
Abstract
Selected inflammatory conditions increase the risk of cancer. An inflammatory component is present also in the micro-environment of tumours epidemiologically unrelated to inflammation. An intrinsic (driven by genetic events that cause neoplasia) and an extrinsic (driven by inflammatory conditions which predispose to cancer) pathway link inflammation and cancer. Smouldering inflammation in the tumour microenvironment contributes to proliferation and survival of malignant cells, angiogenesis, metastasis, subversion of adaptive immunity, response to hormones, and chemotherapeutic agents. Emerging evidence also suggests that cancer-related inflammation promotes genetic instability. Thus, cancer-related inflammation represents a target for innovative diagnostic and therapeutic strategies.
Key words::
Key messages
Inflammation is a key component of the tumour microenvironment.
Cancer-related inflammation can promote tumour progression and is a target for therapeutic intervention
| Abbreviations | ||
| AID | = | activation-induced cytidine deaminase |
| bFGF basic | = | fibroblast growth factor |
| COX2 | = | cyclo-oxygenase 2 |
| CRI | = | cancer-related inflammation |
| DC | = | dendritic cell |
| DcR3 | = | decoy receptor 3 |
| DEN | = | diethyl nitrosamine |
| DSS | = | dextran sulphate sodium salt |
| EGFR | = | epidermal growth factor receptor |
| HCC | = | hepatocellular carcinoma |
| HIF | = | hypoxia-inducible factor |
| IKKβ | = | IkappaB-kinase β |
| IL | = | interleukin |
| IL-1R1 | = | IL-1 receptor, type 1 |
| IL-1ra | = | IL-1 receptor antagonist |
| M2 | = | alternatively activated macrophages |
| MAPK | = | mitogen-activated protein kinase |
| MDSC | = | myeloid-derived suppressor cells |
| MMP | = | matrix metalloproteinase |
| NFκkB | = | nuclear factor kappa-B |
| NO | = | nitric oxide |
| NSCLC | = | non-small cell lung cancer |
| PlDGF | = | placenta-derived growth factor |
| PMN | = | polymorphonuclear cells |
| PTC | = | papillary carcinoma of the thyroid |
| PTEN | = | phosphatase and tensin homologue |
| RANKL | = | receptor activator of NFκB ligand |
| SARM | = | selective androgen receptor modulator |
| SIGIRR | = | single immunoglobulin interleukin 1 receptor-related protein |
| Stat3 | = | signal transducer activator of transcription-3 |
| TAM | = | tumour-associated macrophages |
| TGFβ | = | transforming growth factor β |
| TIR | = | toll-interleukin 1 receptor |
| TIR8 | = | toll-interleukin 1 receptor 8 |
| TLR | = | toll-like receptor |
| TNFα | = | tumour necrosis factor a |
| VEGF | = | vascular endothelial growth factor |
| VEGFR1 | = | vascular endothelial growth factor receptor 1 |
Introduction
Epidemiological studies have revealed that chronic inflammation predisposes to different forms of cancer. The forms of chronic inflammation which increase cancer risk include microbial infections, autoimmune diseases (e.g. inflammatory bowel disease for colon cancer), and inflammatory conditions of uncertain origin (e.g. prostatitis for prostate cancer). Non-steroidal anti-inflammatory agents are associated with protection against various tumours, a finding to a large extent mirroring the finding of inflammation as a risk factor for certain cancers. An inflammatory component is present in the microenvironment of most neoplastic tissues, including those not causally related to an obvious inflammatory process. Key features of cancer-related inflammation include leukocyte infiltration, prominent tumour-associated macrophages (TAM); the presence of cytokines such as tumour necrosis factor (TNF) or interleukin 1 (IL-1), and chemokines such as CCL2; and the occurrence of angiogenesis and tissue remodelling.
Molecular and cellular pathways linking inflammation and cancer have been identified (Citation1). Two pathways link inflammation and cancer (). In the intrinsic pathway, activation of different classes of oncogenes orchestrates the construction of an inflammatory microenvironment. In the extrinsic pathway inflammatory conditions promote cancer development (e.g. colitis-associated cancer of the intestine). Key orchestrators at the intersection of the intrinsic and extrinsic pathway include transcription factors (e.g. nuclear factor kappa-B (NFκB)) (Citation2), cytokines (e.g. TNF), and chemokines. Thus, cancer-related inflammation (CRI) is a key component of the tumour microenvironment and may represent the seventh hall-mark of cancer (Citation3). Here we will review selected pathways linking inflammation and cancer in the context of the tumour microenvironment and emphasize targets for therapeutic intervention.
Figure 1. Pathways linking inflammation and cancer.
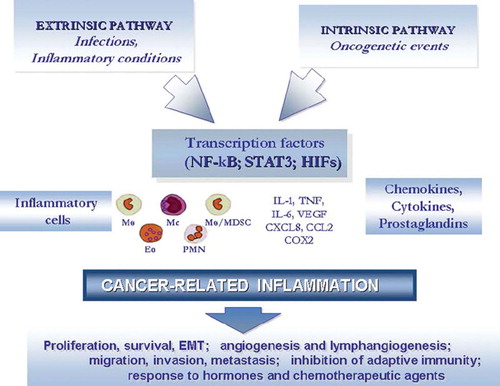
An intrinsic and an extrinsic pathway link inflammation and oncogenes
The occurrence of an inflammatory component in the microenvironment of tumours which are not epidemiologically related to inflammation raised the question whether genetic events causing neoplasia are responsible for the construction of an inflammatory milieu. To address this question in a clinically relevant setting, advantage was taken of papillary carcinoma of the thyroid (PTC), a tumour characterized by the presence of chemokine-guided macrophage and dendritic cell infiltration (Citation4–6). Rearrangements of tyrosine kinase genes are involved in the pathogenesis of this tumour. In particular, the RET/PTC rearrangement represents a frequent, early, causative, and sufficient genetic event in the pathogenesis of PTC. In an appropriate cellular context provided by primary human thyrocytes, RET/PTC activates a genetic programme related to inflammation (Citation4) (Figure 2). In particular, the RET/PTC-activated transcriptome profile includes: colony-stimulating factors (CSFs), which promote leukocyte recruitment and survival; interleukin 1 (IL-1), a primary inflammatory cytokine; cyclo-oxygenase 2 (COX2), frequently expressed in cancer; chemokines attracting monocytes and dendritic cells (CCL2, CCL20); angiogenic chemokines; co-ordinate induction and inhibition of matrix-degrading enzymes and inhibitors; and L-selectin and expression of the chemokine receptor CXCR4 on the initiated cells (Citation7). Key elements of the RET/PTC-activated inflammatory programme were found in biopsy specimens, and patients with lymph node metastasis showed higher levels of the inflammatory molecules in their primary tumours (Citation4,Citation7,Citation8). These results show that an early, causative, and sufficient genetic event (RET/PTC) involved in the pathogenesis of a human tumour directly promotes the build-up of an inflammatory microenvironment to its direct advantage (Citation4).
Members of the epidermal growth factor receptor (EGFR) family have tyrosine kinase activity and are frequently involved in human cancer. EGFR activation in glioma induces COX2 expression via p38-mitogen-activated protein kinase (MAPK) activation of Sp1/Sp3 (Citation9); COX2 is an independent prognostic factor in glioma.
Activated oncogenic components of the ras-raf pathway also induce expression and production of inflammatory mediators. In HeLa cells, ras induces the production of IL-8 (or CXCL8) (Citation10,Citation11), a chemokine which promotes angiogenesis and tumour progression. Adult mice are resistant to K-ras pancreatic carcinogenesis. However, mild chronic pancreatitis, possibly mirroring clinical epidemiology, acts in concert with K-ras mutation to induce pancreatic intra-epithelial neoplasia and invasive ductal carcinoma (Citation12). Along the same lines Braf, frequently activated in malignant melanoma, induces cytokines which contribute to a pro-tumour milieu (Citation13).
Myc is a transcription factor over-expressed in many human tumours—its activation initiates and maintains key aspects of the tumour phenotype. In addition to promoting cell autonomous proliferation, myc instructs remodelling of the extracellular microenvironment with inflammatory cells and mediators playing key roles. In a pancreatic islet tumour system, a first wave of myc-driven angiogenesis is induced by IL-1. The myc-activated genetic programme also includes several CC chemokines which recruit mast cells. Mast cells have long been known to drive angiogenesis and here, after IL-1, they sustain new vessel formation and tumour growth (Citation14).
Tumour suppressor genes have the capacity to regulate the production of inflammatory mediators. The chemokine receptor CXCR4, which is frequently expressed on malignant cells and implicated in cell survival and metastasis, lies downstream of the von Hippel Lindau/hypoxia-inducible factor (HIF) axis, as does the inflammatory cytokine tumour necrosis factor alpha (TNFα) (reviewed in (Citation15)). In non-small cell lung cancer (NSCLC) mutation of the tumour suppressor gene phosphatase and tensin homologue (PTEN) results in up-regulation of HIF-1 activity and in HIF-1-dependent transcription of the CXCR4 gene which promotes metastasis formation. Alpha catenin is more than a tumour suppressor sequestering beta-catenin. Its ablation results in NFκB activation, induction of genes involved in inflammation, cell proliferation, wound-healing, and—ultimately—squamous cell carcinoma (Citation16). The putative tumour suppressor semaphorin 3B triggers an IL-8-mediated pro-metastatic programme (Citation17). P53 has been connected to CXCR4 ligand expression (Citation18). In prostate cancer, the inflammatory cytokines TNF and IL-1 beta induce phosphorylation, ubiquitination, and degradation of the tumour suppressor NKX3.1 (Citation19).
Transforming growth factor beta (TGFβ) is a tumour suppressor frequently involved in human tumours and associated with metastasis, though it can also act under certain conditions as a tumour promoter. In a mammary carcinoma model, deficiency in the type IITGFβ receptor is associated with metastasis and recruitment of myeloid-derived suppressor cells (MDSC), defined as Gr1+Mac1+ (Citation20). The chemokines CXCL5 and CXCL12, acting on the chemokine receptors CXCR2 and CXCR4, respectively, mediate MDSC recruitment. MDSC facilitate metastasis via metalloproteinase activity. Interestingly, chemokine-mediated recruitment in TGFβRII-deficient tumours was associated with increased levels of TGFβ1.
Thus, oncogenes representative of different molecular classes and modes of action (tyrosine kinases, ras-raf, nuclear oncogenes, tumour suppressors) share the capacity to orchestrate pro-inflammatory programmes (). While these may share common elements (e.g. a link to angiogenesis, recruitment of cells of myelo-monocytic origin), the diversity of inflammatory components calls for detailed analysis of essential, common versus tumour/tissue-specific inflammatory players. In the extrinsic pathway, it remains uncertain whether chronic inflammation per se is sufficient for carcinogenesis (Citation1). Reactive oxygen and nitrogen intermediates are obvious inflammation-generated candidate mediators for DNA damage, and evidence obtained in vitro and in vivo is consistent with this view (Citation1). In addition, aberrant expression of activation-induced cytidine deaminase (AID) is induced in gastric epithelium by Helicobacter pylori (Citation21) and by TNF in bile-duct cells (Citation22). Indeed cholangiocarcinoma is associated with liver fluke-induced inflammation. AID is an initiator of somatic hypermutation in B cells and may link chronic inflammation to DNA damage in these tumours.
Masters and commanders: transcription factors orchestrate CRI
In the panoply of molecular players involved in CRI one can identify prime movers (endogenous promoters). These include transcription factors such as NFκB and signal transducer activator of transcription-3 (Stat3), and primary inflammatory cytokines such as IL-1β, IL-6, IL-23, and TNFα (Citation23–26).
NFκB is a key orchestrator of innate immunity and inflammation and has emerged as a major endogenous tumour promoter (Citation23). NFκB is key both in the context of tumour or would-be tumour cells and in the context of inflammatory cells. In both cellular contexts, NFκB can lie downstream of the sensing of microbes or tissue damage by the toll-like receptor (TLR)-MyD88 pathway, the inflammatory cytokines TNFα and IL-1β, and downstream of tissue damage that results in release of alarm signals. In addition, NFκB activation can be the result of cell-autonomous genetic alterations (amplification, mutations, or deletions) in cancer cells.
In both tumour cells and epithelial cells at risk of transformation by carcinogens, as well as in inflammatory cells, NFκB activates the expression of inflammatory cytokines, adhesion molecules, key enzymes in the prostaglandin synthase pathway (COX2), nitric oxide (NO) synthase, and angiogenic factors. NFκB also promotes cell survival by inducing anti-apoptotic genes (e.g. Bcl2), a major function in tumour cells or cells targeted by carcinogenic agents. Accumulating evidence suggests that intersections and compensatory pathways may exist between the NFκB and HIF-1 systems (Citation2,Citation27), linking innate immunity to the hypoxic response.
Strong genetic evidence, including tissue-specific gene targeting of components of the Ikk complex, such as IkappaB-kinase beta (IKKβ), have provided unequivocal evidence for a role of NFκB in tumour promotion in prototypic tissues of CRI (e.g. gastrointestinal tract and liver) (Citation28). Gene targeting in liver epithelial cells can have divergent effects in different models of carcinogenesis possibly depending on the balance between promotion of initiated cell apoptosis and triggering of compensatory cell proliferation (Citation28).
The NFκB pathway is tightly controlled by inhibitors acting at different levels. Toll-interleukin receptor 8 (TIR8), also known as single immunoglobulin interleukin 1 receptor-related protein (SIGIRR), is a fringe member of the IL-1 receptor family with a single Ig domain, a long cytoplasmic tail, and an altered TIR domain. It inhibits TLR and IL-1R signalling and is highly expressed in intestinal mucosa. TIR8 gene deficiency is associated with increased susceptibility to intestinal inflammation and carcinogenesis (Citation29,Citation30) as well as to B cell lymphoproliferation and autoimmunity (Citation31). Thus, balancing inhibitors and activators tunes the action of the NFκB pathway as an endogenous tumour promoter.
NFκB can act in infiltrating leukocytes as well as in cells targeted by carcinogens. Myeloid-specific gene inactivation of IKKβ inhibited CRI and colitis-associated cancer thus providing an unequivocal genetic evidence for the role of inflammatory cells in carcinogenesis in this tissue. In established advanced tumours, where inflammation is typically smouldering (Citation15), tumour-associated macrophages (TAM) have defective and delayed NFκB activation (Citation32). Evidence suggests that p50 homodimer-negative regulator is responsible for the sluggish NFκB activation in TAMs and for their pro-tumour phenotype. Thus, NFκB appears to act as a rheostat tuned at different levels in florid inflammatory conditions predisposing to cancer (e.g. inflammatory bowel disease (IBD)) and in TAMs sustaining the smouldering inflammatory milieu of established metastatic neoplasia. NFκB has also been recently involved in driving the M2 (alternatively activated macrophages) polarization of TAMs (Citation33).
Along with NFκB, Stat3 is a point of convergence for numerous oncogenic signalling pathways (Citation24). This transcription factor is constitutively activated both in tumour cells and in immune cells and plays a role both in oncogenesis and inhibition of apoptosis. Stat3 activation in cancer also enhances tumour immune evasion by inhibiting dendritic cell (DC) maturation and promoting immunosuppression (Citation34).
Inflammatory cytokines in CRI
Studies on TNF demonstrating enhancement of cancer cell invasion ability provided early proof for a pro-tumour function of inflammatory cytokines. In addition, the finding of protection of TNF-deficient mice against skin carcinogenesis offered genetic evidence linking inflammation and cancer (Citation35,Citation36).
TNF-mediated tumour promotion can involve different pathways (Citation37): a direct effect on tumour cells of low concentration of this cytokine, an interplay with the chemokine system with induction of CXCR4, and stimulation of epithelial to mesenchymal transition (Citation38). TAMs promote wnt signalling through TNF in gastric cancer (Citation39). These findings provide a rationale for the development of clinical protocols employing TNF antagonists in cancer therapy (Citation40). Decoy receptor 3 (DcR3) is a member of the TNF receptor superfamily and has been involved in the control of MHC class II in TAMs (Citation41). IL-1, IL-6, TNF, and the related receptor activator of NFκB ligand (RANKL) have long been known to augment the capacity to metastatize by affecting multiple steps in the dissemination and inflammation cascade (Citation1,Citation42,Citation43).
A major source of inflammatory cytokines in the tumour microenvironment are tumour-associated macrophages (TAM) (Citation44,Citation45). TAMs assist tumour cell malignant behaviour in many ways, including producing cytokines, growth factors, and matrix-degrading enzymes (Citation20,Citation46–48). Kim et al. have recently explored the molecular link(s) between tumour cells, macrophages, and metastasis. They found that tumour cell supernatants contained an inducer of cytokine production in macrophages. Unexpectedly, the tumour-derived macrophage activation was identified as a component of the extracellular matrix: versican, which is frequently up-regulated in human tumours. Versican was found to be recognized by toll-like receptor 2 and 6 (TLR2/TLR6) which belong to a family of sensors of microbial moieties and tissue damage. Thus, in the Lewis lung carcinoma model this study identifies a cascade of amplification of metastasis initiated by tumour-derived versican acting on myeloid cells via TLR2/TLR6, leading to inflammatory cytokine production (Citation49).
Early on, IL-1 was shown to augment metastasis, a finding which at the time was related to induction of adhesion molecules in target organs. IL-1R1 (IL-1 receptor, type 1) gene-targeted mice have provided unequivocal evidence for the protumour potential of IL-1 (Citation25,Citation50). In particular, in models of chemical carcinogen-induced tumours, IL-1β, secreted by malignant cells or host cells in the tumour's microenvironment, contributes to increased tumour invasiveness, tumour angiogenesis, and tumour-mediated suppression, and IL-1 receptor antagonist (IL-1ra) negatively controls these processes (Citation51). In a pancreatic islet tumour model, a first wave of myc-driven angiogenesis is induced by the inflammatory cytokine IL-1 (Citation52). In diethyl nitrosamine (DEN)-induced hepatocarcinoma, the unique membrane-associated form of IL-1α acts as pro-tumorigenic mediator: actually, DEN-induced hepatocyte death results in the release of IL-1α and activation of IL-1R signalling, leading to IL-6 induction and compensatory proliferation, processes that are critical for hepatocarcinogenesis (Citation53). However, IL-1α is possibly of importance in 3-methylcholant-rene-induced fibrosarcoma, due to its efficiency in activating anti-tumour innate and specific immune responses, by acting as a focused adjuvant, through binding to IL-1R1 on immunosurveillance cells (Citation54,Citation55). Moreover, small amounts of IL-1α, which is homeostatically expressed in cells, but not secreted, can be poured out from necrotizing cells and serve as a ‘danger signal’ for mounting anti-tumour cell immunity (Citation56).
In addition strong genetic evidence has linked a pro-inflammatory haplotype at the IL-1 gene locus, comprising agonist and antagonist, to the pathogenesis of gastric carcinoma (Citation57). Recent studies have uncovered a novel relationship between sex steroid hormones and cancer. In carcinoma of the prostate, an androgen-dependent tumour, sensitivity to hormonal stimulation is regulated by selective androgen receptor modulators (SARM). The inflammatory cytokine IL-1 produced by macrophages in the tumour microenvironment converts SARMs from inhibitors to stimulators (Citation58).
The signalling by IL-1 receptor and toll-like receptors (TLRs) is under control by negative pathways of regulation (Citation1). There is now evidence that TIR8, an orphan member of the IL-1R family (also known as SIGIRR) inhibits signalling from the IL-1R/TLR complexes, possibly by trapping IRAK-1 and TRAF-6. In a mouse model of intestinal carcinogenesis in response to dextran sulphate sodium salt (DSS) and azoxymethane administration, Tir8-deficient mice exhibited a dramatic susceptibility to inflammation, in terms of weight loss, bleeding, and mortality, and showed increased susceptibility to colon carcinogenesis, associated to increased permeability and local production of prostaglandin E2, pro-inflammatory cytokines (IL-1, IL-6), and chemokines (KC/CXC, JE/CCL2, and CCL3) (Citation29,Citation30). These mediators are downstream of NFκB and have been shown to promote inflammation-propelled neoplasia (Citation23). Thus the lack of a check-point (TIR8) of NFκB activation leads to increased carcinogenesis in the gastrointestinal tract, underlying once more the connection between chronic inflammation and cancer promotion.
IL-6 is a key growth-promoting and anti-apoptotic inflammatory cytokine (Citation59,Citation60) and is also one of the effector signals of activated NFκB in the promotion of neoplasia. Recent studies suggest an alternative pathway of connection between IL-6 and cancer, having NFκB as a link (Citation61,Citation62).
Another example of a role for IL-6 is in hepatocarcinogenesis. Hepatocellular carcinoma (HCC), the most common type of liver cancer, is a frequent outcome after years of chronic inflammation, induced either by chronic infection (HBV, HCV) or sustained alcohol consumption.
Naugler et al. (Citation63) have explored the mechanisms underlying gender difference in HCC. They started from the observation that, in response to injections of the carcinogen DEN, male mice had higher levels of IL-6. The sex difference in susceptibility to cancer was absent in mice deficient in IL-6 and in the adaptor protein MyD88. The latter operates downstream of the toll-like receptors that sense microbial invasion and tissue damage, or (in the form of the IL-1 receptor) act as amplifiers of inflammation. From in vivo studies in mice carcinogen-induced tissue damage results in the release of debris, which in turn causes MyD88-dependent activation of Kupffer cells in the liver. The macrophage-like Kupffer cells produce IL-6, which promotes liver injury, inflammation, compensatory cell proliferation, and carcinogenesis. In females, however, oestrogen steroid hormones act through gene transcription factors (such as NFκB) to inhibit IL-6 production in Kupffer cells, and so protect female mice from cancer (Citation63,Citation64).
The importance of the autocrine IL-6 is confirmed also in other cancer types (Citation65,Citation66) where it is demonstrated that IL-6 is an important activator of oncogenic Stat3 in lung adenocarcinomas and of Jagged-1/Notch signalling in breast tumour mam-mospheres (Citation67).
In an interesting switch, new light was shed on the role of IL-6 in colitis-associated cancer (Citation68,Citation69). It was found that IL-6 produced by myeloid cells is a critical tumour promoter during intestinal carcinogenesis. IL-6 protects normal and pre-malignant intestinal epithelial cells from apoptosis and promotes the proliferation of tumour-initiating cells. These actions are mediated by gp130 and Stat3. Thus, the NFκB/IL-6/Stat3 cascade plays a key role in intestinal carcinogenesis. Interestingly, Stat3 also regulates the balance between IL-12 and IL-23 in the tumour microenvironment (Citation70).
Chemokines in CRI
Chemokines have long been associated with the recruitment of leukocytes in tumours (Citation1,Citation71). Recent results with gene-targeted mice have provided unequivocal evidence for a role of CC chemokines in carcinogenesis. Mice deficient in D6, a decoy and scavenger receptor for inflammatory CC chemokines, show increased susceptibility to skin carcinogenesis and colitis-associated cancer (Citation72).
The contribution of chemokines to angiogenesis and tumour promotion has been the object of intensive investigation. A variety of chemokines, including CCL2, CXCL12, CXCL8, CXCL1, CXCL13, CCL5 (Citation29), CCL17, and CCL22, have been detected in neoplastic tissues as products of either tumour cells or stromal elements. CXCL1 and related molecules (CXCL2, CXCL3, CXCL8, or IL-8) have an important role in melanoma progression by stimulating neoplastic growth, promoting inflammation, and inducing angiogenesis. Strong evidence demonstrates that levels of CCL2 are associated with TAM accumulation (Citation73) and that CCL2 may play an important role in the regulation of angiogenesis. Chemokines are also involved in the rapid turnover of myeloid-derived suppressor cells (Citation74).
Expression of chemokine receptors plays an important role in guiding metastasis. Up-regulation of CXCR4 is downstream of the von Hippel Lindau activation of tyrosine kinase oncogenes and TNF (Citation1). CXCR4 is the most frequently up-regulated chemokine receptor in cancer cells, and it is associated with advanced stages and metastasis (Citation1). Recently, the chemokine receptor CX3CR1 has been reported to be up-regulated in human pancreatic cancer and to be involved in the perineural dissemination of this neoplasia along local nerve terminations (Citation75).
Tumour-associated macrophages as a key cellular component of CRI
In several studies of human cancer, TAM accumulation has been associated with angiogenesis and with the production of angiogenic factors such as vascular endothelial growth factor (VEGF) and platelet-derived endothelial cell growth factor (Citation73,Citation76–85). TAMs accumulate in hypoxic regions of tumours, and hypoxia triggers a pro-angiogenic programme in these cells (Citation86). A number of molecules with possible impact on angiogenesis have been shown to be expressed by macrophages in low-oxygen conditions, such as VEGF, TNFα, basic fibroblast growth factor (bFGF), and CXCL8. Therefore, macrophages recruited in situ represent an indirect pathway of amplification of angiogenesis, in concert with angiogenic molecules directly produced by tumour cells. Strikingly, it was reported that the hypoxia-inducible factor-1 (HIF-1)-dependent chemokine CXCL12 acts as a potent chemoattractant for endothelial cells of different origins bearing CXCR4, and HIF-1 is a participant in angiogenesis that is regulated at the receptor level by VEGF and bFGF In agreement with these observations, the angiogenic programme established by hypoxia relies also on the increased expression of CXCR4 by endothelial cells (Citation1,Citation15). Under hypoxic conditions, tumour-secreted lactic acid promotes a IL-23/IL-17-mediated pro-inflammatory pathway in TAMs (Citation87).
Myeloid cells in the tumour microenvironment are related to the angiogenic switch at different levels. VEGF and the related angiogenic factor placenta-derived growth factor (PlDGF) have long been known to be potent monocyte attractants and to contribute to TAM recruitment (Citation88). VEGF1R+ hematopoietic bone-marrow cells home to tumour-specific premetastatic sites. There, they form a recipient niche which favours secondary localization of cancer. Bv8, also known as prokinetic 2, modulates recruitment of angiogenic Gr+Mac1+ cells from bone-marrow (Citation89) and promotes angiogenesis. Moreover, Gr+Mac1+ cells, presumably MDSC, have recently been shown to mediate resistance to anti-angiogenic therapy in various models (Citation89). Thus TAMs and related cells (MDSC, DC, polymorphonuclear cells (PMN)) represent an important indirect and alternative pathway of angiogenesis. Mast cells and TAMs produce angiogenic factors, such as chemokines and VEGF itself, and are a source of matrix-degrading enzymes (MMP) which mobilize VEGF from extracellular matrix stores. Recent results suggest that TAMs promote tumour angiogenesis also via semaphorin 4D (Citation90). Therefore, a complex reciprocal interaction occurs between M2-polarized myeloid cells and regulation of angiogenesis in the tumour microenvironment (Citation91).
Concluding remarks
Recent efforts have shed new light on molecular and cellular pathways linking inflammation and cancer (Citation1). Two pathways link inflammation and cancer. In the intrinsic pathway, activation of different classes of oncogenes drives the expression of inflammation-related programmes which guide the construction of an inflammatory microenvironment. In the intrinsic pathway inflammatory conditions promote cancer development. Key orchestrators at the intersection of the intrinsic and extrinsic pathway include transcription factors (e.g. NFκB) (Citation23), cytokines (e.g. TNF), and chemokines. Thus, inflammation is a key component of the tumour microenvironment. It should be emphasized that an inflammatory reaction can also have anti-tumour activity (the bright side of the force) (Citation1,Citation92,Citation93). This dual function of inflammatory cells and mediators is reflected by studies on correlations between parameters of CRI and clinical behaviour in different contexts (Citation94–99).
Cytokines are a key component and orchestrator of the inflammatory microenvironment of tumours. As such they represent a prime target in therapeutic efforts aimed at taming tumour-promoting CRI. For many years all efforts to treat cancer have concentrated on the destruction/inhibition of tumour cells. Strategies to modulate the host microenvironment offer a complementary perspective. Initial results in this direction justify continuing efforts (e.g. (Citation40)).
Acknowledgements
The authors are supported by the Italian Association for Cancer Research, Italian Ministry of Health, University and Research, European Commission (INNOCHEM project), Fondazione Cariplo (NOBEL project).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, . NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11.
- Mantovani A. Cancer: Inflaming metastasis. Nature. 2009;457:36–7.
- Borrello MG, Alberti L, Fischer A, Degl’innocenti D, Ferrario C, Gariboldi M, . Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci U S A. 2005;102:14825–30.
- Russell JP, Engiles JB, Rothstein JL. Proinflammatory mediators and genetic background in oncogene mediated tumor progression. J Immunol. 2004;172:4059–67.
- Russell JP, Shinohara S, Melillo RM, Castellone MD, Santoro M, Rothstein JL. Tyrosine kinase oncoprotein, RET/PTC3, induces the secretion of myeloid growth and chemo-tactic factors. Oncogene. 2003;22:4569–77.
- Cerutti JM, Oler G, Michaluart P Jr, Delcelo R, Beaty RM, Shoemaker J, . Molecular profiling of matched samples identifies biomarkers of papillary thyroid carcinoma lymph node metastasis. Cancer Res. 2007;67:7885–92.
- De Falco V, Guarino V, Avilla E, Castellone MD, Salerno P, Salvatore G, . Biological role and potential therapeutic targeting of the chemokine receptor CXCR4 in undifferentiated thyroid cancer. Cancer Res. 2007;67:11821–9.
- Xu K, Shu HK. EGFR activation results in enhanced cycloox-ygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 2007;67:6121–9.
- Masih-Khan E, Trudel S, Heise C, Li Z, Paterson J, Nadeem V, . MlP-1alpha (CCL3) is a downstream target of FGFR3 and RAS-MAPK signaling in multiple myeloma. Blood. 2006;108:3465–71.
- Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, . CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203:941–51.
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, . Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302.
- Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–6.
- Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–8.
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7.
- Kobielak A, Fuchs E. Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A. 2006;103:2322–7.
- Rolny C, Capparuccia L, Casazza A, Mazzone M, Vallario A, Cignetti A, . The tumor suppressor semaphorin 3B triggers a prometastatic program mediated by interleukin 8 and the tumor microenvironment. J Exp Med. 2008;205:1155–71.
- Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66:10671–6.
- Markowski MC, Bowen C, Gelmann EP. Inflammatory cytokines induce phosphorilation and ubiquitination of prostate suppressor protein NKX3.1. Cancer Res. 2008;68:6896–901.
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, . Abrogation of TGFbeta signaling in mammary carcinomas recruits Gr-1 + CD 11 b + myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35.
- Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, . DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25.
- Campregher C, Luciani MG, Gasche C. Activated neutrophils induce an hMSH2-dependent G2/M checkpoint arrest and replication errors at a (CA) 13-repeat in colon epithelial cells. Gut. 2008;57:780–7.
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6.
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51.
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, . IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100:2645–50.
- Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, . IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–5.
- Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, . RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131:309–23.
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, . NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6.
- Garlanda C, Riva F, Veliz T, Polentarutti N, Pasqualini F, Radaelli E, . Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory members of the IL-1 receptor family. Cancer Res. 2007;67:6017–21.
- Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, . The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumori-genesis. Immunity. 2007;26:461–75.
- Lech M, Kulkarni OP, Pfeiffer S, Savarese E, Krug A, Garlanda C, . Tir8/Sigirr prevents murine lupus by suppressing the immunostimulatory effects of lupus autoantigens. J Exp Med. 2008;205:1879–88.
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, . A distinct and unique transcriptional programme expressed by tumor-associated macrophages: defective NF-kB and enhanced IRF-3/STAT1 activation. Blood. 2006;107:2112–22.
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, . ‘Re-educating’ tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–8.
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, . Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21.
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, . Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31.
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–71.
- Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, . Blocking TNF-alpha in mice reduces color-ectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70.
- Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, . The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67:585–92.
- Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, . Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008;27:1671–81.
- Harrison ML, Obermueller E, Maisey NR, Hoare S, Edmonds K, Li NF, . Tumor necrosis factor alpha as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J Clin Oncol. 2007;25:4542–9.
- Chang YC, Chen TC, Lee CT, Yang CY, Wang HW, Wang CC, . Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood. 2008;111:5054–63.
- Giavazzi R, Garofalo A, Bani MR, Abbate M, Ghezzi P, Boraschi D, . Interleukin 1-induced augmentation of experimental metastases from a human melanoma in nude mice. Cancer Res. 1990;50:4771–5.
- Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, . Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690–4.
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
- Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–27.
- Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, . Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56.
- Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22.
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
- Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, . Carcinoma produced factors activate myeloid cells via TLR2 to stimulate metastasis. Nature. 2009;457:102–6.
- Dinarello CA. The paradox of pro-inflammatory cytokines in cancer. Cancer Metastasis Rev. 2006;25:307–13.
- Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, . Interleukin-1 beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007;67:1062–71.
- Shchors K, Shchors E, Rostker F, Lawlor E, Brown-Swigart L, Evan G. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527–38.
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, . Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65.
- Marhaba R, Nazarenko I, Knofler D, Reich E, Voronov E, Vitacolonna M, . Opposing effects of fibrosarcoma cell-derived IL-1 alpha and IL-1 beta on immune response induction. Int J Cancer. 2008;123:134–45.
- Elkabets M, Krelin Y, Dotan S, Cerwenka A, Porgador A, Lichtenstein RG, . Host-derived interleukin-1 alpha is important in determining the immunogenicity of 3-methylcholantrene tumor cells. J Immunol. 2009;182:4874–81.
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6.
- El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, . Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402.
- Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, . Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–29.
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83.
- Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–19.
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, . Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30.
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, . Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44.
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim KH, Elsharkawy AM, . Gender disparity in liver cancer due to sex differences in MyD88-dependent-IL-6 production. Science. 2007;317:121–4.
- Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–7.
- Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, . Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–56.
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, . IL-6 triggers malignant features in mam-mospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002.
- Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9.
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, . IL-6 and stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13.
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, . gp130-Mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102.
- Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, . Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23.
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50.
- Nibbs RJ, Gilchrist DS, King V, Ferra A, Forrow S, Hunter KD, . The atypical chemokine receptor D6 suppresses the development of chemically induced skin tumors. J Clin Invest. 2007;117:1884–92.
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45.
- Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, . Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–66.
- Marchesi F, Piemonti L, Fedele G, Destro A, Roncalli M, Albarello L, . The chemokine receptor CX3CR1 is involved in the neural tropism and malignant behavior of pancreatic ductal adenocarcinoma. Cancer Res. 2008;68:9060–9.
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, . HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20.
- Seandel M, Butler J, Lyden D, Rafii S. A catalytic role for proangiogenic marrow-derived cells in tumor neovascularization. Cancer Cell. 2008;13:181–3.
- Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205.
- Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, . Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–6.
- Kusmartsev S, Eruslanov E, Kubler H, Tseng T, Sakai Y, Su Z, . Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181:346–53.
- Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, . Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–30.
- Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, . Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer Res. 2007;67:6433–41.
- Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, . Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. 2007;67:5708–16.
- Stearman RS, Dwyer-Nield L, Grady MC, Malkinson AM, Geraci MW. A macrophage gene expression signature defines a field effect in the lung tumor microenvironment. Cancer Res. 2008;68:34–43.
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12.
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
- Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, . Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol. 2008;180:7175–83.
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, . Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–75.
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, . Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20.
- Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, . Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med. 2008;205:1673–85.
- Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A. Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev. 2008;27:31–40.
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, . Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
- Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61.
- Shabo I, Stal O, Olsson H, Dore S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. 2008;123:780–6.
- Tamimi RM, Brugge JS, Freedman ML, Miron A, Iglehart JD, Colditz GA, . Circulating colony stimulating factor-1 and breast cancer risk. Cancer Res. 2008;68:18–21.
- Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppa S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res. 2007;13:5784–9.
- Byers RJ, Sakhinia E, Joseph P, Glennie C, Hoyland JA, Menasce LP, . Clinical quantitation of immune signature in follicular lymphoma by RT-PCR-based gene expression profiling. Blood. 2008;111:4764–70.
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, . The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–26.
- Seike M, Yanaihara N, Bowman ED, Zanetti KA, Budhu A, Kumamoto K, . Use of a cytokine gene expression signature in lung adenocarcinoma and the surrounding tissue as a prognostic classifier. J Natl Cancer Inst. 2007;99:1257–69.

