cues that may affect the rate of mutagenesis in cultured hPSCs are poorly studied. Earlier reports suggested that chromosomal abnormalities could be caused by feeder-free culturing (Citation42) or enzymatic passaging of hPSCs (Citation43). Later more comprehensive studies have shown that propensity to undergo genetic changes is attributed to the nature of hPSCs themselves rather than culture conditions (Citation23,Citation24). Importantly, apart from random mutagenesis and common gains of large fragments of chromosomes 12, 17, 20, and X, there is at least one example of small non-random culture adaptation in 20q11.21, which encompasses an anti-apoptotic factor (Citation23). The mutation shows similarity to those found in tumors, and it is too small to be detected by karyotyping. Therefore, a systematic genetic monitoring of cultured clinical-grade hPSCs with a high-resolution method, such as genotyping or whole-genome sequencing, is needed to ensure safety of the cells. Since whole-genome sequencing is still too expensive for routine use, whole-genome single nucleotide polymorphism (SNP) arrays containing a significant number of probes (around 1 million) for SNP and copy number variations (CNVs) can be used to detect SNP, CNV, and loss of heterozygosity (LOH) profiles (Citation24). The method allows detecting small (50 kb) genetic aberrations, but it has some limitations such as inability to detect balanced chromosomal translocations and inversions. Therefore, it should be used along with karyotyping or fluorescent in situ hybridization. One more significant disadvantage of genotyping microarray analysis is the inability to detect genetic aberrations affecting small subpopulations of cells. There is a need for development of new efficient methods with sufficient resolution and sensitivity for adequate monitoring of genetic stability of hPSCs in in vitro cultures. Barbaric et al. (Citation44) showed that some adapted hPSCs have increased motility in comparison with normal cells of the same lines, and time-lapse tracking of single cells can provide an interesting non-invasive approach for the early detection of culture adaptations. However, it is not clear if such a method would have enough accuracy and specificity.
Apart from systematic monitoring, banking of hPSCs at low passages decreases overall time in culture. This would significantly decrease probability of cultural adaptations and increase safety of hPSC in clinical treatments. Thawing of a standard frozen aliquot would give rise to hundreds of kilograms of hPSCs after only 10 passages in culture under xeno-free and chemically defined conditions (Citation7). One important exception is iPSCs that may bear abnormally high number of genetic aberrations immediately after reprogramming (Citation45). Most of de novo mutations that appear during reprogramming are different from those described for culture adaptations in hPSCs and render the affected cells selectively disadvantaged. The mutations affect only some of the cells, and expansion of human iPSCs in culture selects against the mutated cells, driving the lines towards a genetic state resembling hESCs with time. The reprogramming process itself is associated with selection for deletions of tumor-suppressor genes (Citation46) that complicates banking of human iPSC at early passages.
Although no epigenetic marks that correlate with culture adaptation of hPSCs have been reported to date, existing types of pluripotent stem cells exhibit very different epigenetic patterns (). Reprogramming of somatic cells into iPSCs involves epigenomic reconfiguration of the cells. Aberrations in DNA methylation and alterations of gene expression at imprinted regions in iPSCs have been described in many studies (Citation14–17). Although it is not entirely clear whether these epigenetic aberrations arise during reprogramming itself or represent incomplete reversion to pluripotency, they may affect the utility of human iPSCs in regenerative medicine. Indeed, it has been shown that reprogramming-dependent errors in DNA methylation can be transmitted to the differentiated progeny (Citation15), and differentiation of some human iPSCs, but not hESCs, yields only partially functional cardiomyocytes (Citation19). Ma et al. (Citation18) compared genetically matched sets of human ESCs, iPSCs, and NT-ESCs and showed that DNA methylation and transcriptome profiles of NT-ESCs corresponded closely, but not entirely, to those of hESCs; iPSCs, however, differed from ‘gold standard’ hESCs and retained the residual DNA methylation pattern of paternal somatic cells. Further studies on molecular mechanisms of reprogramming are warranted to develop technologies that allow generation of new iPSCs without epigenetic aberrations.
Mammalian parthenogenetic embryos are developmentally incompetent due to abnormal genetic imprinting (Citation47). Parent-specific epigenetic modifications of the genome occur during gametogenesis. In parthenogenetic mammalian embryos, paternal imprinting is absent, and maternally expressed imprinted genes are transcribed from both alleles, leading to aberrant expression levels of the imprinted genes. These properties are inherited by phESCs that are derived from parthenogenetic human embryos. Thus, phESCs are epigenetically aberrant by default.
PSCs exhibit an elevated rate of proliferation and distinct metabolic features compared to somatic cells (reviewed in 48). In vivo, precursors of hESCs from the inner cell mass of blastocysts reside in uterus, which is an oxygen-deprived tissue. In vitro, hPSCs grown under both normoxic and hypoxic conditions demonstrate preference of glycolytic metabolism over oxidative phosphorylation (OXPHOS). Undeveloped mitochondrial network and low mitochondrial activity are features of hPSCs. In somatic cells, high mitochondrial activity is the main source of toxic reactive oxygen species (ROS), which are a common OXPHOS by-product. ROS can lead to oxidative damage of DNA, proteins, lipids, and especially mitochondrial DNA (mtDNA) due to its close proximity to the source of ROS. In humans, mitochondria are inherited from the mother. Thus, prior to derivation mtDNA in hESCs and human NT-ESCs, which inherit the majority of their mitochondria from the oocyte (Citation4), never experience oxidative stress caused by high concentrations of ROS. Mitochondria in iPSCs originate from somatic cells, and therefore mtDNA in iPSCs may contain pre-existing oxidation-induced mutations. To date, the integrity of mtDNA within pluripotent cells has not been adequately investigated. Prigione et al. (Citation49) showed that reprogramming of somatic cells into iPSCs using retroviruses is associated with mutagenesis in mtDNA. Cheng et al. (Citation50) demonstrated much lower incidences of mtDNA mutation in iPSCs generated by non-integrated plasmid expression, suggesting that the reprogramming method may affect mutagenesis in mtDNA. Unexpectedly, some hESC lines have been reported to exhibit large mtDNA deletions, although alteration of the hESCs differentiation potential could not be observed (Citation51).
Tumorigenicity of hPSCs
Gene expression networks that are responsible for maintenance and induction of pluripotency and that are implicated in oncogenesis are partially overlapping. Both pluripotent and tumor cells exhibit glycolytic metabolism (Citation48), high proliferation capacity, DNA repair check-point uncoupling, multipotency, etc. (Citation52). Even genetically normal pluripotent stem cells may form benign teratomas after transplantation into a recipient organism (Citation25). This property is used to confirm pluripotency of hPSCs in in vivo experiments. Existing differentiation methods are not absolutely effective and often generate mixed populations of cells containing residual pluripotent and partially differentiated cells. Therefore, a population of cells aimed for transplantation should be treated to contain no residual pluripotent cells. This may be achieved by purifying desired differentiated cells (Citation53) or by removing the residual pluripotent cells using fluorescent activated cell sorting or magnetic beads coated with antibodies against a particular antigen such as SSEA-5 (Citation54) and Claudin-6 (Citation55). An alternative approach implies selective elimination of residual hPSCs by cytotoxic antibody recognizing podocalyxin-like protein-1 (Citation56) or by a chemical inhibitor of stearoyl-coA desaturase (Citation57), which is the key enzyme in oleic acid biosynthesis. The antibody and the inhibitor do not affect differentiated cells.
As described above, cultural genetic adaptations of hPSCs sometimes show similarity to those found in tumors (Citation23). Närva et al. (Citation24) showed that more than 40% of genes with altered by the mutations levels of expression are functionally linked to cancer. Such cells pose a risk of malignant transformation after transplantation into patients and should not be used in therapeutic procedures. Induction of pluripotency in iPSCs is considered to be associated with additional risk of tumorigenicity because of additional mutation load immediately after generation of iPSCs (Citation46), overexpression of oncogenic transcription factors, and hypomethylation resembling that seen in cancer cells (reviewed in 52).
In homozygous and, to a lesser extent, in heterozygous phESCs, full or partial loss of heterozygosity may be associated with an additional risk of tumorigenicity (Citation9). Many harmful mutations in genomes, such as inactivation of tumor suppressor genes, are masked with expression of normal second alleles. Therefore, lack of the second allele is considered to be detrimental.
Immunological issues of hPSCs in clinical treatments
Immunogenicity of allografts
One of the main hurdles that complicate clinical translation of hPSC-based therapies is the possibility of immune rejection of transplanted cells. The adaptive and innate immune systems are the human organism's defense systems against pathogens with the result that all transplantations from non-identical individuals end in rejection of the grafted cells or organs. Immunological rejection can be divided into three different categories that are ABO blood group antigen, major histocompatibility complex, and minor histocompatibility complex rejections (Citation58).
Acquired immunogenicity of hPSCs
Human ESCs have historically been derived on feeder cells of animal origin in the media supplemented with fetal calf serum (Citation1). The use of animal media or feeder cell components during the derivation represents a risk of transmitting infections as well as xenogenic antigens. Martin et al. (Citation27) showed that hESCs may metabolically incorporate significant amounts of sialic acid (Neu5Gc), which is produced by mouse feeder cells and is a component of some animal-derived cell culture media. Since many humans have circulating antibodies against Neu5Gc, transplantation of hESCs or their progeny pre-exposed to sialic acid would lead to immune response. In agreement with this, it has also been shown that culturing hESCs in a medium containing no animal-derived components substantially reduces the immune response. As described above, the methods of hPSC derivation and culturing under xeno-free (animal component-free) and chemically defined conditions have been developed in order to minimize these risks.
Immunogenicity of hPSCs due to HLA incompatibility
One strategy to overcome the immunological barrier for allogeneic hPSC-based therapies is to create banks of clinical-grade hPSC lines that HLA-match a target population (Citation59,Citation60). However, this approach has certain limitations such as high costs of generation of many clinical-grade hPSC lines under cGMPs to cover a significant proportion of the population and the fact that minor mismatches can still result in immune rejection (Citation61). Generation of HLA-homozygous hPSC lines containing identical alleles for each antigen-presenting protein would facilitate compatibility with recipient individuals. Such cells express a reduced major histocompatibility complex class I (MHC-I, in human also called HLA-I) repertoire that facilitates finding a match for heterozygous recipients. HLA-homozygous individuals are rare, but certain oocyte activation protocols allow efficient derivation of HLA-homozygous phESC (Citation8). Unlike other types of hPSCs, phESCs have abnormal genomic imprinting and loss of heterozygosity that may compromise clinical use of such cells. The other problem of treatments involving HLA-homozygous hPSC lines of any source is possible NK cell-mediated rejection of HLA-homozygous grafts in a HLA-heterozygous host even without a HLA mismatch. Severe NK cell-mediated rejection of the donor cells after transplantation can be caused by missing self MHC-I inhibitory signals to NK cells, a phenomenon called ‘hybrid resistance’ (Citation62). Indeed, even without a MHC mismatch the graft cells are still missing expression of some MHC-I molecules of the heterozygous host. Bone marrow transplantation is the most well-known transplantation type that is highly sensitive to complete MHC overlap. However, solid organ transplantations are slightly less sensitive and even allow partial MHC matching (Citation63). Recently, it has been shown that transplantation of parthenogenetic mouse ESCs into heterozygote immunocompetent mice expressing all MHC-I of the graft (and some more) does not induce NK cell rejection and facilitates teratoma formation (Citation64). This observation provides a strong argument for further development of parthenogenetic ESC approach.
Jacquet et al. (Citation65) described an embryo haplotyping strategy that allows choosing embryos in which HLA haplotypes match a significant proportion of the target population before derivation of the lines. Since the method is based on PCR amplification with sequence-specific primers, it is fast and relatively inexpensive. This strategy significantly reduces costs that are associated with development of the banks of clinical-grade hPSC lines.
Autologous hPSCs
Generation of autologous hPSCs is another way to remove the hurdle of the immune barrier in cell transplantation. Induced pluripotent stem cells have a potential to provide a source of such cells. Earlier report suggested that iPSCs could induce an immune response even in an autologous setting (Citation66). Recent advances in generation of integration-free iPSC lines have successfully solved this problem (Citation67). Nevertheless, staggering costs and the long time of clinical-grade human iPSC lines production in compliance with cGMPs for only one patient suggest that, similar to hESCs, collection of iPSC line banks is a more promising approach for iPSC-based therapies in the close future. Nobel Prize winning Laureate Professor Yamanaka, who has proposed and developed the main principles of the iPSC technology, has also acknowledged this fact (Citation20). It has been calculated that a bank of only 50 HLA-homozygous iPSC lines would match nearly 90% of the Japanese population (Citation68).
Development of human NT-ESCs using SCNT gave hope of generating autologous embryonic stem cells even for patients with hereditary mitochondrial diseases since the mitochondria are mostly inherited from the donor of the oocyte (Citation4). Since the genomic DNA encodes the HLA molecules and comes from the somatic cell donor, SCNT generates HLA-matched pluripotent cells. But it has been very recently reported that the allogeneic mitochondria, which mainly originate from the oocyte, may trigger an adaptive immune response (Citation21). In a mouse teratoma model expression of the allogeneic mitochondrial DNA genes resulted in rejection of transplanted cells. Further studies are warranted to elucidate severity of NT-ESC immunogenicity. One interesting possibility would be generation of NT-ESCs using the oocyte and the somatic cell nucleus from the same donor. Theoretically, such cells should be fully autologous to the donor and, compared to human iPSCs, contain fewer epigenetic abnormalities.
There are oocyte stimulation protocols that allow generation of highly heterozygous parthenogenetic ESCs (Citation69). Some of them are fully immune compatible with the oocyte donors (Citation3,Citation69). The high heterozygosity levels are explained by the extensive exchange of alleles between parental homologous chromosomes providing the alleles an opportunity for germline transmission (Citation69). Similar to human iPSCs, generation of heterozygous phESCs for treatment of only one patient is too expensive and time-consuming to be widely adopted in clinical practice (Citation9).
Prevention of graft rejection
Allogeneic graft rejection is a major problem of medical transplantology. Conventional strategies to reduce the risk of graft rejection, which may be used with hPSC-based therapies, are based on systemic immunosuppression. The treatment implies modulating and/or suppressing the immune system, thereby reducing risk for rejection. The clinical treatment strategies depend on the particular cell type transplanted, and usually a combination of drugs is used (Citation70). The drugs range from conventional cortisone and cytostatic agents to monoclonal antibodies directed against key factors of the immune system. Although effective, this treatment often causes a variety of side effects such as increased susceptibility to infection, decreased cancer immunosurveillance, liver and kidney injury, to name a few.
Generation of immune tolerance using transplantation of hematopoietic stem cells derived from PSCs is, potentially, another approach to suppress the graft rejection. In a proof of concept study, bone marrow-depleted mice have been reconstituted with donor (syngeneic) and recipient (allogeneic) hematopoietic stem cells. This approach has enabled long-term survival of the reconstituted laboratory animals with depletion of donor reactive as well as auto-reactive T-cells (Citation71). Verda et al. (Citation72) showed that transplantation of hematopoietic stem cells derived from ESCs into the irradiated host can successfully prevent autoimmune diabetes mellitus in a mouse model. ESC-derived hematopoietic stem cells are lacking the risk of contamination with donor T-cells, reducing the risk of graft-versus-host disease. Although promising, this approach is yet to be tested in clinical trials.
One more potential approach is to induce immune tolerance of hPSC-derived allogeneic cells by disrupting the co-stimulatory blockade required for T-cell activation, such as the CD28, LFA1, and CD40–CD40L pathways (Citation73). This produces long-term graft tolerance without the side effects of conventional systemic immunosuppression (Citation74).
Other promising approaches for prevention of immune rejection include the generation of autologous pluripotent stem cells (iPSCs or heterozygous parthenogenetic ESCs), transplantation of hPSC into immunoprivileged sites, and genetic modification of hPSCs to reduce the expression of HLA molecules.
Restoration of three-dimensional (3D) organization of cells in vivo
Human PSC-based technologies have reached significant advances in developing differentiation pathways that lead to generation of single cell-type mature somatic cell populations. Recent advances allow generation of mature cell types that not only express specific protein markers, but also exhibit necessary functional properties. For instance, a method reported by Professor Melton's group allows development of hPSC-derived beta cells that are capable of insulin expression in a glucose-dependent manner (Citation75).
However, in order to achieve long-term therapeutic effects in patients, the hPSC-derived somatic cells have to retain sufficient functional properties for many years after the transplantation. In vivo the function of majority of adult cells depends on molecular cues of their natural environment (or niche) that include: 1) interactions with neighboring cells of same or different type, sometimes in a form of syncytium, and 2) interactions with extracellular matrix. For instance, life-long correct function of the insulin-producing beta cells in vivo requires autocrine signaling from various neighboring islet cells, contact with niche-specific extracellular matrix proteins, direct adhesion to blood capillaries, and syncytium-like organization of beta cells, regulated by intrinsic innervation (Citation76,Citation77). Lack of molecular cues of the natural niches may result in cell death (apoptosis), loss of cellular phenotype, loss of function, or malignant transformation (Citation78,Citation79). Therefore, treatment of damaged tissues that enables long-term benefits for the patient implies not only delivery of mature somatic cells of a certain type to the injury site, but also restoration of their natural niches or incorporation of the cells into the pre-existing ones. For instance, reparation of post-infarction heart damage using hPSC-derived cardiomyocytes requires electromechanical incorporation of the cells into pre-existing cardiac syncytium (Citation80). Such long-term functional integration practically never occurs with sufficient efficacy.
An alternative approach is to generate and transplant hPSC-derived 3D organoids, which are small multicellular tissue units comprising the essential cell types and extracellular matrix structures. This allows synergistic cooperation to be established between cells and their niches already in vitro, prior to transplantation. Development of organoids, as opposed to the single cell type-oriented differentiation approach, may be beneficial, since it resembles natural interaction of heterogeneous cell populations in vivo. During the last several years, significant advances in generation of hPSC-derived organotypic cultures of liver, gut, and stomach have been achieved (Citation81–84). However, the hPSC-derived organoids are not a universal cure to all degenerative diseases. In vitro-generated organoids can hardly be useful to repair organs that require syncytium-like cell organization or sophisticated 3D architecture such as the heart.
In order to treat extensive injuries, such as liver diseases or diabetes, there is often a need to transplant many organoids at the same time. Due to their small size, single organoids are not prone to ischemia; however, lack of sufficient vascularization becomes a major problem when millions of them have to survive within a limited space in the body. Use of decellularized matrices (Citation85–87) or bioprinted 3D-artificial vascularized scaffolds may provide a sufficient vascularization and facilitate long-term survival of large artificial organs after transplantation into patients.
Ethical concerns and legal issues of hPSCs in clinical treatments
Ethical issues related to different types of hPSCs are numerous and extensive, especially in the context of variability of traditions, religions, laws, and regulations in different countries and jurisdictions (Citation22,Citation88). But derivation of hESC lines and procurement of unfertilized human oocytes (Citation10) for hPSC line generation (human NT-ESCs and phESCs) are of particular importance and are discussed in detail below.
Embryos for new hESC line derivation are obtained from in vitro fertilization (IVF) clinics with informed consent given by both parents. Only surplus embryos, which could not be used in the infertility treatment, are utilized to derive hESCs. Nevertheless, the derivation often requires destruction of the ex utero embryos (Citation1,Citation89,Citation90), which causes ethical concerns and is prohibited by law in many countries. In a ground-breaking study reported by Professor Lanza's group (Citation5,Citation6), that problem has been addressed by derivation of new hESC lines from single blastomeres. The blastomeres were removed from embryos using a technique similar to that normally carried out to obtain a single cell for preimplantation genetic diagnosis (PGD). PGD is a standard routine in IVF clinics for preimplantation diagnostics in families with certain genetic backgrounds. Although the method has addressed the ethical concerns of many in principle, it relies on the use of animal-derived and batch-to-batch variable components. Recently, our group has reported derivation of new hESCs from PGD biopsy that is done under chemically defined and xeno-free conditions (Citation7). This method may allow derivation of clinical-grade hESC lines free from major ethical concerns. Importantly, such a method can be beneficial for the blastomere donor's family, since close relatives have a better chance for immune compatibility. If the embryo after PGD is used in the infertility treatment, the born child would have a benefit of autologous hESC line.
Generation of phESCs and recently described human NT-ESC lines (Citation4) may also cause certain ethical concerns regarding procurement of unfertilized human oocytes (Citation10). Current IVF clinic routines leave no or small numbers of surplus unfertilized oocytes. The oocytes should be acquired through a special egg donation program involving administration of an ovarian stimulation protocol to the donors. The treatments needed for superovulation is distressful and can have certain side effects (Citation91). Importantly, unless the donor of the oocyte and the donor somatic cell nucleus is the same individual, the SCNT method would not benefit the oocyte donor or her family, because the resulting human NT-ESC lines would not be autologous to her. On the other hand, generation of human NT-ESC lines from oocytes and somatic cells of the same individual would benefit the donor, thus reducing ethical concerns. Moreover, such cells should be fully autologous and carry few epigenetic abnormalities (Citation18).
Compliance with cGMPs is necessary for development of all clinical-grade hPSC lines. One of the main principles of GMP is traceability of all components, including donor materials. This means public availability of the donor's medical history, which may breach privacy of the donor. Therefore, informed consent given by donors of any biological materials is an essential step in development of clinical-grade human ESC, iPSC, pESC, and NT-ESC lines.
The practical implementation of hPSC-based clinical treatments requires compliance with: 1) national and regional regulations, 2) cGMPs, 3) intellectual property rights, 4) current clinical practice of donor consent acquisition and ethical treatment of human tissues, 5) standard clinical routines, and 6) patent protection laws (Citation22). Major practical hurdles arise from the fact that the regulatory requirements may differ from region to region. For instance, different states in the US have practically opposite legislation regarding hESCs. Importantly, the regulatory requirements may principally and abruptly change in the course of time. In 2011, the European Court of Justice banned patent protection for hESC lines, thus jeopardizing the future of biomedical research in Europe. A similar ban exists in Korea but not in the US (Citation22). In 2007, the authorities abruptly banned shipment of genetic material out of the Russian Federation. A similar ban exists in India. At a level of medical regulatory agencies, decisions to allow or forbid certain procedures are based on general principles, rather than on a set of standard technical requirements. The regulatory agencies in each individual case decide whether imperfections of offered solutions could be compensated by public need and current lack of better options. Currently, there is no international regulatory body that could set uniform international standards in the field of hPSC-based therapies. Differences in national regulations complicate the exchange of clinical-grade hPSC cell lines, materials, and technologies between countries, making generation of national banks of clinical-grade hPSC lines significantly more expensive. Andrews et al. (Citation22) proposed the establishment of an international body that would develop and harmonize all technical, ethical, legal, and regulatory aspects of hPSC-based therapies. Such an institution would greatly facilitate hPSC-based regenerative medicine in the whole world.
Current use of hPSCs in medicine
Existing clinical-grade hPSC lines
Generation of clinical-grade hPSC lines for use in patients is done in compliance with cGMPs, varied legislations of specific local jurisdictions, and ethical standards. Current GMPs are sets of regulations defined by the European Medicines Agency, the US Food and Drug Administration, and other national and international regulatory agencies. The aim of cGMPs is to minimize all the above-described risks regarding cells that are used in patients; cGMPs cover both manufacturing and testing of the final cellular product. Hence, substantial investment of time and money is required to generate hPSC lines under cGMP-compliant conditions.
The legislation is still under development, and GMP standards have a tendency to become more stringent with time. Thus, technologies that involve contacts of any animal-derived materials with clinical-grade cells are becoming unacceptable. The first six clinical-grade hESC lines were maintained in a cGMP-manufactured bovine serum albumin (BSA)-based serum-replacement (SR) containing medium (Citation38). That was a compromise solution, because robust methods for derivation and culturing of hPSCs under xeno-free and chemically defined conditions were not available at that time (Citation38). Later clinical-grade lines have been derived and propagated in a xeno-free environment using human feeder cells as a substratum for hESC growth (Citation89,Citation92). In the UK, the Medical Research Council has initiated derivation of clinical-grade hESC lines in several centers across the country (Citation93). Recently, Professor Yamanaka, supported by the Japanese government, has revealed plans to generate a bank of human iPSCs for therapeutic use (Citation94).
Ongoing clinical trials
There are several clinical trials involving hPSCs that have enrolled patients (). The first clinical trial () involving hESCs was conducted by Geron (California, USA) and was designed to test for safety only. Only four (Citation95)—according to other reports, five (Citation96)—of a planned eight patients were enrolled in the study. The trial was stopped in 2011 for financial reasons. No results have been reported aside from a conference presentation at the American Congress of Rehabilitation Medicine in 2011. None of the four patients suffered serious adverse events, but there were also no improvements in the patients’ condition (Citation95). Recently, it has been announced that the trial may be restarted (Citation96).
Table II. Ongoing clinical trials involving hPSCs with enrolled patients.
Schwartz et al. (Citation97) reported data on two prospective phase 1/2 studies on subretinal transplantation of hESC-derived retinal pigment epithelium into 18 patients with macular dystrophy (). The authors used hESC line MA09 to generate a master cell bank according to GMPs. The cells had had ex-vivo exposure with mouse cells and, therefore, were classified as a xenotransplantation product. Prior to differentiation into retinal pigment epithelium (RPE), hESCs were thawed and expanded on mitomycin-C-treated mouse embryonic fibroblasts for three passages. For the differentiation procedure, hESCs were dissociated from the mouse fibroblasts by treatment with 0.05% trypsin-EDTA and seeded into low-attachment plates in minimal essential medium (MEM) based medium with B-27 supplement for embryoid body (EB) formation. Approximately one week later, the EBs were plated on gelatin-coated dishes and were cultured until RPE colonies became visible. RPE was purified by treatment with type IV collagenase and manual isolation with a glass pipette. Purified RPE was seeded into gelatin-coated tissue culture plates, expanded, and cryopreserved at passage 2 for clinical use. RPE cells were assessed for safety (karyotyping, pathogen testing, etc.), expression of RPE markers (RPE-65, PAX-6, MITF, bestrophin), absence of expression of pluripotency markers (OCT-4, NANOG, SOX-2), and ability to engulf bioparticles (phagocytosis) at various time points, including testing of final cell product formulations. On the day of operation, the cryopreserved cells were thawed, formulated, and assessed for viability and the absence of pathogens. A volume of 150 μL was injected into subretinal space in sites that were selected based on the presence of native, albeit compromised RPE and compromised overlying photoreceptors to increase the chances of transplant integration and function rescue. The patients were on a systematic immunosuppression regimen one week prior to and continued for a period of 6 weeks after the surgical procedure. No evidence has been detected of adverse proliferation, transplant rejection, or any other serious safety issues regarding the transplanted cells. In 10 out of 18 patients there is a significant improvement in eye function, seven patients have experienced a modest improvement, and only one patient has suffered a decline in the eye function. These promising data will inspire more clinical studies to be launched in the close future.
A Japanese woman in her 70s with wet-type age-related macular degeneration became the first patient to receive tissues derived from human iPSCs in September 2014. The underlying pathology is caused by damage of the photoreceptors in the eye leading to blindness. The retinal pigment epithelium cells given as an injection into the eye had been derived from an autologous human iPSC line generated from the patient's own skin cells. Data on safety or efficacy have not been reported yet. The study has not been registered in ClinicalTrials.gov.
In November 2014, the first patient was enrolled in a study () involving hESCs conducted by ViaCyte (California, USA). In the phase ½ trial, hESCs are to be differentiated to pancreatic endodermal cells, encapsulated into a semi-permeable cell containment device for immunoprotection, and implanted subcutaneously in patients with type 1 diabetes. According to the company's web page, the differentiation of hESCs is based on a four-stage differentiation protocol starting from hESCs through mesendoderm to definitive endoderm, primitive gut tube, posterior foregut, and finally pancreatic endoderm (Citation98). Different media and growth factors applied at each stage of the protocol have been empirically found based on the expression levels of pancreatic progenitor and endocrine cell markers in the cells at the respective steps. The study has been designed to test for safety and efficacy (changes in C-peptide level). No data have been reported yet. Apart from the studies listed in , there are several registered studies in ClinicalTrials.gov involving hPSCs that have not yet enrolled any patients.
Current trends in therapies involving hPSCs
Current clinical trials with enrolled patients rely on allotransplantation of hPSC-derived cells into immunoprivileged sites (subretinal space) (Citation97) or use of biocompatible semi-permeable cell containment devices to protect the allotransplant from the immune rejection. However, this approach is applicable to a limited subset of diseases. It has been widely accepted that the most realistic method for future routine PSC therapies, including iPSC therapies (Citation20), has to include generation of clinical-grade banks of hPSC lines that HLA-match a target population (Citation9,Citation59). Strategies for development of such banks (Citation65) and strategies for estimation of sufficient number of lines (Citation59,Citation60) have been reported. Establishment of an international body that would harmonize technical, ethical, legal, and regulatory approaches to hPSC therapies would facilitate development of such banks (Citation22).
In this context, generation of homozygous phESCs is of particular interest. Use of HLA-homozygous cells facilitates compatibility with recipient individuals, reducing the number of lines in a cell bank that match a significant proportion of population. Parthenogenetic hESCs exhibit typical hESC morphology and gene expression profile, except for expression of imprinted genes (Citation3,Citation99). But abnormal imprinting and high levels of homozygosity may complicate application of phESC in medicine (Citation9). Therefore, more studies on the safety of HLA-homozygous phESCs-based therapies are required.
The majority of existing clinical-grade hPSC lines have been exposed to animal-derived molecules or animal cells. For instance, differentiated derivatives of hESC line MA09, which are used by Schwartz et al. (Citation97), are classified as a xenotransplantation product and, therefore, subjected to especially rigorous testing before use in patients. Since approval by regulatory authorities is time-taking and very expensive, the methods of hPSC culturing that are used in current clinical trials are old and suboptimal. In general, methods that enable development of cells under animal-component free and chemically defined conditions are most preferred for use in clinical treatments. Such methods have been reported for derivation (Citation7,Citation40,Citation41) and culturing (Citation36,Citation37) of human ESCs and iPSCs. Although human iPSCs are free from major ethical concerns, there are still some technical problems with generation of safe clinical-grade human iPSC lines. Human iPSCs developed using current methods are prone to epigenetic abnormalities (Citation14,Citation15,Citation18), sometimes generate only partially functional differentiated cells (Citation19), and are prone to additional (in comparison with hESCs) risk of tumorigenicity (Citation52). Therefore, clinical-grade iPSC lines may require more rigorous (in comparison with hESCs) testing, although it has not been shown directly. Further studies on molecular mechanisms of reprogramming are warranted to generate better and cheaper iPSCs in the future. Nevertheless, hiPSCs are already used in treatments of patients (), and plans to generate a bank of human iPSCs for therapeutic use have been announced (Citation94).
Human ESCs are the ‘gold standard’ hPSCs so far and are used in ongoing clinical trials (). Moreover, there is a possibility to derive hESC lines in animal-substance free chemically defined conditions from single blastomeres without destroying human embryos (Citation7). Such cells would be free from major technological and ethical issues and, if robust derivation efficacy is achieved, might be used to generate banks of stem cells for clinical treatments in many countries worldwide.
Acknowledgements
Author contributions: O.S., A.D., and P.V.: manuscript writing, collection and assembly of data; S.R.: conception and design, manuscript writing, collection and assembly of data, final approval of manuscript.
Funding: This work was supported by grants to S.R. from Karolinska Institutets Forskningsstiftelser (2014fobi41924); to O.S. from Karolinska University Hospital; and to A.D. from Russian Science Foundation (RSF), Russian Federation (grant 14-15-00712), and from Foundation for Assistance to Small Innovative Enterprises (FASIE), Russian Federation (project Nr. 24026).
Declaration of interest: S.R. is a shareholder in BioLamina, AB. Other authors report no conflicts of interest.
References
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 19986;282:1145–7.
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
- Revazova ES, Turovets NA, Kochetkova OD, Kindarova LB, Kuzmichev LN, Janus JD, et al. Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells. 2007;9:432–49.
- Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153:1228–38.
- Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Human embryonic stem cell lines derived from single blastomeres. Nature. 2006;444:481–5.
- Chung Y, Klimanskaya I, Becker S, Li T, Maserati M, Lu SJ, et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008;2:113–17.
- Rodin S, Antonsson L, Niaudet C, Simonson OE, Salmela E, Hansson EM, et al. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nat Commun. 2014;5:3195.
- Revazova ES, Turovets NA, Kochetkova OD, Agapova LS, Sebastian JL, Pryzhkova MV, et al. HLA homozygous stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells. 2008;10: 11–24.
- Daughtry B, Mitalipov S. Concise review: parthenote stem cells for regenerative medicine: genetic, epigenetic, and developmental features. Stem Cells Transl Med. 2014;3:290–8.
- Isasi RM, Knoppers BM. Monetary payments for the procurement of oocytes for stem cell research: in search of ethical and political consistency. Stem Cell Res. 2007;1:37–44.
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–12.
- Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7: 618–30.
- Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62.
- Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13:541–9.
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73.
- Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–34.
- Ruiz S, Diep D, Gore A, Panopoulos AD, Montserrat N, Plongthongkum N, et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:16196–201.
- Ma H, Morey R, O’Neil RC, He Y, Daughtry B, Schultz MD, et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature. 2014;511:177–83.
- Foldes G, Matsa E, Kriston-Vizi J, Leja T, Amisten S, Kolker L, et al. Aberrant ?-Adrenergic hypertrophic response in cardiomyocytes from human induced pluripotent cells. Stem Cell Reports. 2014;3:905–14.
- Takahashi K, Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013;140:2457–61.
- Deuse T, Wang D, Stubbendorff M, Itagaki R, Grabosch A, Greaves LC, et al. SCNT-derived ESCs with mismatched mitochondria trigger an immune response in allogeneic hosts. Cell Stem Cell. 2015;16:33–8.
- Andrews PW, Cavanagro J, Deans R, Feigel E, Horowitz E, Keating A, et al. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat Biotechnol. 2014;32:724–6.
- Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S, Baharvand H, et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29:1132–44.
- Narva E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–7.
- Cooke MJ, Stojkovic M, Przyborski SA. Growth of teratomas derived from human pluripotent stem cells is influenced by the graft site. Stem Cells Dev. 2006;15:254–9.
- Hovatta O, Rodin S, Antonsson L, Tryggvason K. Concise review: animal substance-free human embryonic stem cells aiming at clinical applications. Stem Cells Transl Med. 2014;3:1269–74.
- Martin MJ, Muotri A, Gage F, Varki A. Human embryonic stem cells express an immunogenic nonhuman sialic acid. Nat Med. 2005;11: 228–32.
- Hovatta O, Mikkola M, Gertow K, Stromberg AM, Inzunza J, Hreinsson J, et al. A culture system using human foreskin fibroblasts as feeder cells allows production of human embryonic stem cells. Hum Reprod. 2003;18:1404–9.
- Inzunza J, Gertow K, Stromberg MA, Matilainen E, Blennow E, Skottman H, et al. Derivation of human embryonic stem cell lines in serum replacement medium using postnatal human fibroblasts as feeder cells. Stem Cells. 2005;23:544–9.
- Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185–7.
- Braam SR, Zeinstra L, Litjens S, Ward-van Oostwaard D, van den Brink S, van Laake L, et al. Recombinant vitronectin is a functionally defined substrate that supports human embryonic stem cell self-renewal via alphavbeta5 integrin. Stem Cells. 2008;26:2257–65.
- Rodin S, Domogatskaya A, Strom S, Hansson EM, Chien KR, Inzunza J, et al. Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511. Nat Biotechnol. 2010;28:611–15.
- Zhang R, Mjoseng HK, Hoeve MA, Bauer NG, Pells S, Besseling R, et al. A thermoresponsive and chemically defined hydrogel for long-term culture of human embryonic stem cells. Nat Commun. 2013;4:1335.
- Villa-Diaz LG, Nandivada H, Ding J, Nogueira-de-Souza NC, Krebsbach PH, O’Shea KS, et al. Synthetic polymer coatings for long-term growth of human embryonic stem cells. Nat Biotechnol. 2010;28:581–3.
- Melkoumian Z, Weber JL, Weber DM, Fadeev AG, Zhou Y, Dolley-Sonneville P, et al. Synthetic peptide-acrylate surfaces for long-term self-renewal and cardiomyocyte differentiation of human embryonic stem cells. Nat Biotechnol. 2010;28:606–10.
- Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M, Kawasaki M, et al. Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat Commun. 2012;3:1236.
- Rodin S, Antonsson L, Hovatta O, Tryggvason K. Monolayer culturing and cloning of human pluripotent stem cells on laminin-521-based matrices under xeno-free and chemically defined conditions. Nat Protoc. 2014;9:2354–68.
- Crook JM, Peura TT, Kravets L, Bosman AG, Buzzard JJ, Horne R, et al. The generation of six clinical-grade human embryonic stem cell lines. Cell Stem Cell. 2007;1:490–4.
- Unger C, Skottman H, Blomberg P, Dilber MS, Hovatta O. Good manufacturing practice and clinical-grade human embryonic stem cell lines. Hum Mol Genet. 2008;17(R1):R48–53.
- Lu HF, Chai C, Lim TC, Leong MF, Lim JK, Gao S, et al. A defined xeno-free and feeder-free culture system for the derivation, expansion and direct differentiation of transgene-free patient-specific induced pluripotent stem cells. Biomaterials. 2014;35:2816–26.
- Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep. 2014;4:3594.
- Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–4.
- Buzzard JJ, Gough NM, Crook JM, Colman A. Karyotype of human ES cells during extended culture. Nat Biotechnol. 2004;22:381–2; author reply 2.
- Barbaric I, Biga V, Gokhale PJ, Jones M, Stavish D, Glen A, et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem Cell Reports. 2014;3:142–55.
- Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Narva E, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62.
- Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–18.
- Kono T. Genomic imprinting is a barrier to parthenogenesis in mammals. Cytogenet Genome Res. 2006;113:31–5.
- Bukowiecki R, Adjaye J, Prigione A. Mitochondrial function in pluripotent stem cells and cellular reprogramming. Gerontology. 2014;60: 174–82.
- Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, et al. Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell-like metabolic reprogramming. Stem Cells. 2011;29:1338–48.
- Cheng L, Hansen NF, Zhao L, Du Y, Zou C, Donovan FX, et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337–44.
- Van Haute L, Spits C, Geens M, Seneca S, Sermon K. Human embryonic stem cells commonly display large mitochondrial DNA deletions. Nat Biotechnol. 2013;31:20–3.
- Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19: 998–1004.
- Chung S, Shin BS, Hedlund E, Pruszak J, Ferree A, Kang UJ, et al. Genetic selection of sox1GFP-expressing neural precursors removes residual tumorigenic pluripotent stem cells and attenuates tumor formation after transplantation. J Neurochem. 2006;97:1467–80.
- Tang C, Lee AS, Volkmer JP, Sahoo D, Nag D, Mosley AR, et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat Biotechnol. 2011;29: 829–34.
- Ben-David U, Nudel N, Benvenisty N. Immunologic and chemical targeting of the tight-junction protein Claudin-6 eliminates tumorigenic human pluripotent stem cells. Nat Commun. 2013;4:1992.
- Choo AB, Tan HL, Ang SN, Fong WJ, Chin A, Lo J, et al. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells. 2008;26:1454–63.
- Ben-David U, Gan QF, Golan-Lev T, Arora P, Yanuka O, Oren YS, et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013;12:167–79.
- Bradley JA, Bolton EM, Pedersen RA. Stem cell medicine encounters the immune system. Nat Rev Immunol. 2002;2:859–71.
- Taylor CJ, Bolton EM, Pocock S, Sharples LD, Pedersen RA, Bradley JA. Banking on human embryonic stem cells: estimating the number of donor cell lines needed for HLA matching. Lancet. 2005;366:2019–25.
- Nakajima F, Tokunaga K, Nakatsuji N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells. 2007;25:983–5.
- Robertson NJ, Chai JG, Millrain M, Scott D, Hashim F, Manktelow E, et al. Natural regulation of immunity to minor histocompatibility antigens. J Immunol. 2007;178:3558–65.
- Hoglund P, Sundback J, Olsson-Alheim MY, Johansson M, Salcedo M, Ohlen C, et al. Host MHC class I gene control of NK-cell specificity in the mouse. Immunol Rev. 1997;155:11–28.
- Chinen J, Buckley RH. Transplantation immunology: solid organ and bone marrow. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S324–35.
- Didie M, Christalla P, Rubart M, Muppala V, Doker S, Unsold B, et al. Parthenogenetic stem cells for tissue-engineered heart repair. J Clin Invest. 2013;123:1285–98.
- Jacquet L, Stephenson E, Collins R, Patel H, Trussler J, Al-Bedaery R, et al. Strategy for the creation of clinical grade hESC line banks that HLA-match a target population. EMBO Mol Med. 2013;5:10–17.
- Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–15.
- Araki R, Uda M, Hoki Y, Sunayama M, Nakamura M, Ando S, et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013; 494:100–4.
- Nakatsuji N, Nakajima F, Tokunaga K. HLA-haplotype banking and iPS cells. Nat Biotechnol. 2008;26:739–40.
- Dighe V, Clepper L, Pedersen D, Byrne J, Ferguson B, Gokhale S, et al. Heterozygous embryonic stem cell lines derived from nonhuman primate parthenotes. Stem Cells. 2008;26:756–66.
- Duncan MD, Wilkes DS. Transplant-related immunosuppression: a review of immunosuppression and pulmonary infections. Proc Am Thorac Soc. 2005;2:449–55.
- Ildstad ST, Sachs DH. Reconstitution with syngeneic plus allogeneic or xenogeneic bone marrow leads to specific acceptance of allografts or xenografts. Nature. 1984;307:168–70.
- Verda L, Kim DA, Ikehara S, Statkute L, Bronesky D, Petrenko Y, et al. Hematopoietic mixed chimerism derived from allogeneic embryonic stem cells prevents autoimmune diabetes mellitus in NOD mice. Stem Cells. 2008;26:381–6.
- Grinnemo KH, Genead R, Kumagai-Braesch M, Andersson A, Danielsson C, Mansson-Broberg A, et al. Costimulation blockade induces tolerance to HESC transplanted to the testis and induces regulatory T-cells to HESC transplanted into the heart. Stem Cells. 2008;26:1850–7.
- Lui KO, Howie D, Ng SW, Liu S, Chien KR, Waldmann H. Tolerance induction to human stem cell transplants with extension to their differentiated progeny. Nat Commun. 2014;5:5629.
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–39.
- Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic beta-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol. 2011;107:224–35.
- Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, et al. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab. 2011;14:45–54.
- Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76:1352–64.
- Marastoni S, Ligresti G, Lorenzon E, Colombatti A, Mongiat M. Extracellular matrix: a matter of life and death. Connect Tissue Res. 2008;49:203–6.
- Poon E, Kong CW, Li RA. Human pluripotent stem cell-based approaches for myocardial repair: from the electrophysiological perspective. Mol Pharm. 2011;8:1495–504.
- Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–4.
- Takebe T, Zhang RR, Koike H, Kimura M, Yoshizawa E, Enomura M, et al. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc. 2014;9:396–409.
- Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–9.
- McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516: 400–4.
- Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, et al. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat Med. 2008;14:213–21.
- Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010;16:814–20.
- Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva I, Ikonomou L, et al. Regeneration and orthotopic transplantation of a bioartificial lung. Nat Med. 2010;16:927–33.
- Hug K, Hermeren G. Do we still need human embryonic stem cells for stem cell-based therapies? Epistemic and ethical aspects. Stem Cell Rev. 2011;7:761–74.
- Stephenson E, Jacquet L, Miere C, Wood V, Kadeva N, Cornwell G, et al. Derivation and propagation of human embryonic stem cell lines from frozen embryos in an animal product-free environment. Nat Protoc. 2012;7:1366–81.
- Strelchenko N, Verlinsky O, Kukharenko V, Verlinsky Y. Morula-derived human embryonic stem cells. Reprod Biomed Online. 2004;9:623–9.
- Talaulikar VS, Arulkumaran S. Maternal, perinatal and long-term outcomes after assisted reproductive techniques (ART): implications for clinical practice. Eur J Obstet Gynecol Reprod Biol. 2013;170:13–19.
- Tannenbaum SE, Turetsky TT, Singer O, Aizenman E, Kirshberg S, Ilouz N, et al. Derivation of xeno-free and GMP-grade human embryonic stem cells—platforms for future clinical applications. PLoS One. 2012;7:e35325.
- Murdoch A, Braude P, Courtney A, Brison D, Hunt C, Lawford-Davies J, et al. The procurement of cells for the derivation of human embryonic stem cell lines for therapeutic use: recommendations for good practice. Stem Cell Rev. 2012;8:91–9.
- Cyranoski D. Stem-cell pioneer banks on future therapies. Nature. 2012;488:139.
- Baker M. Stem-cell pioneer bows out. Nature. 2011;479:459.
- Hayden EC. Funding windfall rescues abandoned stem-cell trial. Nature. 2014;510:18.
- Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase studies. Lancet. 2015;385:509–16.
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–52.
- Sritanaudomchai H, Ma H, Clepper L, Gokhale S, Bogan R, Hennebold J, et al. Discovery of a novel imprinted gene by transcriptional analysis of parthenogenetic embryonic stem cells. Hum Reprod. 2010;25:1927–41.