Abstract
The concept of chimeric antigen receptors (CARs) as molecules able to redirect T lymphocytes toward tumor cells is currently being exploited in the field of cancer immunotherapy. Despite promising preliminary results, some clinical trials evidenced limitations for this technology that must be overcome for more extensive application of CARs in tumor immunotherapy. We describe here the fundaments of these molecules in terms of structure, function, possible targets and pre-clinical and clinical applications. We also discuss strategies that can potentially overcome the limitations seen so far, paving the road to a wider application of this exciting new technology.
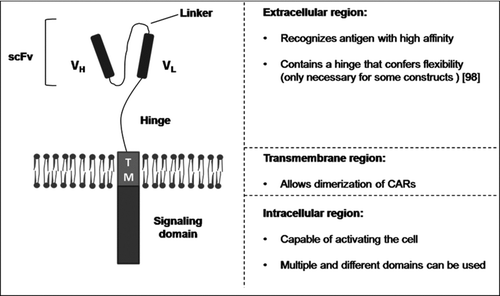
The adoptive transfer of antigen-specific T cells is a promising new treatment in the field of cancer immunotherapy. After isolation and ex vivo expansion, these cells are able to recognize and kill the tumor, sometimes obtaining response rates far greater than the standard therapy. It has been shown that transfer of autologous tumor infiltrating lymphocytes (TILs) to patients with advanced metastatic melanoma after severe lymphodepletion achieves objective responses in 72% of the cases [Citation1]. However, most tumors do not present TILs capable of recognizing and killing tumor cells and their self-origin difficult discerning between healthy and abnormal cells. A strategy to overcome these limitations was first proposed by Eshhar's group, consisting of redirecting the immune response through the introduction of chimeric antigen receptors (CARs) into the T cells surface [Citation2, 3]. Currently, most CARs consist of an extracellular binding moiety—comprising the light (VL) and heavy (VH) variable chains derived from a monoclonal antibody joined by a flexible linker (scFv)-, a transmembrane region, and a signaling endodomain ().
FIGURE 1 Chimeric antigen receptor. Schematic representation of the standard structure of a chimeric antigen receptor. VH, heavy-chain variable domain; VL, light-chain variable domain; scFv, single chain fragment variable.
TUMOR-ASSOCIATED TARGETS
The characterization of tumor-associated antigens (TAAs) allowed the development of immunotherapies based on genetic engineered T lymphocytes. TAAs are nonmutated self-antigens derived from proteins expressed by the tumor and normal tissue [Citation4]. There are three types of antigen panels: (1) tissue-specific antigens, like MAGE, BAGE, GAGE, and NY-ESO1, that are reactivated in tumor cells (these antigens are not typically expressed by normal tissues, with the exception of testis and placenta, but the low level of MHC expression on these tissues impairs efficient antigen recognition by the immune system); (2) the differentiation antigens, such as melanoma-related gp100, Mart-1/Melan-A, and pMel-17 molecules, that are expressed by both the tumor and the healthy equivalent tissue, but not by other cells; and (3) the ubiquitously expressed antigens, like survivin, hTERT, and PRAME, found at healthy tissues, but frequently upregulated in tumor cells. Even though the antigen aberrant expression promotes a major number of peptides presented by MHC molecules, the self-origin of TAAs implies that the patient reactive lymphocytes consist of low avidity clones due to mechanisms of central tolerance, and are susceptible to peripheral tolerance by different mechanisms, including inhibition by regulatory T cells (Tregs).
CHIMERIC ANTIGEN RECEPTOR (CAR) DOMAINS
Antigen Recognition Domain
scFv Domains for Antigen Recognition
CAR-based immunotherapies can overcome most of the limitations of TIL-based immunotherapies due to the ability of CARs to bind the target antigen with high affinity without being susceptible to central tolerance mechanisms. Since most CARs are composed by a scFv-derived extracellular domain, they recognize the target antigen on the tumor surface in a MHC-independent fashion, overcoming important mechanisms of tumor evasion, such as MHC downregulation or functional defects in the antigen processing machinery. This important feature allows CAR-expressing cells to attack tumors that are poorly immunogenic. Moreover, the same CARs can be used for therapy in patients with different HLA-haplotypes, favoring the clinical application of this technology. Since most CARs use a scFv derived from antibodies, it is possible to design CARs specific for biochemically different tumor specific targets, including protein-derived peptides or nonprotein molecules, such lipids and carbohydrates.
These advantages led to the development of CARs directed against several relevant tumor antigens. An example is the receptor against CD33 in acute myeloid leukemia [Citation5] and CD23 in chronic lymphocytic leukemia [Citation6]. More recently, clinical trials used chimeric receptors against molecules found on tumor surface (), such as CAIX for treatment of renal cell carcinoma [Citation7], CD20 to treat mantle cell lymphoma [Citation8], GD2 in neuroblastoma [Citation9], CD19 in lymphomas [Citation10] and chronic lymphocytic leukemia [Citation11], and the CAR against ErbB2 for treatment of colon carcinoma [Citation12] (further described in the next section). An extensive list of ongoing clinical trials can be consulted at public databases (www.clinicaltrials.gov and www.wiley.com/legacy/wileychi/genmed/clinical). The chimeric receptors can be combined with up to three signaling endodomains, including the γ chain derived from FcεRI and the ζ chain derived from CD3 complex, as well as intracellular domains from co-stimulatory molecules (i.e., CD28 or 4-1BB).
TABLE 1 Published clinical trials
scFv Recognizing MHC/Peptide Complexes
Another strategy to retarget T lymphocytes consists of introducing MHC-restricted CARs that recognize peptide/MHC complexes [Citation13–15]. Studies performed in vitro have shown that human T lymphocytes transduced with scFvs derived from selected peptide/MHC-specific Fab fragments and fused to CD3γ [Citation14] or CD3ζ [Citation15] chains joined to CD28 co-stimulatory endodomain could promote cytokine production and cytotoxic response against peptide pulsed target cells. The use of peptide/MHC-specific Fab fragments is advantageous, since it allows targeting of intracellular peptides, thus expanding the array of therapeutic targets. These receptors are likely to show higher binding affinity and promote vigorous in vitro expansion. An advantage of using MHC-restricted CARs is the potential to respond against phosphopeptides. Studies have shown that phosphorylated antigens are indeed recognized by TCRs with variable affinities [Citation16, 17]. Because many phosphopeptides are differently expressed in normal and tumor tissues, they are potential targets for immunotherapy. Certain phosphoproteins, such as Mart-1 and tensin-3 expressed in melanomas, contain either MHC I- and MHC II-restricted epitopes, which raises the possibility of treatment approaches combining both MHC I- and MHC II-restricted phosphopeptides [Citation18]. However, the capacity of the endogenous TCR to recognize the phosphopeptide varies, depending on the phosphoantigens encountered during thymic selection, a limitation that does not apply to CARs. Nevertheless, the MHC-dependent response is subject to some of the common tumor evasion mechanisms, such as downregulation of MHC expression, and is restricted to patients carrying certain HLA haplotypes.
Zetakine
Most CARs usually recognize antigens on the tumor surface. CARs constructs can also utilize ligands that interact with their respective receptors at the tumor membrane, such as heregulin [Citation19] or IL-13. For glioblastomas, an innovative targeting approach was developed consisting of genetic engineered IL13Rα2-specific T lymphocytes that express a chimeric receptor called zetakine. The glioblastoma-specific zetakine incorporates an IL-13 mutant protein (for selective binding to IL13Rα2) joined to the CD3ζ endodomain. IL-13 zetakine-modified T lymphocytes specifically lyse IL13Rα2-positive target cells, proliferating and releasing IFN-γ, GM-CSF, and TNF-α upon IL13Rα2+ cell recognition. The infusion of 106 IL-13 zetakine T cells promoted regression of established glioblastoma xenografts in NOD-scid mice and progression-free survival up to 100 days [Citation20], indicating that this novel approach can be used in vivo.
Transmembrane Domains
It is thought that dimerization of CARs plays an important role in its signaling capacity. Domains derived from CD8 or CD28 are commonly used in CARs, but it is not known how these domains affect/contribute to the activation of T cells. Recently, Bridgeman and colleagues showed that the dimerization of an anti-CEA CAR through its CD3ζ transmembrane domain is required to the efficient activation of Jurkat and primary human T lymphocytes. This domain also allows the interaction with the endogenous TCR complex [Citation21]. Nevertheless, the best strategy to build a CAR is still being debated. Signaling through dimerization of CARs and interaction with endogenous TCR is a feature that contributes to activation of the lymphocyte and should be taken into consideration in future designs. Aspects that may contribute to the design of an optimal structure have been extensively reviewed by Bridgeman and collaborators [Citation22].
Intracellular Domains for CARs
First-generation CARs
Eshhar and colleagues proposed the activation of T cells by chimeric receptors by fusing an immunoglobulin-like scFv with the intracellular domains of an FcεRI receptor (γ chain) or CD3 complex (ζ chain), the so-called first-generation CARs [Citation3]. The intracellular signaling domain is important to promote T-cell activation and induces effector functions. Studies performed in vitro and in vivo demonstrated that activation induced by first-generation CARs could promote calcium influx, T-cell activation, and initial cytotoxic response, yet low or absent IL-2 production. [Citation23, 24]. Furthermore, studies have shown superior tumor eradication by T cells bearing CD3ζ CAR-based endodomain in comparison to FcεRIγ receptor-transduced T lymphocytes [Citation25–27]. The most likely hypothesis to explain the stronger signal observed is the presence of three ITAMs at the CD3ζ chain, in contrast to a unique ITAM displayed in the FcεRIγ chain, even though a recent report showed that abrogation of the phosphorilation of two of the ITAMs in the CD3ζ chain reduces apoptosis after T-cell transduction, leading to a longer expression of the transgene [Citation28]. Moreover, early studies showed that the stability of CARs expression is dependent on the signaling domain used. The CARs containing CD3ζ had a lower expression than CARs bearing FcεRIγ endodomains, despite the former being more efficient in activating T cells [Citation27].
NOD/scid mice are the most used model for in vivo assessment of lymphocytes modified with CARs. As they do not have T and B cells, this model is optimal, since it allows the transfer of human cells (tumor and/or lymphocytes) to the host without significant rejection. Several groups have developed CARs directed against important tumor antigens such as GD3 (melanoma) [Citation29], CAIX (renal carcinoma) [Citation7], PSMA (prostate cancer) [Citation30], and CD20 (B-cell lymphoma) [Citation8]. The first-generation CARs showed promising results in some studies using models of colon carcinoma [Citation26] and prostate cancer [Citation31]. However, evidence shows that naïve T cells need more than one signal for efficient activation and tumor eradication [Citation32]. The stimulation only through the TCR pathway does not induce proliferation in most primary T cells, and these cells become insensitive to a new stimulus, a property known as anergy [Citation33]. The T-cell co-stimulation is an important aspect of the adoptive transfer of antigen-specific cells, since the provided signals are crucial in determining the threshold of activation, the type of response, and survival of cells. As tumor cells usually do not express co-stimulatory molecules, the development of an anti-tumor response may be compromised. Indeed, in a study using a transgenic mouse expressing CARs in all cells, the first-generation receptors were not able to induce proliferation of naïve T cells [Citation23]. To circumvent this problem, some studies have induced the expression of CARs in anti-EBV or anti-influenza lymphocytes. Since these infections are recurrent, the modified lymphocytes are constantly restimulated in vivo by the endogenous TCR, increasing the persistence and antitumoral activity. This approach showed promising results in preclinical models [Citation34, 35] and was recently used in clinical trials [Citation9] (see next sections).
Second- and Third-generation CARs
The addition of co-stimulatory domains in chimeric receptors, such as CD28 and 4-1BB, led to an increase in the effector activity of lymphocytes. The CD28 molecule has an important role in regulating the proliferation and survival of lymphocytes, being crucial for the effector function and establishment of memory cells. These effects occur due to recruitment of molecules such as PI3K, Grb2, and Lck, which, in turn, regulate the activity of crucial transcription factors such as NFκB and increase the production of IL-2 and Bcl-xL [Citation36]. The TNF family receptors, like 4-1BB, also provide signals to sustain T-cell responses, playing a key role in T-cell survival and CD8 T-cell memory. This receptor recruits TRAF1 and TRAF2 adaptors, activating downstream JNK, p38 MAPK, and NFκB signaling pathways [Citation37].
Sadelain's group first demonstrated, in 2003, the relevance of co-stimulatory signals to sustain CARs activation. Peripheral blood T lymphocytes were genetic modified to express the 19z chimeric receptor and expanded in vitro through co-culture with CD19+CD80+ APCs in the presence of IL-15. Under these conditions, transduced T cells were able to persist in vivo and migrate to the bone marrow of SCID mice bearing Raji tumor, eradicating tumor cells [Citation38].
Further evidence of co-stimulatory signaling improved effect was the work by Kowolik and collaborators, using the human cell line Daudi in mice. In this study, lymphocytes were modified with a CAR containing the signaling domain of CD28 and showed increased production of IL-2 and IFN-γ and augmented expression of Bcl-xL anti-apoptotic protein. In addition, the lymphocytes showed higher persistence in the host and increased antitumoral activity [Citation39].
Another study using T lymphocytes transduced with CD80, 4-1BBL, and a CAR against PSMA reported the rejection of systemic tumors in immunodeficient mice without need of APC-mediated stimuli. The presence of CD80 and 4-1BBL co-stimulatory ligands induces auto- and trans-co-stimulation of activated T lymphocytes, resulting in a rapid expansion of the T cells, leading to tumor eradication and increased survival of tumor-bearing mice [Citation30].
Subsequent studies compared the addition of co-stimulatory domains, such as CD28 or 4-1BB, or the so-called third-generation receptors containing two co-stimulatory domains (CD28 plus 4-1BB), achieving different conclusions. In an in vivo model of leukemia using the human cell line NALM-6, CARs containing a CD28 domain had higher antitumoral effect when compared with receptors containing 4-1BB domain [Citation32]. However, more recent studies conducted on different models demonstrated that CARs containing 4-1BB or 4-1BB plus CD28 showed greater persistence and antitumoral activity in vivo when compared to receptors containing only the CD28 domain [Citation28, Citation40–42]. These enhanced responses with CARs bearing dual co-stimulatory domains are probably due to additive effects leading to upregulation of antiapoptotic proteins like Bcl-xL [Citation28] and superior activity of the Akt pathway [Citation42]. Another study showed that the combination of CD28 and OX40 co-stimulatory domains is also effective in an in vivo melanoma model [Citation43]. These results illustrate the stronger antitumor responses obtained by new generation CARs. The reason for the differences observed by different groups when comparing different co-stimulation domains (e.g., 4-1BB vs. CD28) is not clear yet and may involve characteristics of the experimental systems, such as CAR design, immunotherapy protocol, and tumor model, including levels of target antigen expression or tumor microenvironment.
A recent publication of Houston's group compared in parallel CARs anti-CD19 containing the zeta-chain endodomain alone or in addition to CD28 domain. The infusion of cells carrying first-generation CARs and cells carrying second-generation CARs in the same patient demonstrated that lymphocytes expressing the second-generation CARs were much more persistent than lymphocytes bearing CARs with the zeta-chain signal alone. However, this study was not designed to detect differences in anti-tumor response between these two cell populations [Citation44].
CHASING THE PERFECT SIGNAL COMBINATION
Besides the addition of co-stimulatory domains, other approaches have been used with the aim of increasing the antitumoral activity of CARs. In a recent study, T cells were engineered to express a CAR containing the CD28 and ζ domains and to produce IL-15, a cytokine crucial for homeostasis and survival of lymphocytes [Citation45]. This combination resulted in a major expansion and survival of lymphocytes, both in vitro and in vivo, and in greater antitumoral activity in vivo. Importantly, these lymphocytes were also modified to express a suicide gene (iCasp9) to limit T-cell expansion and toxicity, since this stimulatory signals combination can pose a risk of uncontrolled toxicity and proliferation. Another good candidate cytokine for gene modification of T lymphocytes expressing CARs is IL-12. Indeed, some experimental data in animal models with T lymphocytes modified to express CARs and IL-12 are in progress with promising results. In this study, T cells expressing CARs against CD19 and producing IL-12 are able to overcome local effector cell inibition by Tregs, showing much stronger anti-tumor responses than their counterparts not secreting the transgenic IL-12 (R. Brentjens, personal communication).
Another approach being tested is the combination of the expression of chemokine receptors with CARs. The work by Stasi and collaborators used Hodgkin lymphoma as a model, since this tumor secretes large amounts of TARC/CCL17 and MDC/CCL22 chemokines. Both interact specifically with the CCR4 receptor expressed by regulatory T cells and Th2 CD4+ lymphocytes, recruiting these cells to the tumor microenvironment and creating an immunosuppressive milieu. Thus, the authors modified CD8+ T lymphocytes to express CCR4 in combination with an anti-CD30 CAR containing CD28 and CD3ζ domains. After infusion in mice grafted with Hodgkin lymphoma cells, human CD8+ T lymphocytes overexpressing CCR4 showed enhanced migration to the tumor site and greater antitumoral activity than cells expressing CARs alone [Citation46]. Similar results were obtained by combining an anti-GD2 CAR with the CCR2b receptor in an in vivo model of neuroblastoma [Citation47]. A recent paper using a model of malignant pleural mesothelioma also demonstrated that CCR2b transduction of lymphocytes bearing anti-tumor CAR enhances the cytotoxicity against tumor and the number of CAR+ T lymphocytes infiltrated in the tumoral milieu. The rationale of increasing the trafficking of CAR+ T lymphocytes to tumor site through expression of CCR2 is due to the fact that CCL2 chemokine is highly secreted by these tumor cells [Citation48].
CHOOSING THE POPULATION OF EFFECTOR CELLS
Initial work with CARs used total T lymphocytes as target cells for gene transfer. However, recent studies suggest that central memory T lymphocytes are more persistent in vivo and have higher anti-tumor activity [Citation49, Citation50], and some groups are already performing CAR transfer to this subset of cells [Citation51]. These cells are characterized by expression of CD62L and CCR7 and a low proliferation and effector function in vitro [Citation52]. Recent studies have shown that the protocols used for transduction and expansion of lymphocytes with CARs are able to generate this subpopulation [Citation53, 54]. In this regard, a clinical trial is under way using central memory-enriched CD8+ T cells transduced to express a CD19-specific CAR for patients with B-lineage non-Hodgkin lymphoma [Citation55]. It has also been shown recently that adoptive transfer of naïve T cells is associated with an increased cytokine production, a more effective anti-tumor activity, and a prolonged in vivo persistence [Citation56], although the feasibility of this approach remains to be proven.
Besides the use of T lymphocytes, some authors have proposed the use of other lymphocyte populations. Some studies have used NK cells modified with CARs, showing a high cytotoxic activity against target cells [Citation57, 58]. Another study used CIK (cytokine-induced killer) cells, a population generated in vitro after stimulation with IFN-γ, OKT3 antibody, and IL-2 and enriched in NKT cells, showing good in vitro results [Citation59].
LIMITATIONS OF PRECLINICAL MODELS
The major limitation of using mice as preclinical models for CAR studies is that these animals do not express the target antigen with the physiological distribution found in humans, impairing direct testing of CARs against human molecules for their off-target effects in vivo. The use of mice models expressing human tumor antigens under the control of their own regulatory elements can partially recapitulate the panel of physiological expression of the target molecules. Such animal models are currently being used for mAb studies, such as for mice expressing the human CD20 [Citation60] or carcinoembryonic antigen (CEA) [Citation61], and can be used to anticipate major acute responses when using CARs. One major limitation is that the expression of the transgene can be different from that found in humans, not covering all the unanticipated ectopic expression of the antigen due to differences in gene expression regulation mechanisms in mice. Under these conditions, the same CAR to be used in patients can be previously tested and validated in mice.
An alternative to this approach is to generate CARs against the murine equivalent tumor-associated antigen, such as for the CAR anti-mouse CD19. This approach, although not generating a parallel between affinities of human- and mouse-specific CARs, can be very helpful to anticipate serious acute and chronic affects, as demonstrated for anti-mouse CD19 CARs in vivo in mice [Citation62]. Another study used the A20 mouse lymphoma cells in immunocompetent mice and a first-generation CAR against murine CD19. The T cells modified with this CAR showed anti-tumor activity, but also induced a temporary depletion of normal B cells. Moreover, the transferred lymphocytes showed decreased persistence [Citation63].
Apart from the forced expression of the antigen, which can validate CAR usage in immunocompetent mice, most of the animals models used so far include immunodefficient mice, such as NOD/SCID or NSG mice [Citation64], with human tumors (both primary or cell line derived). These immunodeficient mouse models fail to generate part of the tumor microenvironment based in immune cells. Some efforts are being made to partially recapitulate such tumor environments. One example is the infusion of human Tregs redirected to the tumor using anti-CD19zeta CARs in a model of the human lymphoma Raji cell line. These CARs guide Tregs to the tumor microenvironment and mimic the important role of this subpopulation of cells in the tumor, impairing non-Treg anti-CD19-CAR T-cell anti-tumor function [Citation65]. Lymphodepletion with cyclophosphamide in this system restores the function of anti-tumor CAR+ cells, reinforcing the relevance of previously chemo-conditioning the animals with functional anti-tumor impairment.
Methods for CAR Expression in T Lymphocytes—Benefits and Risks
The present gold standard method of genetic modification of T lymphocytes involves the use of retroviral or lentiviral vectors and hence integration of the provirus into the genome of the cells. Despite advantages, such as stable expression of the transgene and high rates of transduction, there is the theoretical risk of insertional mutagenesis (provirus integration near a proto-oncogene, resulting in aberrant patterns of expression) and initiation of a transformation process [Citation66]. Recent papers described the use of transposon-based vectors, like Sleeping Beauty or piggyBac, for the modification of primary T cells with CARs [Citation51, Citation67–69]. This system consists of two plasmids, one encoding a transposase and the other containing the transgene flanked by inverted repeats (IR/DRs). After electroporation of target cells, the transposase recognizes the IR/DRs repeats and integrates the transgene at random AT sites in the genome. Despite being a nonviral method, it has a reasonable level of transfection and a reduced cost when compared to viral methods. However, like other integrating methods, it has a potential risk of insertional mutagenesis.
No insertional mutagenesis has been reported in clinical trials with gene-modified T lymphocytes so far [Citation70, 71], but because of this possibility, methods avoiding gene disruption or even targeted insertion of the transgene are highly desirable. In this regard, the development of zinc-finger nuclease (ZFN) technology is quite promising. These enzymes are generated by fusing zinc-finger-based DNA binding domains to an independent catalytic domain of FokI, allowing targeted integration of the co-transfected transgene. Recent work has demonstrated that the integration site of adeno-associated vírus (AAV) in the human genome, the AAVS1 locus on chromosome 19 containing the PPP1R12C gene, is a “safe harbor” for ZFN-driven gene addition, as no adverse effects on the cell resulting from its disruption were observed [Citation72, 73]. Future work will explore the potential of this new technology in the genetic modification of T cells.
In a recent report, T lymphocytes were modified by electroporation of CAR-encoding RNA [Citation74]. Transgene expression was detected for up to 6–7 days and multiple infusions were necessary to obtain anti-tumor effects, showing the feasibility of this approach. Since transgene expression lasts only a few days using this methodology, this technique may also induce less chronic toxicity if the CAR recognizes healthy tissues, an important advantage of this approach that will be further discussed in the next sections.
Clinical Trials Using CARs
The first clinical trials to use T cells modified with chimeric molecules were designed to make T cells capable of recognizing HIV-infected cells by expressing the extracellular and transmembrane domains of CD4 molecules fused to the ζ chain of the TCR complex [Citation75]. The rationale was that CD4 on the surface of T cells would recognize an HIV protein in the membrane of HIV-infected cells, activating cytotoxic lymphocytes expressing the chimeric molecule, allowing the elimination of infected cells. Twenty-four patients were enrolled and received mixed populations of CD4 and CD8 gene-modified T cells. Of those, 11 patients received additionally IL-2, while 13 patients received only the transduced lymphocytes (2–3 × 1010 cells).
Transduced cells represented 1–3% of circulating cells in patients after 8 weeks of follow-up, but no significant reduction in HIV loads was observed. A recent update of these results by the same group at the 2010 ATTACK meeting held in Montpellier reported that, after 10 years, circulating cells expressing the CD4-ζ construct (less than 0.1% of the cells) could still be found, showing that gene-modified T lymphocytes can be detected years after the infusion even in the absence of selective pressure (C. June, personal communication).
Regarding anti-tumor-directed CAR molecules, some of the early clinical trials showed no benefit after the infusion of T lymphocytes carrying CARs against CD20 for follicular lymphoma [Citation76] and CD171 for neuroblastoma [Citation77]. In 2006, Lamers and collaborators reported a clinical trial using T cells expressing a scFv CAR against carbonic anhydrase IX (CAIX), an antigen expressed in tumors of renal cell carcinoma patients [Citation7]. After the infusion of transduced cells in 3 patients, hepatic toxicity was observed and biopsies revealed lymphocytic infiltration of the liver and expression of CAIX in bile duct epithelial cells. This was the first report of an off-target function of CAR-bearing cells and was clinically managed by blocking T-cell–CAIX interaction through the administration of a mAb against CAIX. During the follow-up of patients, humoral immune responses against the CAR were also observed, showing that mouse-derived scFv can be immunogenic in humans. This clinical protocol was modified to include the administration of the anti-CAIX mAb prior to CAR+ T-cell infusions. The aim was to saturate liver CAIX molecules while leaving renal cell carcinoma cells only partially saturated and still amenable to T-cell recognition. The interruption of the original clinical trial precludes any evaluation of effectiveness on the treated patients.
Also in 2006, Kershaw and collaborators reported a series of 14 patients with ovarian cancer receiving T cells modified with a CAR (scFv and γ chain of the Fc receptor) against the α-folate receptor (FR). No clinical benefit was observed; transduced T cells were detected only transiently in patient samples and an inhibitory factor was evidenced that avoided T-cell function against FR-positive cells [Citation78].
In 2008, Till and colleagues reported a clinical trial using T cells electroporated with plasmids encoding CARs against CD20 for the treatment of 7 non-Hodgkin lymphoma patients. Persistence of modified T cells up to 9 weeks was reported. Two patients maintained a previous complete response; 1 achieved a partial response and 4 remained with stable disease, showing potential benefit of the therapy [Citation8].
The first impacting clinical results with CARs for cancer treatment were obtained by the group of Malcolm Brenner using CARs against diasialoganglioside (GD2) for neuroblastoma treatment in children [Citation9]. The main difference of this trial was the use of Epstein-Barr virus (EBV)-specific T cells as the target cell population for CAR transduction. Since these cells are physiologically maintained in vivo through the engagement of their natural TCR and co-stimulation by EBV+ cells, the in vivo expected survival and anti-tumor response mediated by the anti-GD2 CAR+ cells should be increased. The clinical results showed that EBV-specific CAR+ cells were found in the circulation of patients up to more than 6 weeks after the infusion, while for non-EBV-specific cells this period was as short as 1 week. Six out of 11 treated patients showed tumor regression or tumor necrosis 6 weeks after the treatment.
There are many currently ongoing clinical trials and clinical benefits are being systematically reported. Recently, Rosenberg's group at the NCI reported the case of one lymphoma patient treated with a CAR against CD19 carrying the CD28 and ζ chain stimulating domains [Citation10]. The patient showed the elimination of CD19+ lymphoma cells; however, this effect was accompanied by the persistent elimination of peripheral B cells for as much as 39 weeks, showing that the off-target effect against healthy mature B cells is still one potential major drawback for clinical application of this approach.
Two phase I clinical trials targeting autologous T cells with second-generation CD19-specific chimeric antigen receptors (19-28z) have been initiated at the Memorial Sloan-Kettering Cancer Center to treat patients with chemotherapy-refractory chronic lymphocytic leukemia (CLL) [Citation79] and relapsed acute lymphoblastic leukemia (ALL) [Citation80]. So far, 8 patients have been treated and these trials are still ongoing. A recent report at the 2010 ATTACK meeting described a marked reduction of peripheral lymphadenopathy at 3 months following CD19-targeted T-cell infusion in 1 of 3 patients with CLL who received cyclophosphamide chemotherapy prior to infusion, while a second patient exhibited mixed response by CT scan at 2 months post-treatment. The third patient with CLL was not evaluable and died 2 days post-T-cell infusion as a likely consequence of sepsis unrelated to infusion of T cells [Citation11]. Importantly, the first treated patient with ALL demonstrated a persistent B-cell aplasia for at least 5 weeks following 19-28z T-cell infusions. The FDA approved reinfusion of CD19 targeted T cells in patients with progressing disease on a case-by-case basis (I. Rivière, personal communication).
At the same meeting, some other clinical advances were reported. Bruce Levine, at the University of Pennsylvania, reported encouraging results of CLL patients treated with second-generation (expressing the domains of 4-1BB and ζ chain) anti-CD19 CARs leading to outstanding reductions in lymphocyte counts after T-cell infusions (B. Levine, personal communication). Some trials led by Rosenberg and Kochenderfer at the NCI are also using second-generation CARs (CD28 and ζ chain) against CD19 for the treatment of CLL and lymphoma patients [Citation10], with one trial reporting recently that clinical responses have been achieved, although the trial still has to be concluded (data updated by J. Yang at the ATTACK meeting, personal communication).
Morgan and colleagues reported recently a serious adverse event following the administration of T cells transduced with a CAR-recognizing ErbB2. The protocol was designed to treat colon cancer and the patient received 1010 cells expressing anti-ErbB2 third-generation CARs (CD28-4-1BB-ζ). The patient presented respiratory distress and died after 5 days despite supportive care. High levels of cytokines were detected and CAR-modified T cells were found infiltrating the lungs. The proposed mechanism for the lung failure observed includes the administration of high numbers of CAR+ lymphocytes, the pattern of circulation of the transferred cells (that first circulate through the lung and then migrate to other tissues), and the expression of the target antigen in the lung. This report represents the best-documented off-target serious adverse event in clinical trials with CAR+ T lymphocytes until now [Citation12].
Despite the encouraging results reported so far, some aspects of the treatments using CARs should still be improved to make the therapy as safe and effective as possible. The serious severe adverse effects reported recently outline the deleterious potential of off-target responses. The development of alternative targets or safety switches is mandatory for further improvement in clinical results.
Other aspect relevant to the maintenance of CAR+ cells in vivo is the target subpopulation of T cells selected for gene modification. At least one clinical trial is pursuing this issue, as mentioned before [Citation55]. The potential immune responses to transduced CARs sequences are another point of concern, since some immune responses against CARs have been reported [Citation7].
The issue of acute toxicity due to CAR activation soon after infusion is a major concern, which has been raised earlier and was reinforced by recent adverse effects. Given the observed severe adverse events (SAEs) and the number of ongoing trials, a recent symposium of the U.S. Recombinant DNA Advisory Comitee (RDAC) discussed methods and guidelines to improve safety and efficacy of CAR-based treatment [Citation81]. Among the proposed controls are inter- or intrapatient dose-escalation schemes. Also proposed is a combination of dose escalation with first- and second- (or third-) generation CARs, moving to the next-generation CAR when no toxicity is observed in the first- or second-generation molecule. For direct testing of third-generation CAR, it is advised to first test very conservative dose-escalating strategies. One alternative approach mentioned in the report would be to split T-cell dose over 2 or more days, constantly monitoring for indications of toxicity to prevent further infusions that could induce massive responses.
It is clear that suicide genes will not be able to prevent the tissue damaging generated by rapid acute off-target toxicities, since they take some time to be activated and the acute toxicity occurs within minutes [Citation12, Citation81]. This approach can be useful for late toxicities instead. Although not mentioned in this report, preventively blocking the target antigen with mAb can be an alternative to saturate potential targets, while leaving tumor-associated high-level target expression available for CAR recognition when a tumor expresses higher levels of the target antigen [Citation7] One last approach that will be discussed in detail in the last part of this review is the use of conditional response based on panels of antigens and cell signaling provided by more than one CAR.
Strategies to Circumvent CAR Therapy Limitations
The recent adverse effects reported for the ErbB2 clinical trial outline the relevance of careful T-cell dose escalation for phase I trials. Not only the total number of infused T lymphocytes must be considered, but also the possibility of fragmentation of the total number of T cells administered. Performing repeated infusions with a lower number of cells could play a role in reducing the deleterious effects of massive T-cell activation in specific organs, as seen in the lung of a patient who died in the trial using the CAR against ErbB2 [Citation12]. The strategy of blocking the availability of the antigen in tissues expressing lower levels of the antigen, as performed in the CAIX clinical trial, represents an interesting attempt to lower off-target side effects and could be exploited in other contexts like ErbB2 clinical trials.
In any case, unpredicted deleterious effects after CAR+ T-cell infusions are always a concern and the field is rapidly moving toward the inclusion of suicide gene systems in CAR-transduced cells. Different suicide gene systems have been proposed, such as the HSV-TK gene [Citation82], already in phase III trial; the CASPASE 9 safety switch, currently under clinical validation [Citation83]; and the CD20/rituximab suicide system [Citation84, 85], with recent encouraging preclinical validation of this particular system [Citation86].
One of the most important aspects of CAR-mediated gene therapy is the choice of the target antigen. Although the perfect target surface antigen seems almost impossible to find, some antigens should present less pronounced off-target clinical effects than others. The B-cell-restricted CD20 surface antigen, for example, should be a less deleterious off-target antigen than ErbB2, since B-cell depletion is not a life-threatening condition, while CAR recognition of the ErbB2 in vital target organs can lead to severe side effects. Proposed first by Kershaw and colleagues [Citation87] and, more recently, by Varela-Rohena and colleagues [Citation88], the possibility of combining different target antigens for restricting the range of target cells seems promising. A recent study proposed to enhance the specificity for the tumor tissue by modifying T cells with two separate CARs able to recognize different targets and containing either the cytoplasmic domain FceRI or both CD3z and CD28 [Citation89]. The amount of IFN-g produced by the modified T cells and the cytotoxic effects observed were increased in the lymphocytes expressing two CARs when incubated with antigen double-positive target cells in comparison to single-positive target cells. Our group is exploiting a similar approach by combining activating and inhibitory CARs [Citation90]. Our preliminary data indicates that in Jurkat cells, engagement of inhibitory anti CD20 CARs bearing CTLA-4, PD-1, or BTLA signaling domains is capable of inhibiting the massive NFAT-mediated transactivation of luciferase promoted by the engagement of a second-generation activating CAR (anti-CD19-4-1BBzeta).
Some efforts are being made to make a conditional response based on different thresholds of activation due to complementary signal domain triggering or enhanced signaling [Citation89; Sodre and Bonamino, unpublished data], as described in . These efforts have shown few results so far but represent one of the promising approaches to limit acute toxicities and narrowing anti-tumor responses to tumor cells. We are currently testing one CAR carrying the zeta chain and a second CAR bearing CD28, 4-1BB, or OX40 signaling domains alone or in combination.
FIGURE 2 Strategies to circumvent off-target effects. The off-target effects can be minimized through (a) a suicide gene system to delete remaining cells that recognize the target antigen; (b) splitting the activation endodomains (signal 1 and signal 2) in two different CARs to enable a complete activation response only after interaction of both CARs with their target antigens at the tumor surface. A similar approach was published by Duong et al. and consisted of using two different CARs to promote more potent responses against target cells expressing two tumor antigens [Citation89]; (c) the use of inhibitory CARs, besides an activating receptor, to impair T-cell activation after interaction with the target antigen at the normal tissues; (d) fine tuning of CAR affinity based on the target antigen level of expression. In an ideal situation, it is possible that a lower affinity would lead to recognition of tumor cells expressing high levels of antigen, sparing normal cells with low expression of the same antigen.
![FIGURE 2 Strategies to circumvent off-target effects. The off-target effects can be minimized through (a) a suicide gene system to delete remaining cells that recognize the target antigen; (b) splitting the activation endodomains (signal 1 and signal 2) in two different CARs to enable a complete activation response only after interaction of both CARs with their target antigens at the tumor surface. A similar approach was published by Duong et al. and consisted of using two different CARs to promote more potent responses against target cells expressing two tumor antigens [Citation89]; (c) the use of inhibitory CARs, besides an activating receptor, to impair T-cell activation after interaction with the target antigen at the normal tissues; (d) fine tuning of CAR affinity based on the target antigen level of expression. In an ideal situation, it is possible that a lower affinity would lead to recognition of tumor cells expressing high levels of antigen, sparing normal cells with low expression of the same antigen.](/cms/asset/a826426d-aad6-4bb4-b86c-7348297e9115/iiri_a_595855_f0002_b.gif)
Another interesting possibility is to fine tune the CAR affinity by performing screening on mutation libraries so the response, which is antigen dose dependent [Citation91], will mainly happen when the antigen expression at the surface of the cell is above a certain threshold (). This approach will probably be valid at certain ranges of CAR affinity, as it has been demonstrated that affinities above a specific threshold lead to loss of selectivity [Citation92]. This strategy could be useful in the context of the ErbB2 trial-related adverse event or when the antigen expression levels are very different between tumor and healthy cells (as in the case of the CAIX-based trial), but has the disadvantage of potentially losing target tumor cells expressing low levels of antigen. However, this approach may still induce acute toxicity, as observed in some published trials [Citation11, 12].
Regarding the reported immune responses against CARs in human patients, a straightforward approach to circumvent this limitation is humanizing the CAR sequence. Many groups are adopting this strategy for the new clinical trials [Citation8, Citation10, Citation12]. Humanization of mAbs or CAR sequences generally results in modifications in the target affinity requiring selection of higher affinity clones by random mutagenesis. This often generates a collection of molecules with wide affinity constants and these CAR molecules with different affinities could be used to validate the fine-tuning affinity approach mentioned above.
One last strategy that could be considered is the utilization of T cells transfected with in vitro transcribed RNA instead of the stable retrovirus transduction of the T cells. Carl June reported recently at the 2010 ATTACK meeting that 1010 cells could be generated in a single transfection with this approach. The CAR+ cells can be frozen for further utilization and cells generated in this way can promote relevant clinical responses. The advantage of this approach is that the transient expression of the CAR could theoretically preclude long-term toxicities, probably avoiding the need of suicide genes in the transferred T cells, since CAR expression should be self-limited.
One strategy is to modify T cells with T-cell receptors (TCRs) specific for tumor-associated antigens (TAAs) as a way to generate tumor-specific T cells. This strategy proved successful in preclinical studies, and two clinical trials from Rosenberg's group reported its use for the treatment of metastatic melanoma. The first study used an anti-MART-1 TCR to treat 17 patients and obtained an objective response rate of 12% [Citation93]. In the second study, the patients were treated with T cells modified with a high-affinity anti-MART-1 TCR or an anti-gp100 TCR, obtaining response rates of 30% (20 patients) and 19% (16 patients), respectively [Citation94]. These observed response rates are lower than those reported by the same group using TILs, which achieved responses up to 72% [Citation1]. This fact might be explained by the polyclonal nature of TILs, which might enhance the response and restrict the emergence of escape variants.
The use of this strategy is associated with several disadvantages. HLA restriction requires the isolation of TCRs matched to at least the most common haplotypes and the HLA typing for each patient. The nature of TCR allows only the recognition of proteins, excluding carbohydrates and lipids from the possible tumor targets. Downregulation of HLA expression is commonly observed in tumors, impairing the recognition by T lymphocytes. Tumor cells rarely express co-stimulatory ligands, leading to an incomplete activation of transgenic TCR expressing cells. Finally, there is a risk of mispairing between the transgenic TCR and the endogenous TCR, creating a hybrid TCR with unknown specificity that could result in an autoimmune disease. A recent paper demonstrated that this can happen in animal models [Citation95], although it has not been observed so far in clinical trials [Citation96]. The design of CARs allows it to overcome most of these disadvantages, although CARs only recognize membrane antigens. Some of these disadvantages can be overcome by new strategies, such as the one developed by Stewart-Jones and colleagues, that have recently created a Fab fragment of mAbs against HLA*0201/NY-ESO1157-165 MHC–peptide complex [Citation15]. Using the Fab domain of these new mAbs in CARs could expand the pattern of recognition of CARs to target intracellular epitopes such as NY-ESO1. This particular antigen has presented very safe clinical profiles after gene therapy clinical protocols with transferred CTLs or even TCR genes without any noticeable off-target effects against this molecule (J. Yang, personal communication). A recent clinical trial with lymphocytes carrying a TCR against NY-ESO1 showed regressions of metastatic melanomas and synovial cell sarcomas without adverse events related to T-cell infusions [Citation97]. NY-ESO1 should be the first target to be tested using CARs specific for HLA peptide. The possibility of fine tuning the response to phosphorylated proteins could add a new layer of specificity, and perhaps safety, to this approach, further widening clinical CAR application.
CONCLUSIONS
Chimeric antigen receptors have already been shown to be useful tools for cancer therapy in proof of principle clinical trials. Although the field still awaits consensus regarding some aspects of CAR design and signaling domains, published data on clinical trials and preliminary data reported for ongoing trials support an extended clinical validation of these molecules and the design of new approaches to enhance CAR selectivity, anti-tumor activity, and safety. Future clinical studies should validate, through careful study designs, new approaches comparing different aspects of CAR-based therapy, such as signaling domains, lymphocyte subpopulations for gene transfer, and additional approaches to enhance lymphocyte function that can be used to further potentiate the effects of CAR gene transfer.
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Dudley ME, Yang JC, Sherry R, Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239.
- Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. 1989;21(1 Pt 1):127–130.
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724.
- Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia. 2007;21(9):1859–1874.
- Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172(1):104–113.
- Giordano Attianese GM, Marin V, Hoyos V, In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood. 2011;117(18):4736–4745.
- Lamers CH, Sleijfer S, Vulto AG, Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–e22.
- Till BG, Jensen MC, Wang J, Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–2271.
- Pule MA, Savoldo B, Myers GD, Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270.
- Kochenderfer JN, Wilson WH, Janik JE, Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102.
- Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18(4):666–668.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851.
- Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8(21):1601–1608.
- Willemsen RA, Ronteltap C, Chames P, Debets R, Bolhuis RL. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol. 2005;174(12):7853–7858.
- Stewart-Jones G, Wadle A, Hombach A, Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A. 2009;106(14):5784–5788.
- Li Y, Depontieu FR, Sidney J et al. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J Mol Biol. 2010;399(4):596–603.
- Petersen J, Wurzbacher SJ, Williamson NA, Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U S A. 2009;106(8):2776–2781.
- Depontieu FR, Qian J, Zarling AL, Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc Natl Acad Sci U S A. 2009;106(29):12073–12078.
- Altenschmidt U, Kahl R, Moritz D, Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clin Cancer Res. 1996;2(6):1001–1008.
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166.
- Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938–6949.
- Bridgeman JS, Hawkins RE, Hombach AA, Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther. 2010;10(2):77–90.
- Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181(5):1653–1659.
- Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000;96(5):1999–2001.
- Roberts MR, Cooke KS, Tran AC, Antigen-specific cytolysis by neutrophils and NK cells expressing chimeric immune receptors bearing zeta or gamma signaling domains. J Immunol. 1998;161(1):375–384.
- Haynes NM, Snook MB, Trapani JA, Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol. 2001;166(1):182–187.
- Heuser C, Hombach A, Losch C, Manista K, Abken H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003;10(17):1408–1419.
- Zhao Y, Wang QJ, Yang S, A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183(9):5563–5574.
- Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans RP. Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia. 2000;2(5):449–459.
- Stephan MT, Ponomarev V, Brentjens RJ, T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440–1449.
- Gade TP, Hassen W, Santos E, Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65(19):9080–9088.
- Brentjens RJ, Santos E, Nikhamin Y, Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426–5435.
- Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol. 2010;11(1):21–27.
- Cooper LJ, Al-Kadhimi Z, Serrano LM, Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105(4):1622–1631.
- Savoldo B, Rooney CM, Di Stasi A, Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110(7):2620–2630.
- Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3(12):939–951.
- Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285.
- Brentjens RJ, Latouche JB, Santos E, Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3): 279–286.
- Kowolik CM, Topp MS, Gonzalez S, CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66(22):10995–11004.
- Carpenito C, Milone MC, Hassan R, Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–3365.
- Milone MC, Fish JD, Carpenito C, Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464.
- Zhong XS, Matsushita M, Plotkin J, Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–420.
- Yvon E, Del Vecchio M, Savoldo B, Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15(18):5852–5860.
- Savoldo B, Ramos CA, Liu E, CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5): 1822–1826.
- Hoyos V, Savoldo B, Quintarelli C, Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–1170.
- Di Stasi A, De Angelis B, Rooney CM, T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392-6402.
- Craddock JA, Lu A, Bear A, Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–788.
- Moon EK, Carpenito C, Sun J, Expression of a functional CCR2 receptor enhances tumor localization and eradication by human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. Published online May 24, 2011, doi: 10.1158/1078-0432.CCR-11-0351.
- Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305.
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102(27):9571–9576.
- Singh H, Manuri PR, Olivares S, Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68(8):2961–2971.
- Gattinoni L, Klebanoff CA, Palmer DC, Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115(6):1616–1626.
- Yang S, Luca G, Liu F, In vitro generated anti-tumor T lymphocytes exhibit distinct subsets mimicking in vivo antigen-experienced cells. Cancer Immunol Immunother. 2011;60(5):739–749.
- Neeson P, Shin A, Tainton KM, Ex vivo culture of chimeric antigen receptor T cells generates functional CD8+ T cells with effector and central memory-like phenotype. Gene Ther. 2010;17(9):1105–1116.
- Popplewell L. Genetically engineered lymphocyte therapy after peripheral blood stem cell transplant in treating patients with high-risk, intermediate-grade, B-cell non-Hodgkin lymphoma. National Clinical Trials [Internet]. 2011 Mar 16; NCT01318317. Available from: clinicaltrials.gov/ct2/show/ record/NCT01318317
- Hinrichs CS, Borman ZA, Cassard L, Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106(41):17469–17474.
- Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–383.
- Muller T, Uherek C, Maki G, Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57(3):411–423.
- Marin V, Dander E, Biagi E, Characterization of in vitro migratory properties of anti-CD19 chimeric receptor-redirected CIK cells for their potential use in B-ALL immunotherapy. Exp Hematol. 2006;34(9):1219–1229.
- Gong Q, Ou Q, Ye S, Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005;174(2):817–826.
- Chan CH, Stanners CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol Ther. 2004;9(6):775–785.
- Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875–3886.
- Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural expression of the CD19 antigen impacts the long-term engraftment but not antitumor activity of CD19-specific engineered T cells. J Immunol. 2010;184(4):1885–1896.
- Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7(2):118–130.
- Lee JC, Hayman E, Pegram HJ, In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011;71(8):2871–2881.
- Pike-Overzet K, van der Burg M, Wagemaker G, van Dongen JJ, Staal FJ. New insights and unresolved issues regarding insertional mutagenesis in X-linked SCID gene therapy. Mol Ther. 2007;15(11):1910–1916.
- Huang X, Guo H, Kang J, Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. 2008;16(3):580–589.
- Manuri PV, Wilson MH, Maiti SN, piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21(4):427–437.
- Jin Z, Maiti S, Huls H, The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011.
- Bonini C, Grez M, Traversari C, Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9(4):367–369.
- Newrzela S, Cornils K, Li Z, Resistance of mature T cells to oncogene transformation. Blood. 2008;112(6):2278–2286.
- DeKelver RC, Choi VM, Moehle EA, Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20(8):1133–1142.
- Zou J, Sweeney CL, Chou BK, Oxidase deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease mediated safe harbor targeting. Blood. 2011.
- Zhao Y, Moon E, Carpenito C, Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061.
- Mitsuyasu RT, Anton PA, Deeks SG, Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96(3):785–793.
- Wang J, Press OW, Lindgren CG, Cellular immunotherapy for follicular lymphoma using genetically modified CD20-specific CD8+ cytotoxic T lymphocytes. Mol Ther. 2004;9(4):577–586.
- Park JR, Digiusto DL, Slovak M, Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833.
- Kershaw MH, Westwood JA, Parker LL, A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115.
- Brentjens R, Riviere I. Laboratory-treated T cells with or without cyclophosphamide in treating patients with refractory chronic lymphocytic leukemia. National Clinical Trials [Internet]. 2007 Apr 25; NCT00466531. Available from: clinicaltrials.gov/ct2/show/NCT00466531
- Brentjens R. Precursor B cell acute lymphoblastic leukemia (B-ALL) treated with autologous T cells genetically targeted to the B cell specific antigen CD19. National Clinical Trials [Internet]. 2010 Jan 6; NCT01044069. Available from: clinicaltrials.gov/ct2/show/NCT01044069
- Ertl HC, Zaia J, Rosenberg SA, Considerations for the clinical application of chimeric antigen receptor T cells: observations from a Recombinant DNA Advisory Committee symposium held June 15, 2010. Cancer Res. 2011;71(9):3175–3181.
- Bonini C, Ferrari G, Verzeletti S, HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724.
- Di Stasi A, Tey SK, Fujita Y, CASPALLO: Phase I clinical trial of allodepleted T cells transduced with inducible caspase 9 suicide gene after haploidentical stem cell transplantation. ASH Annual Meeting Abstracts 2010;116(21):559–.
- Serafini M, Bonamino M, Golay J, Introna M. Elongation factor 1 (EF1alpha) promoter in a lentiviral backbone improves expression of the CD20 suicide gene in primary T lymphocytes allowing efficient rituximab-mediated lysis. Haematologica. 2004;89(1):86–95.
- Serafini M, Manganini M, Borleri G, Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther. 2004;15(1):63–76.
- Vogler I, Newrzela S, Hartmann S, An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther. 2010;18(7):1330–1338.
- Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5(12):928–940.
- Varela-Rohena A, Carpenito C, Perez EE, Genetic engineering of T cells for adoptive immunotherapy. Immunol Res. 2008;42(1-3):166–181.
- Duong CP, Westwood JA, Berry LJ, Darcy PK, Kershaw MH. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy. 2011;3(1):33–48.
- ESGCT Poster presentations. XVIII Annual Congress of the European Society of Gene and Cell Therapy. Hum Gene Ther. 2010; 21:1397–1488.
- Turatti F, Figini M, Balladore E, Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. 2007;30(7):684–693.
- Chmielewski M, Hombach A, Heuser C, T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647–7653.
- Morgan RA, Dudley ME, Wunderlich JR, Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129.
- Johnson LA, Morgan RA, Dudley ME, Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546.
- Bendle GM, Linnemann C, Hooijkaas AI, Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565–570, 1p following 70.
- Rosenberg SA. Of mice, not men: no evidence for graft-versus-host disease in humans receiving T-cell receptor-transduced autologous T cells. Mol Ther. 2010;18(10):1744–1745.
- Robbins PF, Morgan RA, Feldman SA, Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924..
- Guest RD, Hawkins RE, Kirillova N, The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211.