
Abstract
Transthyretin (TTR) amyloidosis (ATTR amyloidosis) is a multisystemic, multigenotypic disease resulting from deposition of insoluble ATTR amyloid fibrils in various organs and tissues. Although considered rare, the prevalence of this serious disease is likely underestimated because symptoms can be non-specific and diagnosis largely relies on amyloid detection in tissue biopsies. Treatment is guided by which tissues/organs are involved, although therapeutic options are limited for patients with late-stage disease. Indeed, enthusiasm for liver transplantation for familial ATTR amyloidosis with polyneuropathy was dampened by poor outcomes among patients with significant neurological deficits or cardiac involvement. Hence, there remains an unmet medical need for new therapies. The TTR stabilizers tafamidis and diflunisal slow disease progression in some patients with ATTR amyloidosis with polyneuropathy, and the postulated synergistic effect of doxycycline and tauroursodeoxycholic acid on dissolution of amyloid is under investigation. Another therapeutic approach is to reduce production of the amyloidogenic protein, TTR. Plasma TTR concentration can be significantly reduced with ISIS-TTRRx, an investigational antisense oligonucleotide-based drug, or with patisiran and revusiran, which are investigational RNA interference-based therapeutics that target the liver. The evolving treatment landscape for ATTR amyloidosis brings hope for further improvements in clinical outcomes for patients with this debilitating disease.
ATTR amyloidosis is a serious disease with substantial morbidity and a poor prognosis, although it is likely underdiagnosed due to the non-specific symptoms of the disease and a heavy reliance on biopsy testing.
Currently available treatments for ATTR amyloidosis are designed to reduce levels of mutant TTR, or stabilize the normal confirmation of the TTR protein, although these therapies are not effective in all patients.
A range of novel treatment options that target both mutant and wild-type TTR are under development (e.g. antisense oligonucleotide-based drugs, RNA interference therapies), which are expected to improve outcomes for patients with ATTR amyloidosis.
Introduction
Transthyretin (TTR) amyloidosis (ATTR amyloidosis) is characterized by the deposition of amyloid fibrils, derived from TTR protein, in various organs and tissues (Citation1). TTR is predominantly produced by hepatocytes and circulates as a homotetrameric complex that functions as a transporter for thyroxine and vitamin A (Citation2). However, TTR monomers can undergo a major conformational transformation to aggregate in a highly ordered and abnormal amyloid fibril form (Citation3); TTR in its amyloid conformation is referred to as ATTR throughout this article. Deposition of wild-type (wt) ATTR typically occurs in older patients, giving rise to wt-ATTR amyloidosis, formerly known as senile systemic amyloidosis (Citation4). Mutations in the TTR gene give rise to variants that destabilize the tetramer such that TTR can more readily undergo the conformational change to amyloid, and these genetic changes underlie the various hereditary ATTR amyloidosis (h-ATTR amyloidosis) clinical syndromes. Some 100 amyloidogenic TTR mutations have been recognized, the distribution of which varies in geography and disease presentation (Citation5,Citation6).
ATTR is derived either wholly from wt TTR in non-hereditary disease or from a mix of variant and wt TTR in hereditary forms. However, in both cases the deposition of this protein at various sites within the body gives rise to a multisystemic disorder. Hereditary ATTR amyloidosis has traditionally been described according to whether the predominant clinical manifestation is neuropathy (h-ATTR amyloidosis with polyneuropathy) or cardiomyopathy (h-ATTR amyloidosis with cardiomyopathy). In clinical practice, however, a wide range of overlapping phenotypes are recognized, not only among patients with different mutations but also among those with the same mutation. Indeed, the majority of TTR mutations give rise to a mixed clinical phenotype, where both neurologic and cardiac impairments are present (Citation6). The disease phenotype has an impact on mortality: patients with dominant neuropathy have a median survival from diagnosis of 5–15 years (Citation7–9), whereas h-ATTR amyloidosis with cardiomyopathy is associated with a poorer survival of 2.5–4 years (Citation10). Furthermore, substantial impairment in quality of life (QoL) is associated with both disease phenotypes (Citation11,Citation12).
The purpose of this article is to review the key aspects of the diagnosis and natural history of ATTR amyloidosis and, with reference to clinical studies of both approved and investigational therapies, describe the evolving treatment landscape for this disease.
Search strategy and selection criteria
References for this review were identified by a hand search of the PubMed database (January 1981–September 2014). The search terms used were ‘transthyretin amyloidosis’, ‘TTR amyloidosis’, ‘TTR-FAP’, ‘TTR-FAC’, ‘TTR amyloidosis treatment’, ‘diflunisal’, and ‘tafamidis’. The abstract listings of the XIIIth (2012) and XIVth (2014) International Symposia on Amyloidosis and the European Society of Cardiology Congress 2014 were searched for data on treatments for ATTR amyloidosis. References were also hand chosen from the bibliographies of identified articles. Only papers published in English or with English translations were reviewed.
Diagnosis and disease assessment
Diagnosis of ATTR amyloidosis is hindered by a lack of awareness among clinicians, and further complicated by the presentation of this disease with a variety of non-specific clinical features. For example, thickened ventricular walls identified on echocardiography in older patients with cardiac symptoms are readily attributed to hypertrophic cardiomyopathy caused by a sarcomeric disease or hypertension, and the possibility of ATTR amyloidosis may not be pursued. The type of amyloidosis may also be misdefined, as characteristic echocardiographic appearances in patients with ATTR amyloidosis may be attributed to immunoglobulin light chain (AL) amyloidosis if monoclonal gammopathy is present. Monoclonal gammopathy is a common incidental finding in older patients, thus increasing the likelihood of this misdiagnosis (Citation13). Polyneuropathy itself is associated with numerous conditions, including diabetes mellitus, and the heterogeneity of clinical phenotypes that mimic other causes of peripheral neuropathy is a further diagnostic challenge (Citation14,Citation15). The difficulties in recognition of the disease can lead to a considerable delay in the diagnosis of ATTR amyloidosis; a mean interval of 4 years from symptom onset to diagnosis has been reported for h-ATTR amyloidosis with polyneuropathy (Citation16), and periods of up to 8 years have been described in patients with h-ATTR amyloidosis with cardiomyopathy (Citation17,Citation18).
Patients with progressive axonal peripheral neuropathy of unknown origin, especially those with autonomic neuropathy (e.g. erectile dysfunction, orthostatic hypotension, or constipation alternating with diarrhea) in the absence of diabetes, should raise suspicion of ATTR amyloidosis with polyneuropathy. Investigations of the autonomic and somatic nervous system by orthostatic tests, sympathetic skin tests, or heart rate variability, as well as quantitative sensory testing and electrophysiological analysis, have value for tracking disease progression, but are less frequently used for initial diagnosis (Citation1). The gold standard for diagnosis is identification of amyloid deposits in a tissue sample, which can be detected by histologic staining with Congo red. Fat pad or labial salivary gland biopsies have been promoted for diagnosis, with reported sensitivity in expert hands of 73%–83% (Citation19,Citation20) and up to 92% in Portuguese patients with h-ATTR amyloidosis with polyneuropathy (Citation21). Biopsies of the gastrointestinal tract are similarly informative for this form of ATTR amyloidosis. However, non-cardiac tissues often lack amyloid deposits in patients with suspected ATTR amyloidosis with cardiomyopathy, in whom endomyocardial biopsy may be the only way to gain histologic confirmation (Citation22). Patients with amyloid diseases can have patchy distribution of amyloid deposits in their tissues, notably within the nerves in cases with significant neuropathy, such that examination of multiple samples may be necessary before a confident diagnosis of ATTR amyloidosis can be made (Citation22,Citation23). As mentioned above, it is important to distinguish ATTR amyloidosis from other amyloid diseases, especially AL amyloidosis, which can be achieved through identification of the precursor protein using techniques such as Western blotting of amyloid tissue extract, immunohistochemical staining of tissue sections, or proteomic analyses of laser-dissected amyloid material (Citation24–26). Analysis of ATTR deposits has also revealed an association between fibril subunit size and clinical features: in patients with ATTR amyloidosis with the TTR Val30Met mutation, full-length TTR subunits were associated with neuropathy but not cardiomyopathy, whereas a mixture of truncated and full-length peptides was present in patients with cardiomyopathy (Citation25). Truncated peptides were also detected in most patients with ATTR amyloidosis and cardiomyopathy who had mutations other than Val30Met, and in all patients with wt-ATTR amyloidosis tested to date (Citation27).
Sequencing of the TTR gene can reliably detect the mutations associated with h-ATTR amyloidosis (Citation28), and genetic testing is therefore a crucial component in supporting a diagnosis. Genetic testing alone, however, is not sufficient to make a diagnosis, given that a positive result neither confirms the presence of amyloid nor excludes the individual from having a different type of amyloid disease. The specificity of gene testing depends, in part, on the prevalence and penetrance of the trait in the population, which is generally poorly defined. However, a high prevalence in certain geographic areas and populations (e.g. northern Sweden and individuals of African descent (Citation29,Citation30)) makes the broad application of this diagnostic approach questionable. Prenatal and adult predictive genetic testing can easily and accurately be performed in individuals at risk of h-ATTR amyloidosis, but expert genetic counseling is mandatory (Citation28). Pre-implantation genetic testing has been developed in major endemic regions (Citation31,Citation32). However, individuals with a positive genetic test should be made aware of the variable penetrance of the disease, which is generally lower in non-endemic regions, and the differences in age of symptom onset (Citation33–35).
A simple clinical staging system for h-ATTR amyloidosis with polyneuropathy was developed in an endemic area of Portugal (familial amyloidosis with polyneuropathy (FAP) staging system), which classifies patients as stage 0 (asymptomatic), stage I (those with symptoms who are ambulatory but do not require assistance), stage II (those who are ambulatory but require assistance), and stage III (wheelchair- or bed-bound patients with advanced disease). The polyneuropathy disability (PND) score is another simple staging system whereby patients are categorized into four stages according to their degree of walking disability (Citation36): stage I (sensory disturbances but with preserved walking capability); stage II (impaired walking ability without need for a stick); stage III (walking only with the help of one stick (IIIA) or two sticks (IIIB)); and stage IV (wheelchair- or bed-bound).
Several non-invasive tests are useful to support the diagnosis of ATTR amyloidosis with cardiomyopathy. These include echocardiography, electrocardiography (ECG), cardiac magnetic resonance imaging (CMRI), and nuclear scintigraphy. Ventricular wall thickening (i.e. intraventricular septum thickness > 12 mm) with normal ejection fraction and reduced longitudinal strain identified by echocardiography and/or low QRS voltage recorded by ECG are strong signals for cardiac amyloidosis (Citation37,Citation38), although in some cases the ECG may be normal or even show increased voltage (Citation39). CMRI has a much greater diagnostic yield than echocardiography for recognizing cardiac amyloidosis, as it produces a characteristic appearance on late gadolinium enhancement due to the infiltration of myocardial interstitial space by amyloid deposits. Recently, CMRI with T1 mapping was shown to be highly effective in the identification of both AL and ATTR amyloid in the heart (Citation40). Another sensitive and complementary imaging method that is gaining widespread use is scintigraphy, with the technetium isotopes technetium- 99m-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) (Citation41–43) and technetium-99m-pyrophosphate (99mTc-PYP) (Citation44). These techniques were originally developed for bone assessment but have demonstrated remarkable diagnostic accuracy in patients with ATTR amyloidosis with cardiomyopathy. Both of these tracers localize strongly to the heart in this type of amyloidosis, whereas localization is usually weaker and less frequent in cardiac AL and other types of amyloidosis. In light of current knowledge, a 99mTc-DPD or 99mTc-PYP scan that shows no or minor uptake in the heart argues strongly against a patient having clinically significant cardiac ATTR amyloid deposition.
Disease characteristics and natural history
The prevalence of h-ATTR amyloidosis with polyneuropathy is estimated at approximately 10,000 patients worldwide, or one in 1 million habitants (Citation45), with onset of this disease occurring at any stage of life from early adulthood (Citation6). Val30Met is the most common TTR mutation in patients with h-ATTR with polyneuropathy, especially in countries with the highest prevalence, for example Portugal, France, Sweden, and Japan (Citation46); prevalence may reach 1 in 1000 in endemic areas in Portugal, Japan, and Sweden. In the early stages of h-ATTR with polyneuropathy, neuropathy manifests as neuropathic pain or numbness in the hands and feet. As the disease progresses, the sensory deficit extends to the arms and thighs, and then to the anterior trunk (Citation1,Citation47). Hand involvement, including weakness, sensory impairment, and reduction in fine motor skills, was also observed in the vast majority of patients with h-ATTR amyloidosis with polyneuropathy in a recent study (Citation15). Disease characteristics can vary according to the specific mutation involved; for example patients with Tyr78Phe tend to present with predominant motor neuropathy (Citation48). Age at onset and gender also influence disease characteristics (Citation49,Citation50). With prolonged disease duration, motor skills deteriorate such that walking becomes increasingly difficult (Citation51), and patients with late-stage disease generally display significant and often life-threatening autonomic neuropathy, with gastrointestinal impairment, weight loss, and cachexia.
Hereditary ATTR amyloidosis with polyneuropathy can progress quickly following the onset of symptoms, with a rapid increase in Neuropathy Impairment Score (NIS) frequently observed. In the phase III clinical trial of diflunisal, mean NIS had increased by 10.1 points and 23.2 points at years 1 and 2, respectively, in patients receiving placebo (Citation52). Supporting these data, a larger, multinational, cross-sectional study of 283 patients with h-ATTR with polyneuropathy (stage I–III FAP, with median NIS of 32) reported a progression rate of 14.3 points/year (Citation23). In the latter study, the increase in NIS correlated with PND score and FAP stage, validating it as a measure of disease progression (Citation23). NIS was also used to assess patient impairment according to time of disease onset: median NIS was higher in the cohort of patients with late-onset versus early-onset Val30Met h-ATTR amyloidosis with polyneuropathy (59.6 versus 9.0, respectively) (Citation23). However, the difference between these cohorts is likely related to the emphasis placed by the NIS score on motor impairment, and the scoring system has thus been subsequently refined for h-ATTR with polyneuropathy by development of the modified NIS plus seven nerve tests (mNIS + 7) (Citation47). Analyses of patient characteristics have also extended from monitoring of disease burden to the investigation of symptoms that are associated with survival. In this regard, modified body mass index (mBMI; BMI multiplied by serum albumin level to compensate for edema) value has demonstrated a strong correlation with survival in Swedish patients with ATTR amyloidosis with Val30Met, and also with survival after liver transplantation (Citation36,Citation53). In addition, mean heart rate (Citation54) and the combination of septal thickening and diastolic dysfunction (Citation55) have been independently identified as prognostic indicators in patients with ATTR amyloidosis with the Val30Met mutation.
The prevalence of h-ATTR amyloidosis with cardiomyopathy is estimated at over 40,000 patients worldwide, although it is likely that both wt- and h-ATTR cardiac amyloidosis are currently underdiagnosed. Indeed, an autopsy study in elderly Finnish patients revealed that amyloid deposits were present in 25% of patients aged 80–85 years (Citation56). Cardiac ATTR was also found on autopsy in 5% of patients with heart failure and preserved ejection fraction (Citation57), suggesting that amyloidosis is not an uncommon causative factor for this very prevalent clinical syndrome. Wt-ATTR amyloidosis with cardiomyopathy is most often diagnosed in elderly men (Citation58), but it can occur in women and occasionally in individuals younger than 60 years. Accumulation of amyloid in the heart leads to ventricular wall thickening and diastolic dysfunction, arrhythmias, and ultimately intractable heart failure (Citation59). Val122Ile TTR is the most common genetic variant globally associated with h-ATTR amyloidosis with cardiomyopathy, although it is largely restricted to the black population. Atrial and ventricular arrhythmia, bradycardia, advanced heart block, and congestive heart failure (CHF) may be especially frequent in the black population (Citation18). Clinical features may differ according to the TTR genotype, although confirmatory studies to date have been relatively small. For example, normal QRS voltages appear to be more common in patients with wtATTR rather than h-ATTR amyloidosis with cardiomyopathy. Data on the rate of progression of ATTR amyloidosis with cardiomyopathy are limited, although rapid disease progression can occur. Significant increases in myocardial mass were reported over 1 year of observation in one study (Citation8), and the Transthyretin Amyloidosis Cardiac Study (TRACS) reported decreases in 6-minute walking distance (mean reduction of 25.8 m every 6 months) and left ventricular (LV) ejection fraction over 18 months (Citation10).
The genotype of patients with ATTR amyloidosis with cardiomyopathy may also be associated with outcome, with poorer survival observed in patients with variant TTR versus those with wt-ATTR amyloidosis. In TRACS, patients with h-ATTR amyloidosis with cardiomyopathy and the Val122Ile mutation had shorter median survival than those with disease associated with wt TTR (25.6 versus 43.0 months, respectively) (Citation10). A more recent single-center observational study supported the deleterious impact of the TTR Val122Ile mutation, with earlier-onset disease and a shorter time from diagnosis to death or orthotopic heart transplant reported in patients with variant versus wt TTR (36.4 versus 66.5 months, respectively) (Citation60). The impact of non-Val122Ile mutations on outcomes may be less severe, with an overall median survival of approximately 41 months among a series of patients with the TTR Thr60Ala mutation (Citation7). Other factors that have been associated with prognosis in patients with h-ATTR amyloidosis with cardiomyopathy include N-terminal pro-brain natriuretic peptide (BNP) concentration, New York Heart Association (NYHA) stage, cardiac output, and pericardial effusion (Citation61).
Evolution of therapies
Symptomatic relief
Relief of symptoms remains of utmost importance. For example, in patients with h-ATTR amyloidosis with polyneuropathy, control of peripheral (e.g. pain, trophic ulceration) and autonomic (e.g. gastrointestinal symptoms, hypotension) neuropathy is key to improving QoL (Citation62). Prophylactic pacemaker implantation may also be valuable for patients with h-ATTR with polyneuropathy and conduction disorders, with major cardiac events prevented in a quarter of patients during a 45-month mean follow-up (Citation63). Symptomatic relief and stabilization of fluid balance are extremely important in patients with ATTR amyloidosis with cardiomyopathy because heart transplantation may not be a treatment option in these often older patients. A key goal of supportive care in patients with cardiac involvement is the reduction of filling pressure, which can be achieved with loop diuretics. However, caution is necessary as excessive reduction in filling pressure results in reduced cardiac output and hypotension; current recommendations suggest prescription of loop diuretics at very low doses either daily or every other day. Other cardiac manifestations include dysrhythmias, for example tachycardia or bradycardia, and atrial fibrillation, which can be relieved pharmacologically or through insertion of a permanent pacemaker (Citation18). Many drugs commonly used in other types of heart failure may not be beneficial in amyloidosis, and may actually exacerbate symptoms. Special care is recommended for agents (including digoxin, dihydropyridine calcium channel blockers, and vasodilators) whose distribution may be affected by binding to amyloid fibrils (Citation64,Citation65). Caution should also be taken with inhibitors of cyclic guanosine monophosphate used for erectile dysfunction, as these may worsen hypotension and could potentially prompt syncope, arrhythmias, or cardiac arrest (Citation66).
Liver transplantation (1990s onwards)
Liver transplantation remains the first-line treatment option offered to patients with early-stage h-ATTR amyloidosis with polyneuropathy in Japan (Citation1), although not recently in France or Portugal (Citation67). The rationale for this procedure is to remove the major source of genetically variant TTR in the plasma, replacing it with the wt protein of the donor. The concentration of variant TTR has been reduced by 98% in patients following orthotopic liver transplantation (Citation68,Citation69). This surgical intervention has been shown to increase patient survival significantly (Citation53,Citation70,Citation71), and prevents progression of neuropathy in the majority of patients with the TTR Val30Met mutation (Citation68,Citation72). Follow-up studies have indicated regression of visceral amyloid deposits (Citation73,Citation74), but the course of amyloid deposition in nerves is less clear. The outcome of liver transplantation is influenced by various patient characteristics, including age, severity of disease at the time of surgery, and mutation type. For example, the 5- and 10-year survival rates are markedly worse among patients with mutations other than Val30Met compared with the Val30Met change (Citation75,Citation76). In addition, the utility of liver transplantation is limited by restricted donor availability, patient eligibility (including age < 70 years), and the risk of adverse events (AEs).
The capacity of liver transplantation to halt progressive neuropathy is best supported in patients with the Val30Met mutation (Citation68,Citation77), although this is not necessarily accompanied by an improvement in QoL (Citation1). Deposition of wtATTR in peripheral tissues may still continue following liver transplantation (Citation78,Citation79), which is thought to account for the progression of cardiomyopathy that occurs in many patients, especially those with non-Val30Met mutations (Citation80–82). Progression of cardiac amyloidosis following liver transplantation does not seem to occur in patients with Val30Met TTR mutations and early-onset disease; it has been proposed that this favorable situation is associated with ATTR amyloid fibrils that are composed of full-length TTR peptides (Citation83). Unfortunately, this phenomenon may be restricted to patients with this particular mutation, in whom cardiac involvement is unusual other than in late-onset cases. Liver transplantation does not prevent progression of amyloid in the eye or in the central nervous system because TTR is produced locally at these particular sites (Citation84).
Liver transplantation is associated with a variety of risks, with 1-year mortality rates of 7%–25% reported for patients with h-ATTR amyloidosis with polyneuropathy (Citation62,Citation72,Citation85–87). In particular, the risk of hepatic artery thrombosis appears greater in transplant patients with h-ATTR amyloidosis with polyneuropathy, with an incidence approximately 8-fold higher than in patients with other indications (Citation88). The requirement for long-term immunosuppressive agents, for example ciclosporin and tacrolimus, can also be problematic for patients after liver transplantation. Of note are the increased risk of post-surgical infection (Citation68,Citation72) and long-term issues with chronic renal failure and diabetes, which are observed in 30% and 10%–20% of patients, respectively, 10 years after liver transplantation (Citation62). Contrasting data have been published with respect to renal function, with a long-term study in Swedish patients with h-ATTR amyloidosis with polyneuropathy suggesting that liver transplantation may in fact arrest the progression of kidney damage (Citation89). Post-transplant lymphoproliferative disorder can also be a fatal complication (Citation90), but overall the long-term outcomes for patients with ATTR following liver transplantation are comparable to those for patients with other diseases in which this procedure is performed (Citation53).
Combined heart and liver transplantation is a possibility for patients with severe h-ATTR amyloidosis with cardiomyopathy and mutations associated with extra-cardiac amyloid disease (Citation91), although this procedure has only been performed in a small number of patients. Such surgery carries substantial risk, yet some studies have reported outcomes similar to those obtained followed isolated heart transplantation for other indications (Citation92,Citation93). Of note, cardiac transplantation alone for patients with wt or Val122Ile-mutant TTR amyloidosis with cardiomyopathy has generated encouraging long-term outcomes, with 5-year survival similar to that observed after general heart transplantation in a UK study (Citation17,Citation94).
Era of targeted therapies and new and potential medicines
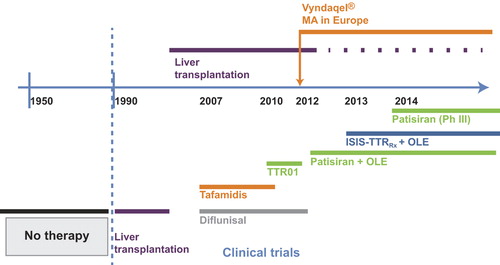
The outcome of liver transplantation has ultimately proved disappointing for patients with Val30Met TTR and more severe disease, as well as for the majority of patients with other TTR mutations. A new era of targeted medicines was initiated approximately 10 years ago (), and these novel therapeutic strategies are discussed below.
Figure 1. Timeline of access to anti-amyloid therapies for patients with hereditary transthyretin amyloidosis with polyneuropathy. OLE = open-label extension; Ph = phase; Vyndaqel = trade name for tafamidis.
TTR tetramer stabilizers
TTR tetramer stabilizers are agents designed to stabilize the normal circulating tetrameric form of TTR, and hence are hypothesized to prevent the protein from dissociating and undergoing conformational change that leads to its aggregation as amyloid. Tafamidis and diflunisal, which have both been shown to stabilize TTR through interaction with the thyroxine binding site of the tetramer, are discussed.
Tafamidis
Tafamidis has been demonstrated to increase the stability of the TTR protein in vitro, based on data from an immunoturbidimetric assay. Immunoturbidimetry involves measuring the TTR tetramer concentration in plasma samples before and after denaturation; these data are then extrapolated to generate percent TTR stabilization. This is a complex, non-physiologic assay that is unsuitable for monitoring the pharmacodynamics of tafamidis in the clinic. More recently a subunit exchange technique has been proposed for quantifying the stability of endogenous TTR in plasma, which has demonstrated a correlation between TTR tetramer stability and tafamidis concentration (Citation95).
Tafamidis treatment for h-ATTR amyloidosis with polyneuropathy was assessed in the randomized, double-blind, placebo-controlled phase II/III Fx-005 trial, which included patients with the TTR Val30Met mutation and stage I neuropathy () (Citation96). In patients with evaluable data at 18 months, treatment response (< 2-point increase in NIS in the lower limbs (NIS-LL; range 0–88)) was significantly higher in the tafamidis arm compared with the placebo arm (60% versus 38%, respectively; P = 0.041). Least-squares mean change from baseline in Norfolk QoL–Diabetic Neuropathy total (TQoL) score was also significantly better for tafamidis versus placebo (0.1 versus 8.9, respectively; P = 0.045). However, in the intention-to-treat population, the differences in treatment response (45% in the tafamidis arm versus 30% in the placebo arm) and TQoL (2.0 versus 7.2, respectively) were not statistically significant between the study groups (study did not meet its co-primary end-points). However, a trend towards benefit in the tafamidis group remained in the extension study. The discontinuation rate was higher than expected in this trial, primarily due to patients undergoing liver transplantation, which impacted on the ability of the trial to achieve statistical significance in the co-primary end-points. Tafamidis was generally well tolerated, with a similar incidence of AEs between the active and control treatment groups. However, urinary tract infections and diarrhea were more common in the tafamidis treatment group than the placebo group.
Table I. Key clinical data for novel agents in clinical development of TTR amyloidosis.
This study provided no indication that the disease process could be reversed by tafamidis treatment. Indeed, the summated seven nerve tests normal deviate score, NIS-LL score, and muscle weakness increased steadily in the 12-month open-label extension of this study, indicating disease progression (Citation107). Similar data were also recently reported from a Portuguese single-center 12-month study of tafamidis in 103 patients with stage I FAP. Here, the response rate (NIS change < 2 points across 1 year) was 64%, although an increase in mean NIS score from baseline to 6 and 12 months was observed across the whole study population (Citation108).
While tafamidis may slow disease progression in the majority of patients with the Val30Met TTR mutation and early-stage disease, this treatment may be less effective in more advanced disease. In an early-access program for tafamidis in patients with Val30Met-mutant late-onset disease, 77% of whom had walking disability, deterioration of either NIS score or disability was observed in 93% of patients after 1 year of treatment (Citation99). Indeed, the rate of increase in NIS-LL was unaltered during the first 6 months of tafamidis treatment compared to before treatment.
Tafamidis has also been investigated in 21 patients with non-Val30Met TTR mutations in a single-arm open-label study (Citation97). This study met its primary end-point of biochemical stabilization of TTR in 95% of patients. Exploratory analyses demonstrated no clinical worsening of health-related QoL or mBMI, although worsening of neurologic function (i.e. increases in NIS, NIS-LL, and upper leg NIS) was observed at 1 year of treatment (Citation97). Survival rates at 15 years with tafamidis have been estimated as 87% and 78% for patients with Val30Met and non-Val30Met mutations, respectively (Citation109).
Based on the results of the Fx-005 trial, tafamidis at a dose of 20 mg administered orally once daily was approved in Europe for the treatment of stage I FAP. Tafamidis has become the first-line treatment option for patients with early-stage h-ATTR amyloidosis with polyneuropathy in France (Citation67). However, the US Food and Drug Administration (FDA) did not grant regulatory approval for tafamidis based on inadequate evidence of effectiveness (FDA evaluation of tafamidis (Citation110)).
Tafamidis has also been assessed in patients with wt- or Val122lle-TTR associated ATTR amyloidosis with cardiomyopathy in a single-arm, open-label phase II study (Citation98). The proportion of patients with cardiac function worsening (NYHA criteria) increased after month 36, with deterioration in approximately 60% of patients by month 42. The estimated 5-year survival rates were 86% from symptom onset and 45% from the first dose of tafamidis. While the efficacy of tafamidis cannot be determined in this single-arm study, tafamidis was generally well tolerated in this relatively elderly patient population. An international, placebo-controlled, phase III trial of tafamidis in patients with ATTR with cardiomyopathy is ongoing (NCT01994889). Participants eligible for this study have a history of heart failure, evidence of cardiac involvement by ECG, and the presence of non-amyloid A or -AL amyloid deposits in biopsy tissues. Patients who have received a prior heart transplant are excluded. The primary outcome measure is a combination of all-cause mortality and the frequency of cardiovascular-related hospitalization after 30 months’ treatment with tafamidis (20 or 80 mg) compared with placebo.
The impact of tafamidis on other body systems in patients with h-ATTR amyloidosis with polyneuropathy has also been assessed in a single-center Portuguese study suggesting that this agent may reduce progression of nephropathy. Among 98 patients with Val30Met-mutant stage I FAP who received tafamidis for 12 months, no significant changes were recorded in creatinine concentration, and no new cases of albuminuria or augmentations of the urinary albumin:creatinine ratio (UACR) were observed (Citation111). Additionally, in the 12 patients who had proteinuria at baseline (UACR > 300 mg/g), UACR decreased significantly and a stable estimated glomerular filtration rate (eGFR) was maintained. These data contrast with the anticipated progression of nephropathy in untreated patients; however, this was an uncontrolled study and thus precludes any definitive judgment of the benefits of tafamidis treatment on renal involvement.
Diflunisal
Diflunisal is a non-steroidal anti-inflammatory drug (NSAID) that has been administered orally twice daily at a dose of 250 mg in clinical trials to patients with h-ATTR amyloidosis with polyneuropathy. Although it is not licensed for the treatment of ATTR amyloidosis, many patients with polyneuropathy have lately been offered this agent off-label (Citation112). Diflunisal has been shown to increase the stability of TTR tetramers in vitro (Citation113) and in healthy subjects (Citation114), but, as for tafamidis, it is not possible routinely to measure the magnitude of its pharmacodynamic effects in patients.
In a randomized, placebo-controlled, phase III trial in 130 patients with h-ATTR amyloidosis with polyneuropathy (the Diflunisal Trial), 24 months’ diflunisal treatment was associated with a significantly smaller deterioration in NIS + 7 (primary end-point) compared with placebo () (Citation52). Furthermore, a greater proportion of patients in the diflunisal group showed neurologic stability (< 2-point increase in NIS + 7) at 2 years (29.7% versus 9.4%). Patients’ QoL, assessed using mean Medical Outcomes Study Short-Form (36-item) Health Survey physical and mental scores, also improved with diflunisal treatment, but worsened in patients in the placebo arm (Citation52). Compared with the FX-005 tafamidis study, the diflunisal trial included patients with more advanced disease (only 37.7% of the population in PND stage I) and patients with Val30Met or non-Val30Met mutations. Among patients randomized to receive diflunisal, only 58% completed the 2-year study treatment, and this high discontinuation rate introduces some uncertainty about interpretation of the results. Discontinuation was primarily due to disease progression (11/37 cases), which was less common with diflunisal than placebo. Diflunisal was generally well tolerated, although events from the musculoskeletal and general disorders classes occurred more frequently in the active treatment group compared with the placebo group. Separately, isolated cases of deterioration in renal function or thrombocytopenia have been reported in small studies of long-term diflunisal treatment in patients with h-ATTR amyloidosis with polyneuropathy (Citation115).
The tolerability of NSAIDs has mostly been assessed in patients with rheumatic disease (i.e. patients without amyloidosis). This large body of data indicates that NSAID usage is associated with an increased risk of gastrointestinal AEs, cardiotoxicity, and/or renal AEs (Citation116–118). Indeed, NSAIDs are contraindicated in patients with severe congestive heart failure (CHF) or renal insufficiency, which may limit its value in patients with ATTR amyloidosis with cardiac involvement. In the phase III study of patients with h-ATTR amyloidosis with polyneuropathy, the incidences of cardiac or renal events were similar in the diflunisal and placebo groups (Citation52), although two patients in the diflunisal group discontinued due to gastrointestinal bleeding and CHF, respectively. As some toxicities associated with NSAIDs are more pronounced with prolonged treatment (Citation119), long-term safety data for diflunisal are needed in populations with ATTR amyloidosis.
The impact of diflunisal on cardiac measures was also assessed in the phase III trial (Citation52) of patients with h-ATTR amyloidosis with polyneuropathy (Citation120). Among the patients who had cardiac involvement at baseline (approximately 50% of the total number of patients included), 24 months’ diflunisal treatment did not significantly reduce LV wall thickness or LV longitudinal strain compared with placebo. However, in a subgroup analysis of patients with abnormal BNP levels, the rate of increase in LV wall thickness did appear to be slower with diflunisal treatment compared with placebo. The efficacy and safety of diflunisal in patients with cardiac amyloidosis have been explored in a single-arm, open-label study of a small cohort of 13 patients with wt, or TTR-variant cardiac ATTR amyloidosis (Citation100). Diflunisal treatment was generally well tolerated, and no significant changes in cardiac structure were identified at a mean of 0.9 years’ follow-up. A 6% reduction in glomerular filtration rate was observed in this study with prolonged diflunisal treatment, and controlled studies are evidently required to determine whether diflunisal slows progression of cardiac amyloidosis in some patients.
Doxycycline plus tauroursodeoxycholic acid
The therapeutic potential of doxycycline and tauroursodeoxycholic acid (TUDCA) has been studied in a transgenic Val30Met TTR mouse model, with the combination of these agents resulting in a reduction in TTR amyloid deposition and associated tissue markers (Citation121). Although neither this nor any other experimental system created to date has proved to be fully representative of human ATTR amyloidosis, the safety of both of these long-used oral drugs has been established in a variety of clinical settings, providing confidence for investigating this therapeutic approach in patients with ATTR amyloidosis.
Results of an open-label phase II study of doxycycline (100 mg twice daily) plus TUDCA (250 mg three times a day) in patients with ATTR amyloidosis (NCT01171859) were presented by Obici et al. at the XIVth International Symposium on Amyloidosis (Citation101). In this study, 60% of patients (24/40) completed 12 months of treatment; stable cardiac disease was observed in 75% of these patients (18/24) and stable neuropathy in 46% of patients (6/13). The treatment was generally well tolerated, although persistent mild skin redness was reported. Four patients had discontinued by 12 months due to doxycycline-related gastrointestinal events. Additionally, an open-label, single-arm, phase I/II study of doxycycline plus TUDCA in patients with either wt- or h-ATTR amyloidosis with cardiomyopathy is currently recruiting participants (NCT01855360).
TTR-lowering investigational treatments
Reduction in the supply of the respective amyloid fibril precursor protein is a rationale for treatment in all types of amyloidosis. This approach has been validated extensively in amyloid A and AL types, in which it is associated with preservation or improvement in amyloidotic organ function and improved survival (Citation13,Citation122–124). In h-ATTR amyloidosis, liver transplantation has been used to suppress production of the genetically variant amyloid-forming protein, but the inherent amyloidogenicity of the wt TTR protein that replaces it has greatly limited its success. There are two distinct pharmacologic methodologies in clinical development that aim to reduce total TTR production: antisense oligonucleotide (ASO)-based therapy, and RNA interference (RNAi) therapeutics.
ISIS-TTRRx
ASOs are short synthetic oligonucleotides that bind directly to TTR messenger RNA (mRNA), leading to its degradation by RNAase H. ISIS-TTRRx is an ASO-based therapy that causes destruction of both wt and mutant forms of the TTR transcript (Citation125). In a mouse model expressing a mutant human form of TTR, ISIS-TTRRx treatment reduced hepatic TTR mRNA and serum TTR protein levels by up to 80% (Citation125). Similar results were observed in non-human primate models, with TTR mRNA and serum TTR levels reduced by approximately 80% after 12 weeks’ treatment with ISIS-TTRRx (Citation126).
Currently, all reported clinical data for ISIS-TTRRx are from studies in healthy volunteers. In a phase I study, single doses of ISIS-TTRRx were well tolerated up to 400 mg, with the most common AEs reported to be somnolence and injection-site pain (Citation126). Multiple dosing elicited a dose-dependent decrease in plasma TTR levels, with reductions of approximately 80% observed after 4 weeks’ treatment with the 300 and 400 mg doses (Citation102). TTR levels increased following cessation of ISIS-TTRRx dosing, with subjects monitored for up to 92 days after the first drug administration (Citation102).
ISIS-TTRRx has transitioned from phase I testing in healthy volunteers to patients with h-ATTR amyloidosis with polyneuropathy in an ongoing, randomized, double-blind, placebo-controlled, phase II/III trial to evaluate the efficacy and safety of prolonged treatment with ISIS-TTRRx (NCT01737398). Patients with stage I or II FAP will be randomized to receive ISIS-TTRRx 300 mg or placebo for 65 weeks (administered subcutaneously three times in the first week, on alternate days, and then once weekly for 64 weeks). Eligible patients have an NIS of 10–100 and require the aid of only one walking stick. Other key inclusion criteria relate to liver function (aspartate transaminase (AST) and alanine transaminase (ALT) levels < 1.9 × upper limit of normal (ULN), and bilirubin < 1.5 × ULN), renal function (eGFR ≥ 45 mL/min/1.73 m2), and platelet count (> 100 cells/mm3). The primary outcome measures are changes from baseline in mNIS + 7 and TQoL score at 65 weeks. An open-label extension study to assess the long-term safety and efficacy of this agent is also planned (NCT02175004). ASO-based TTR-lowering therapy is not currently in development for cardiac ATTR amyloidosis.
Patisiran (ALN-TTR02) and revusiran (ALN-TTRsc)
Patisiran and revusiran are investigational RNAi therapeutics in clinical development for the treatment of ATTR amyloidosis. The activity of these agents is based on delivery of a small interfering RNA (siRNA) that binds to a conserved sequence in the TTR mRNA common to both wt and all documented variants of TTR. Through engagement of the RNAi-induced silencing complex, the target mRNA is site-specifically degraded. Thus, synthesis of both the mutant and wt TTR protein is reduced, providing the potential to prevent amyloidosis and promote clearance of these deposits. In preclinical models, significant regression of TTR deposits in peripheral tissues was observed following RNAi-mediated TTR suppression, with regression correlated to serum TTR concentration (Citation127). The regression was superior to that achieved with the TTR stabilizer tafamidis, which demonstrated minimal effect (Citation127). The major differences between patisiran and revusiran relate to their route of administration and formulation for delivery of RNAi to the liver, which are discussed in more detail below.
Patisiran (ALN-TTR02). Patisiran consists of an siRNA encapsulated in a lipid nanoparticle (LNP). Following intravenous administration, the LNPs are opsonized by apolipoprotein E (ApoE) and are delivered to the liver through interaction with ApoE receptors expressed on hepatocytes (Citation103). Patisiran was generally well tolerated and demonstrated strong knockdown (up to 94% achieved) in a phase I study in healthy volunteers (Citation103). This high level of TTR knockdown is comparable to the reduction in mutant TTR achieved following liver transplantation. Mild-to-moderate infusion-related reactions were reported, but no significant changes in liver, renal, or hematologic assessments were observed.
Patisiran was assessed in 29 patients with stage I or II h-ATTR amyloidosis with polyneuropathy in a phase II open- label, multidose, dose-escalation study (Citation104). Patients with various TTR mutations were recruited, with the Val30Met mutation the most common (76% of patients). The maximum mean reduction in TTR level was 87% in patients who received patisiran 0.3 mg/kg every 3 weeks, with a maximum knockdown of 96% attained in one individual in this dose group. The majority of AEs reported were of mild or moderate intensity, and there were no clinically relevant changes in liver function tests, renal function, or hematologic parameters. Serious AEs were reported in two patients during the phase II study, with one patient discontinuing the trial after a severe episode of nausea/vomiting. Patients receiving TTR tetramer stabilizers were also eligible for this study, which demonstrated that co-administration of tafamidis or diflunisal did not compromise the knockdown efficiency of patisiran. Patients in the phase II trial were eligible to continue treatment in an open-label extension study, which is ongoing (NCT01961921) (Citation128). Initial analyses of the extension phase showed that patisiran treatment achieved a sustained mean serum TTR knockdown at the 80% level for approximately 16 months, with mean knockdown up to 88% attained between doses, and that extended patisiran treatment was generally well tolerated. Neuropathy impairment scores (e.g. NIS and mNIS + 7 scores) and cardiac measures (e.g. LV wall thickness and ejection fraction) were stable over 12 months of treatment in the extension study (Citation105). In fact, mean mNIS + 7 actually decreased by 2.5 points with 12 months’ patisiran treatment (Citation105), in contrast to the 12- to 18-point increases in this measure at 12 months estimated from previous studies of patients with h-ATTR amyloidosis with polyneuropathy and a similar baseline NIS (Citation23,Citation52,Citation129).
A randomized, double-blind, placebo-controlled, phase III trial of patisiran for h-ATTR amyloidosis with polyneuropathy (APOLLO; NCT01960348) has been initiated and is currently recruiting patients. Patients will be randomized (2:1) to 18 months’ treatment with patisiran 0.3 mg/kg or placebo every 3 weeks. Eligible patients will have an NIS of 5–130 and may have Val30Met or other TTR mutations. Patients must also have adequate liver function (AST and ALT ≤ 2.5 × ULN and total bilirubin within normal limits), adequate renal function (serum creatinine ≤ 1.5 × ULN), and a platelet count ≥ 50,000 cells/mm3. Prior use of diflunisal or tafamidis is also permitted. The primary end-point of the study is the change from baseline in mNIS + 7 at 18 months.
Revusiran (ALN-TTRsc). Revusiran is administered subcutaneously and consists of a TTR-targeting siRNA conjugated to a triantennary N-acetylgalactosamine (GalNAc) ligand. The GalNAc ligand binds to the asialoglycoprotein receptor expressed on hepatocytes, such that this agent targets hepatocytes (Citation130,Citation131). In non-human primate models, repeat dosing of revusiran reduced TTR protein expression by approximately 80% at doses as low as 2.5 mg/kg, although the agent was well tolerated up to 300 mg/kg (Citation130). In preclinical assessments of multiple doses in mice, a more potent and rapid knockdown of TTR was achieved with twice-weekly revusiran 10 mg/kg compared with twice-weekly TTR ASO 10 mg/kg, with a maximum knockdown of almost 100% observed in 3–10 days for revusiran versus 70% in 24 days following TTR ASO treatment (Citation127). Additionally, despite revusiran being present at 2–3-log lower levels in the liver and kidney compared with the TTR ASO at the same dose level, it achieved superior TTR knockdown, which may have positive implications for the long-term safety profile of this agent.
In a phase I study in healthy volunteers, the safety and tolerability of revusiran were assessed following a single dose and multiple dosing up to 5 weeks (Citation130). Multiple dosing up to 10 mg/kg was generally well tolerated, although transient, mild-to-moderate injection-site reactions were more frequent with revusiran treatment than placebo. The mean reduction in serum TTR levels with weekly dosing of revusiran 10 mg/kg was 92%, although up to 94% reduction of TTR was recorded in one subject in this dose cohort. The effects of revusiran in patients with ATTR amyloidosis with cardiomyopathy are under assessment in a phase II pilot study (NCT01981837) with an open-label extension phase, and in a phase III study in patients with h-ATTR amyloidosis with cardiomyopathy that was initiated at the end of 2014. Preliminary results for 26 patients in the phase II trial revealed that serum TTR levels were reduced by approximately 90% with multiple dosing of revusiran over 5 weeks (Citation106). The level of TTR knockdown was similar in patients with h-ATTR versus non-h-ATTR amyloidosis, and indeed comparable reductions in wt and variant TTR were observed in patients with the Val122Ile mutation. In accordance with the data from the phase I trial in healthy volunteers, revusiran was generally well tolerated in these patients, with mild injection-site reactions the most common treatment-emergent AEs. Transient mild elevations in liver function tests were reported in 15% of patients, but were resolved with continued dosing, and no changes in any other laboratory parameters were observed.
A pivotal, phase III, randomized, double-blind, placebo-controlled trial of revusiran in patients with h-ATTR amyloidosis with cardiomyopathy has recently been initiated (NCT02319005). In this study (ENDEAVOUR), patients with a TTR mutation and biopsy-confirmed amyloid deposition will be randomized 2:1 to receive subcutaneous revusiran 500 mg (once daily for 5 days then once weekly thereafter) or placebo for 18 months. The co-primary end-points are the change from baseline in 6-minute walk distance and the percentage reduction in TTR burden between placebo- and revusiran-treated patients at 18 months. All patients who complete this trial will be eligible to enroll in an extension study.
Monoclonal antibodies
Endogenous antibodies against TTR have been reported in patients with Val30Met-mutant ATTR amyloidosis. Higher antibody concentrations in patients with late-onset disease raise the possibility that an immune response to misfolded TTR protein may protect against amyloid formation (Citation132). Amyloid deposits can also be targeted by exogenous therapeutic antibodies. Serum amyloid P component (SAP), which is present as a non-fibrillar constituent of all amyloid deposits, is therefore an attractive target in all types of amyloidosis. In preclinical murine models, antibodies to SAP triggered the rapid clearance of extensive amyloid deposits through a complement-dependent macrophage-mediated mechanism (Citation133). A phase I study of an anti-SAP antibody (GSK2398852), given in combination with an agent designed to deplete SAP from the plasma (GSK2315698), has been initiated in patients with systemic amyloidosis (NCT01777243). GSK2315698 cross-links pairs of SAP molecules in the serum, prompting removal by the liver, so enabling the GSK2398852 antibody to target SAP within amyloid deposits. Promisingly, the GSK2315698 SAP depleter has also been administered as monotherapy to patients with various types of systemic amyloidosis for prolonged periods in an open-label study, and no accumulation of amyloid was identified in any patient receiving this treatment (Citation134). A phase I, dose-escalation, open-label study of NEOD001 (NCT01707264), a humanized monoclonal antibody that is claimed to target various elements in the amyloidogenesis pathway, is also ongoing in patients with AL amyloidosis.
Antibody-mediated clearance of amyloid deposits may also hold potential for patients with ATTR amyloidosis, and this approach could complement the other developmental treatments that have the potential to inhibit ongoing amyloid formation.
Combination therapy
Based on the promising data reported with individual agents, the additive, or synergistic potential of combination therapies is tantalizing. Although liver transplant and pharmacotherapies could be used in conjunction (e.g. drug therapy before or after transplantation), there is a paucity of data regarding patients who have received a transplant and other treatments. Prior liver transplant has been an exclusion criterion in recent clinical trials of some therapies (Citation52,Citation96), although small studies (2–3 patients) suggest that tafamidis and diflunisal are tolerated after transplantation (Citation135,Citation136). Analyses of pharmacotherapies in a liver transplantation setting are therefore required.
The combination of tafamidis or diflunisal with patisiran appears generally well tolerated in patients with h-ATTR amyloidosis with polyneuropathy, and knockdown of TTR is not compromised (Citation104). Reduction in TTR production in combination with TTR stabilization is therefore an enticing prospect. Another sequence of therapy could be reduction of the amyloid load with therapeutic monoclonal antibodies, accompanied or followed by inhibition of TTR synthesis with RNAi- or ASO-based agents. Any such combinations or treatment strategies will require extensive testing in clinical trials.
Summary
ATTR amyloidosis is a rare hereditary, multisystemic, multigenotypic disease that severely compromises patients’ QoL and is ultimately fatal. Current treatments are limited, and there is an unmet medical need for new treatments that can halt or reverse progression of amyloidosis and are effective across all genotypes and disease stages. The current treatment pathway for patients with ATTR amyloidosis with polyneuropathy is depicted in ; yet, considering the progressive nature of this disease, the schematic highlights the paucity of therapeutic options available for patients with advanced symptoms. Of the currently used treatments, liver transplantation is only recommended in patients with early-stage h-ATTR amyloidosis with polyneuropathy and does not improve neurologic symptoms. Data suggest that TTR-stabilizing drugs may reduce progression of polyneuropathy, although there are limited data to describe their effects in patients with ATTR amyloidosis with cardiomyopathy. Furthermore, tafamidis may be less effective in patients with late-stage disease, and has yet to definitively demonstrate efficacy in patients with non-Val30Met TTR mutations. Doxycycline and TUDCA combination treatment is currently in clinical development.
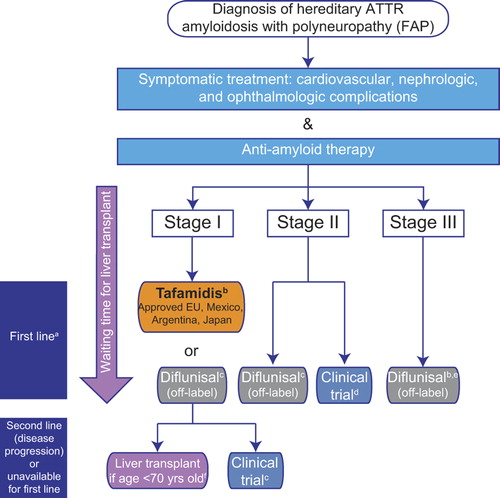
Figure 2. Current treatment pathway for patients with ATTR amyloidosis with polyneuropathy. Disease is classified in stages according to Coutinho et al. (Citation51) at initial diagnosis. a First-line anti-amyloid therapy is initiated according to stage of the disease and approval of the medicine in the country, in parallel with symptomatic treatment, to prevent disease progression and improve patient quality of life. Currently approved treatments may stabilize disease (liver transplantation (Citation53)), or slow progression of the disease (tafamidis, diflunisal (Citation52,Citation96)); treatment options are very limited for patients with stage II and III. The pathway described is followed irrespective of TTR gene mutation at stage I, and initiation of treatment for non-neurologic symptoms (renal, cardiac) may also need to be considered in affected patients. bMostly performed in patients with early-onset FAP with Val30Met mutation. Approval based on pivotal phase II/III study in patients with FAP stage I with Val30Met mutation (Citation96) and an open-label phase II study in patients with FAP stage I with non-Val30Met mutations (Citation97). cDiflunisal should be used with caution in patients with a history of gastrointestinal bleeding or ulceration, renal impairment, or heart failure. dPreliminary data from a phase II open-label extension study with patisiran, a RNAi investigational agent, demonstrate a mean 2.5-point decrease in mNIS + 7 score; this treatment has the potential to halt progression of neuropathy (Citation105). Of ongoing recruiting trials, none specify the acceptance of patients with late-stage (FAP stage > II) ATTR amyloidosis with polyneuropathy (clinicaltrials.gov, accessed on 15 May 2015). eThere are limited published data for diflunisal treatment of patients with FAP stage III (4 patients with PND stage IV received diflunisal in the Diflunisal Trial (Citation52)). fLiver transplant is proposed according to eligible criteria (health status, no evolutive cancer, compliance in the anti-rejection treatment). Best outcome recorded for early-onset (≤ 50 years of age) Val30Met patients. ATTR = transthyretin amyloidosis; FAP = familial amyloidosis with polyneuropathy; PND = polyneuropathy disability; TTR = transthyretin.
Other therapies have focused on preventing the production of the amyloidogenic TTR fibril precursor protein. ASO-based (ISIS-TTRRx) and RNAi-based (patisiran and revusiran) therapeutics have elicited potent, sustained knockdown of both mutant and wt TTR in clinical trials. This is particularly encouraging since substantial reduction in production of serum light chains using chemotherapy has been shown to significantly improve survival in AL amyloidosis (Citation13). In addition, the ability readily to measure serum TTR concentration in patients provides a simple measure of activity compared with other treatments, such as TTR stabilizers. In preclinical comparisons with other treatments, RNAi elicited superior regression of TTR protein deposits compared with tafamidis, and more rapid and potent TTR knockdown versus ASO-based treatment. The level of serum TTR was shown to correlate with TTR protein regression in preclinical models of h-ATTR with polyneuropathy (Citation127), highlighting the strong potential of this approach.
The evolution of targeted treatment options, potentially including combinations of TTR stabilizers with TTR-lowering agents and amyloid-clearing therapies, holds much promise for improving the outcomes of patients with this debilitating disease.
Acknowledgements
Editorial assistance was provided by Adelphi Communications (Bollington, UK).
Funding: Editorial assistance was funded by Alnylam Pharmaceuticals (Cambridge, MA, USA).
Declaration of interest: P.N.H. has received honoraria from and is participating in clinical trials sponsored by Alnylam, and is a director of Pentraxin Therapeutics Ltd, which owns intellectual property licensed to GlaxoSmithKline. A.D. is participating in clinical trials sponsored by Celgene, Millennium, Janssen, Alnylam, and Pfizer. D.A. has received honoraria for conferences and symposia from Pfizer, and for consultancy from Alnylam and ISIS. He has also received financial support to attend scientific meetings from Alnylam and Pfizer. O.B.S. is participating in clinical trials sponsored by Alnylam and Pfizer and has acted as a speaker at an educational meeting sponsored by Pfizer. Y.A. and A.G.-D. have no conflicts of interest to disclose.
References
- Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31–8.
- Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995; 268:1039–41.
- Pepys MB. Amyloidosis. Annu Rev Med. 2006;57:223–41.
- Westermark P, Sletten K, Johansson B, Cornwell GG. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87:2843–5.
- Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–23.
- Coelho T, Maurer MS, Suhr OB. THAOS - The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyreting amyloidosis. Curr Med Res Opin. 2013;29:63–76.
- Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33:1120–7.
- Benson MD, Teague SD, Kovacs R, Feigenbaum H, Jung J, Kincaid JC. Rate of progression of transthyretin amyloidosis. Am J Cardiol. 2011;108:285–9.
- Koike H, Tanaka F, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152–8.
- Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, et al. Prospective evaluation of the morbidity and mortality of wild-type and Val122Ile mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164:222–8.
- Stewart M, Lenderking W, Loftus J, Murphy B, Alvir J, Shaffer S, et al. Evaluating the quality of life and burden of illness in an ultra-rare disease in the U.S.: transthyretin familial amyloid polyneuropathy (TTR-FAP) patients and caregivers. IXth International Symposium on Familial Amyloidotic Polyneuropathy (ISFAP), Rio de Janeiro, Brazil; 10–13 November 2013 [Poster No. E-16].
- Stewart M, Loftus J, Lenderking W, Murphy B, Alvir J, Shaffer S, et al. Characterizing disease burden in an ultra-rare disease in the U.S.: transthyretin (TTR) amyloidosis patients and caregivers. ISPOR 16th Annual European Congress, Dublin, Ireland; 4–6 November 2013 [Poster No. PSY49].
- Lachmann HJ, Gallimore R, Gillmore JD, Carr-Smith HD, Bradwell AR, Pepys MB, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122:78–84.
- Adams D, Lozeron P, Lacroix C. Amyloid neuropathies. Curr Opin Neurol. 2012;25:564–72.
- Dohrn MF, Rocken C, De Bleecker JL, Martin JJ, Vorgerd M, Van den Bergh PY, et al. Diagnostic hallmarks and pitfalls in late-onset progressive transthyretin-related amyloid-neuropathy. J Neurol. 2013;260: 3093–108.
- Planté-Bordeneuve V, Ferreira A, Lalu T, Zaros C, Lacroix C, Adams D, et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology. 2007;69:693–8.
- Hamour IM, Lachmann HJ, Goodman HJ, Petrou M, Burke MM, Hawkins PN, et al. Heart transplantation for homozygous familial transthyretin (TTR) Val122Ile cardiac amyloidosis. Am J Transplant. 2008;8:1056–9.
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765–74.
- Ikeda S, Sekijima Y, Tojo K, Koyama J. Diagnostic value of abdominal wall fat pad biopsy in senile systemic amyloidosis. Amyloid. 2011;18:211–15.
- van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54:2015–21.
- Do AB, Coelho T, Sousa A, Guimaraes A. Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid. 2009;16:232–8.
- Fine NM, Arruda-Olson AM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kyle RA, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol. 2014;113:1723–7.
- Adams D, Coelho T, Obici L, Merlini G, Mincheva Z, Suanprasert N, et al. Rapid progression of familial amyloid polyneuropathy: a multinational natural history study. Neurology. 2015;85:675–82.
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol. 2011;4:47–54.
- Ihse E, Ybo A, Suhr O, Lindqvist P, Backman C, Westermark P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin Val30Met amyloidosis. J Pathol. 2008;216:253–61.
- Aljaroudi WA, Desai MY, Tang WH, Phelan D, Cerqueira MD, Jaber WA. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: state of the art review and focus on emerging nuclear techniques. J Nucl Cardiol. 2014;21:271–83.
- Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20: 142–50.
- Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther. 2014;39:225–33.
- Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336: 466–73.
- Olsson M, Jonasson J, Cederquist K, Suhr OB. Frequency of the transthyretin Val30Met mutation in the northern Swedish population. Amyloid. 2014;21:18–20.
- Carvalho F, Sousa M, Fernandes S, Silva J, Saraiva MJ, Barros A. Preimplantation genetic diagnosis for familial amyloidotic polyneuropathy (FAP). Prenat Diagn. 2001;21:1093–9.
- Morris M, Nicholes W, Benson M. Prenatal diagnosis of hereditary amyloidosis in a Portugese family. Am J Med Genet. 1991;39:123–4.
- Hellman U, Alarcon F, Lundgren HE, Suhr OB, Bonaiti-Pellié C, Planté-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15:181–6.
- Misu K, Hattori N, Nagamatsu M, Ikeda S, Ando Y, Nakazato M, et al. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features. Brain. 1999;122(Pt 10):1951–62.
- Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97.
- Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235:479–85.
- Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–12.
- Lindqvist P, Olofsson BO, Backman C, Suhr O, Waldenstrom A. Pulsed tissue Doppler and strain imaging discloses early signs of infiltrative cardiac disease: a study on patients with familial amyloidotic polyneuropathy. Eur J Echocardiogr. 2006;7:22–30.
- Dungu J, Sattianayagam PT, Whelan CJ, Gibbs SD, Pinney JH, Banypersad SM, et al. The electrocardiographic features associated with cardiac amyloidosis of variant transthyretin isoleucine 122 type in Afro-Caribbean patients. Am Heart J. 2012;164:72–9.
- Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7:157–65.
- Rapezzi C, Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1, 2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging. 2011;38:470–8.
- Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46:1076–84.
- Hutt DF, Quigley AM, Page J, Hall ML, Burniston M, Gopaul D, et al. Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging. 2014;15:1289–98.
- Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6:195–201.
- Kato-Motozaki Y, Ono K, Shima K, Morinaga A, Machiya T, Nozaki I, et al. Epidemiology of familial amyloid polyneuropathy in Japan: identification of a novel endemic focus. J Neurol Sci. 2008;270:133–40.
- Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62:1057–62.
- Suanprasert N, Berk JL, Benson MD, Dyck PJ, Klein CJ, Gollob JA, et al. Retrospective study of a TTR FAP cohort to modify NIS + 7 for therapeutic trials. J Neurol Sci. 2014;344:121–8.
- Riboldi G, Del Bo R, Ranieri M, Magri F, Sciacco M, Moggio M, et al. Tyr78Phe transthyretin mutation with predominant motor neuropathy as the initial presentation. Case Rep Neurol. 2011;3:62–8.
- Hornsten R, Pennlert J, Wiklund U, Lindqvist P, Jensen SM, Suhr OB. Heart complications in familial transthyretin amyloidosis: impact of age and gender. Amyloid. 2010;17:63–8.
- Koike H, Misu K, Ikeda S, Ando Y, Nakazato M, Ando E, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59:1771–6.
- Coutinho P, Martins da Silva A, Lopes Lima J, Resende Barbosa A. Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: Glenner GG, Pinho e Costa P, Falcao de Freitas A, editors. Amyloid and amyloidosis. Amsterdam: Excerpta Medica; 1980. p. 88–93.
- Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–67.
- Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Rodrigues Pena J, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–54.
- Ramalho A, Cortez-Dias N, Guimaraes T, Francisco AG, Nobre Menezes M, Lima Silva G, et al. Prognostic analysis of heart rate variability assessment in familial amyloid polyneuropathy. Eur Heart J. 2014;35(Suppl):933 [Abstract P5268].
- Ramalho A, Cortez-Dias N, Lima Silva G, Placido R, Guimaraes T, Francisco AG, et al. Progression of cardiomyopathy in familial amyloid polyneuropathy – impact on prognosis. Eur Heart J. 2014;35(Suppl.):1058 [Abstract P5971].
- Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40:232–9.
- Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2:113–22.
- Dharmarajan K, Salomon S, Helmke S, Tereyua S, Rapezzi C, Falk R, et al. Genotype and phenotypic characteristics of persons with cardiac amyloidosis from the Multinational Transthyretin Amyloidosis Survey (THAOS) registry. J Cardiac Fail. 2014;117(Suppl):S69 [Abstract 221].
- Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126:1286–300.
- Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild-type and Val122Ile transthyretin in older adults referred to an academic medical center. Aging Health. 2013;9: 229–35.
- Damy T, Jaccard A, Guellich A, Lavergne D, Rappeneau S, Deux JF, et al. Comparison and identification of early clinical, biological and echocardiographic prognostic markers in transthyretin and AL cardiac amyloidosis. Eur Heart J. 2014;35(Suppl):668 [Abstract P3655].
- Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6:129–39.
- Algalarrondo V, Dinanian S, Juin C, Chemla D, Bennani SL, Sebag C, et al. Prophylactic pacemaker implantation in familial amyloid polyneuropathy. Heart Rhythm. 2012;9:1069–75.
- Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63:1285–8.
- Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55:1645.
- Piccirillo G, Nocco M, Lionetti M, Moise A, Naso C, Marigliano V, et al. Effects of sildenafil citrate (viagra) on cardiac repolarization and on autonomic control in subjects with chronic heart failure. Am Heart J. 2002;143:703–10.
- Adams D, Lozeron P, Theaudin M, Mincheva Z, Cauquil C, Adam C, et al. Regional difference and similarity of familial amyloidosis with polyneuropathy in France. Amyloid. 2012;19(Suppl 1):61–4.
- Adams D, Samuel D, Goulon-Goeau C, Nakazato M, Costa PM, Feray C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7): 1495–504.
- Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40:242–6.
- Suhr OB, Ando Y, Holmgren G, Wikström L, Friman S, Herlenius G, et al. Liver transplantation in familial amyloidotic polyneuropathy (FAP). A comparative study of transplanted and non-transplanted patient’s survival. Transpl Int. 1998;11(Suppl 1):S160–3.
- Yamashita T, Ando Y, Okamoto S, Misumi Y, Hirahara T, Ueda M, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637–43.
- Yamamoto S, Wilczek HE, Nowak G, Larsson M, Oksanen A, Iwata T, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant. 2007;7:2597–604.
- Tsuchiya A, Yazaki M, Kametani F, Takei Y, Ikeda S. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl. 2008;14:563–70.
- Rydh A, Suhr O, Hietala SO, Ahlstrom KR, Pepys MB, Hawkins PN. Serum amyloid P component scintigraphy in familial amyloid polyneuropathy: regression of visceral amyloid following liver transplantation. Eur J Nucl Med. 1998;25:709–13.
- Wilczek HE, Larsson M, Ericzon BG; FAPWTR. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid. 2011;18(Suppl 1):193–5.
- Herlenius G, Wilczek HE, Larsson M, Ericzon BG; Familial Amyloidotic Polyneuropathy World Transplant Registry. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 2004;77:64–71.
- Shimojima Y, Morita H, Kobayashi S, Takei Y, Ikeda S. Ten-year follow-up of peripheral nerve function in patients with familial amyloid polyneuropathy after liver transplantation. J Neurol. 2008; 255:1220–5.
- Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75:324–7.
- Ihse E, Suhr OB, Hellman U, Westermark P. Variation in amount of wild-type transthyretin in different fibril and tissue types in ATTR amyloidosis. J Mol Med (Berl). 2011;89:171–80.
- Olofsson BO, Backman C, Karp K, Suhr OB. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002; 73:745–51.
- Yazaki M, Tokuda T, Nakamura A, Higashikata T, Koyama J, Higuchi K, et al. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun. 2000;274:702–6.
- Stangou AJ, Hawkins PN, Heaton ND, Rela M, Monaghan M, Nihoyannopoulos P, et al. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation. 1998;66:229–33.
- Gustafsson S, Ihse E, Henein MY, Westermark P, Lindqvist P, Suhr OB. Amyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosis. Transplantation. 2012;93:1017–23.
- Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85.
- Sá Couto P, Barros F, Esteves S, Aragão I. Comparison of outcome data after liver transplant: familial amyloid polyneuropathy (Portuguese type) recipients versus classical indications for liver transplant. Eur J Anaesthesiol. 2010;27:13 [Abstract 1AP3-6].
- Barreiros AP, Post F, Hoppe-Lotichius M, Linke RP, Vahl CF, Schafers HJ, et al. Liver transplantation and combined liver-heart transplantation in patients with familial amyloid polyneuropathy: a single-center experience. Liver Transpl. 2010;16:314–23.
- Takei Y, Ikeda S, Ikegami T, Hashikura Y, Miyagawa S, Ando Y, et al. Ten years of experience with liver transplantation for familial amyloid polyneuropathy in Japan: outcomes of living donor liver transplantations. Intern Med. 2005;44:1151–6.
- Bispo M, Marcelino P, Freire A, Martins A, Mourao L, Barroso E. High incidence of thrombotic complications early after liver transplantation for familial amyloidotic polyneuropathy. Transpl Int. 2009; 22:165–71.
- Nowak G, Suhr OB, Wikstrom L, Wilczek H, Ericzon BG. The long-term impact of liver transplantation on kidney function in familial amyloidotic polyneuropathy patients. Transpl Int. 2005;18:111–15.
- Jain A, Nalesnik M, Reyes J, Pokharna R, Mazariegos G, Green M, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg. 2002;236:429–36.
- Rapezzi C, Perugini E, Salvi F, Grigioni F, Riva L, Cooke RM, et al. Phenotypic and genotypic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid. 2006;13:143–53.
- Nelson LM, Penninga L, Sander K, Hansen PB, Villadsen GE, Rasmussen A, et al. Long-term outcome in patients treated with combined heart and liver transplantation for familial amyloidotic cardiomyopathy. Clin Transplant. 2013;27:203–9.
- Raichlin E, Daly RC, Rosen CB, McGregor CG, Charlton MR, Frantz RP, et al. Combined heart and liver transplantation: a single-center experience. Transplantation. 2009;88:219–25.
- Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23:1142–53.
- Rappley I, Monteiro C, Novais M, Baranczak A, Solis G, Wiseman RL, et al. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry. 2014;53:1993–2006.
- Coelho T, Maia LF, Martins da Silva A, Waddington CM, Planté-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–92.
- Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6:1011–20.
- Maurer MS, Judge DP, Rosas GR, Mandel FS, Aarts J. Interim analysis of long-term, open-label tafamidis treatment in transthyretin amyloid cardiomyopathy after up to 5 years of treatment. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Presentation No. OP-66].
- Lozeron P, Théaudin M, Mincheva Z, Ducot B, Lacroix C, Adams D, et al. Effect on disability and safety of tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2013; 20:1539–45.
- Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18:315–19.
- Obici L, Cortese A, Lozza A, Palladini G, Perlini S, Gobbi M, et al. A phase II study of doxycycline plus tauroursodeoxycholic acid in transthyretin amyloidois. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP–68].
- Crooke ST. ISIS Pharmaceuticals. J P Morgan Healthcare Conference. Available at: http://www.google.co.uk/url?sa =trct = j&q = &esrc = s&source = web&cd = 3&cad = rja&uact = 8&sqi = 2&ved = 0CD8QFjAC&url = http%3A%2F%2Fwww.buenavistainv.com%2Fdbimages%2FJP%2520Morgan%2520Healthcare%2520%2520Presentation%25201-2013.pdf&ei = TLEqVIK5BoTB7gb87oCYCw&usg = AFQjCNFZE5NPGhbaAN8RwLwZ0K9kbziyZQ&bvm = bv.76477589 (accessed 16 January 2015).
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819–29.
- Gollob J. Summary data from Phase II ALN-TTR02 RNAi treatment of ATTR. IXth International Symposium on Familial Amyloidotic Polyneuropathy (IFSAP). Rio de Janeiro, Brazil; 10–13 November 2013.
- Coelho T, Suhr O, Conceicao I, Wadington-Cruz M, Schmidt H, Buades J, et al. Phase 2 open-label extension study of patisiran, an investigational RNAi therapeutic for the treatment of familial amyloid polyneuropathy. 67th Annual Meeting of the American Academy of Neurology (AAN), Washington, DC, USA; 18–25 April 2015 [Abstract S9.003].
- Gillmore J, Falk R, Maurer M, Hanna M. Interim results from pilot phase 2 trial of revusiran (ALN-TTRsc). American Heart Association Annual Meeting Scientific Sessions. Chicago, IL, USA; 15–19 November 2014.
- Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260: 2802–14.
- Coelho T, Silva AM, Valdrez K, Cardoso M, Monteiro C, Ferreira H, et al. Familial amyloid polyneuropathy treatment with tafamidis – evaluation of one year treatment at Porto, Portugal. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP-67].
- Merlini G, Waddington CM, Stewart M, Rosas GR, Huertas PE, Mandel FS, et al. Survival in patients with transthyretin familial amyloid polyneuropathy receiving tafamidis treatment. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April– 1 May 2014 [Abstract OP-65].
- US Food and Drug Administration. Vyndaqel (tafamidis meglumine) – Proposed indication: treatment of transthyretin (TTR) familial amyloid polyneuropathy. Available at: http://www.fda.gov/downloads/AdvisorycommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM304830.pdf (accessed 16 January 2015).
- Rocha A, Silva AM, Cardoso M, Monteiro C, Beirão I, Alves C, et al. Treatment of ATTR Val30Met with tafamidis: successful decrease of proteinuria. XIVth International Symposium on Amyloidosis (ISA). Indianapolis, IN, USA; 27 April–1 May 2014.
- National Amyloidosis Centre. ATTR Amyloidosis. Available at: http://www.amyloidosis.org.uk/introduction-to-attr-amyloidosis/ (accessed 16 January 2015).
- Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–52.
- Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–49.
- Sekijima Y, Tojo K, Morita H, Koyama J, Yazaki M, Ikeda S. Safety and efficacy of long-term diflunisal administration in familial amyloid polyneuropathy – Summary of ten years therapeutic experience. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP-70].
- Hernández-Díaz S, Rodríguez LA. Association between nonsteroidal anti-inflammatory drugs and upper gastrointestinal tract bleeding/perforation: an overview of epidemiologic studies published in the1990s. Arch Intern Med.2000;160:2093–9.
- Singh BK, Haque SE, Pillai KK. Assessment of nonsteroidal anti- inflammatory drug-induced cardiotoxicity. Expert Opin Drug Metab Toxicol. 2014;10:143–56.
- Reddy KS, Roy A. Cardiovascular risk of NSAIDs: time to translate knowledge into practice. PLoS Med. 2013;10:e1001389.
- Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol. 2005;3:55–9.
- Quarta CC, Falk RH, Soloman SD, Suhr OB, Obici L, Perlini S, et al. The prevalence of cardiac amyloidosis in familial amyloidotic polyneuropathy with predominant neuropathy: The Diflunisal Trial. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP-69].
- Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74–84.
- Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN. Amyloid load and clinical outcome in amyloid A amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet. 2001;358:24–9.
- Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica. 2014;99:209–21.
- Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1:1331–41.
- Benson MD, Kluve-Beckerman B, Zeldenrust SR, Siesky AM, Bodenmiller DM, Showalter AD, et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006;33:609–18.
- Ackermann EJ, Guo S, Booten S, Benson M, Hughes S, Monia BP. Clinical development of an antisense therapy for the treatment of hereditary transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis (ISA), Groningen, The Netherlands; 6–10 May 2012 [Abstract OP 73].
- Butler JS, Chan A, Costelha S, Fishman S, Milstein S, Willoughby JS, et al. Preclinical evaluation of RNAi therapeutics for the treatment of ATTR: an update. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract PC-9].
- Suhr O, Adams D, Coelho T, Conceicao I, Waddington CM, Schmidt H, et al. Further analysis of phase II trial of patisiran, a novel RNAi therapeutic for the treatment of familial amyloidotic polyneuropathy. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP-72].
- European Medicines Agency. Tafamidis EMA assessment report. Available at: http://wwwemaeuropaeu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002294/WC500117838 pdf (accessed 16 January 2015).
- Zimmermann T, Karsten V, Harrop J, Chan A, Chiesa J, Peters G, et al. Phase I first-in-human trial of ALN-TTRsc, a novel RNA interference therapeutic for the treatment of familial amyloidotic cardiomyopathy (FAC). Heart Failure Society of America 17th Annual Meeting; Orlando, FL, USA; 22–25 September 2013 [Poster].
- Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136:16958–61.
- Obayashi K, Tasaki M, Jono H, Ueda M, Shinriki S, Misumi Y, et al. Impact of antibodies against amyloidogenic transthyretin (ATTR) on phenotypes of patients with familial amyloidotic polyneuropathy (FAP) ATTR Valine30Methionine. Clin Chim Acta. 2013;419:127–31.
- Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–7.
- Gillmore JD, Tennent GA, Hutchinson WL, Gallimore JR, Lachmann HJ, Goodman HJ, et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br J Haematol. 2010;148:760–7.
- Planté-Bordeneuve V, Guendouz S, Damy T, Hulin A. Use of tafamidis in 3 transplanted amyloidosis ATTR-FAP patients: safety and association with immunosuppressive drugs. IXth Congress of Physiology, Pharmacology and Therapeutics, Poitiers, France; 22–24 April 2014 [Abstract PM1-043].
- Yoshinaga T, Yazaki M, Sekijima Y, Kametani F, Ikeda S. Regression of gastroduodenal mucosal amyloid deposits in FAP patients after combined therapy with oral intake of diflunisal followed by liver transplantation. XIVth International Symposium on Amyloidosis (ISA), Indianapolis, IN, USA; 27 April–1 May 2014 [Abstract OP-77].